Introduction
All-trans retinoic acid (RA) is a derivative
of vitamin A, which is required for development, vision and immune
function (1–3). As a signaling molecule, RA affects
target gene transcription through retinoid receptor-mediated
mechanisms (4). There are two
families of nuclear retinoid receptors: retinoic acid receptors
(RARs) and retinoid X receptors (RXRs). RXR/RAR heterodimers and
RXR homodimers exist; these respective complexes bind to defined
retinoic acid response elements (RAREs) in the promoter regions of
retinoid target genes, as reviewed (4). In the absence of RA-treatment, these
receptors basally associate with an inhibitory co-repressor complex
and upon RA-treatment a stimulatory co-activator complex is
recruited that leads to chromatin remodeling and retinoid target
gene transcription, as reviewed (4).
In addition to its physiological roles, RA is also
used as therapy for acute promyelocytic leukemia (APL) (4,5).
RA-treatment of APL is a successful example of differentiation
therapy. The majority of clinical APL cases exhibit a balanced
chromosomal translocation t(15;17), resulting in a fusion protein
between the promyelocytic leukemia (PML) and the retinoic acid
receptor-α (RARα) gene products (6,7).
This fusion protein retains the ability to bind to an RARE, but
also has a strong association with its co-repressor complex, as
reviewed (7). Physiological
retinoid levels are not able to dissociate the co-repressor
complex, resulting in transcriptional repression of retinoid target
genes. Since these target genes are critical for induced cellular
differentiation, basal repression of transcription of these species
can block maturation of immature promyelocytes, leading to APL
(7). In contrast, pharmacological
concentrations of RA can overcome the inhibitory association
between the co-repressor complex and the PML/RARα fusion protein
and can recruit a stimulatory co-activator complex that leads to
the transcription of retinoid target genes. One consequence is
retinoid-induced degradation of the PML/RARα fusion protein, as
reviewed (8). Together, these
pathways contribute to the maturation of APL cells and clinical
remission of APL patients.
Clinical use of retinoids is limited by toxicity and
resistance (4,7). In a search for retinoid target genes
that could serve as candidate therapeutic targets in APL, the G0/G1
switch gene 2 (G0S2) was found. G0S2 is one of the most rapid and
prominently-induced RA target genes in APL (9,10).
G0S2 is a small basic protein with 103 amino acids (11). It does not have apparent homology
to other proteins and its functions are under intensive study. The
G0S2 gene was discovered in a screen to identify species regulated
in the lectin-induced G0 to G1 cell cycle change of human
peripheral blood mononuclear cells (11). However, its precise role in cell
cycle regulation has been elusive (11). G0S2 is expressed in white and brown
adipose tissue; it is highly expressed in the liver, heart and
skeletal muscle (12,13).
G0S2 is a regulator of lipolysis (13). It is a target of the
peroxisome-proliferator-activated receptor γ (PPARγ) in adipocytes
and G0S2 is upregulated in adipogenesis (12). G0S2 is also known to regulate
adipose lipolysis through its inhibition of adipose triglyceride
lipase (ATGL) activity (13). In
settings of high metabolic demand, ATGL mediates hydrolysis of
triglyceride (TAG) stored in lipid droplets of adipocytes to
diglyceride (DAG) and free fatty acid (FFA) for subsequent energy
use. It is through its hydrophobic domain (HD) that G0S2 binds to
ATGL, which can inhibit lipolysis (13). As expected, G0S2 knockdown was
found to enhance lipolysis in adipocytes, whereas G0S2
overexpression reduced lipolysis; this resulted in TAG accumulation
and an increase in lipid droplet size (13).
G0S2 is involved in diverse cellular activities. For
example, G0S2 is upregulated after treatment with the lymphocyte
mitogen lectin and downregulated in peripheral blood mono-nuclear
cells by treatment with the immunosuppressive agent cyclosporine
(11,14). G0S2 is also upregulated in
peripheral blood or bone marrow-derived mononuclear cells isolated
from patients with different autoimmune diseases, including
psoriasis, rheumatoid arthritis, vasculitis and lupus (15–17).
Although engineered G0S2 transgenic mice did not exhibit evidence
for an autoimmune disease, these mice did have autoimmunity-related
antibodies in their serum (17).
Together, these findings implicated a role for G0S2 in immune
regulation.
G0S2 was proposed to act as a tumor suppressor. This
hypothesis came about from evidence for hypermethylation of the
G0S2 promoter that conferred its silencing in head and neck
squamous cell carcinomas (18,19)
and squamous cell lung carcinomas (20,21).
G0S2 overexpression also augmented apoptosis in lung and colon
cancer cells by interacting with Bcl-2, which in turn antagonized
the formation of anti-apoptotic Bcl-2/Bax heterodimers (22). These studies were consistent with a
tumor suppressive role for G0S2.
In this study, G0S2 was shown to be induced in APL
cells after treatment with RAR, but not RXR agonists. A previously
unrecognized function of G0S2 was uncovered. G0S2 was found to
repress exogenous gene expression and reporter activity. Yet, G0S2
did not affect endogenous expression of the examined species. These
inhibitory effects were not restricted to APL cells, but were also
detected in diverse cellular contexts, including those that were
retinoid differentiation-responsive or not. The studies reported
here indicate that these inhibitory G0S2 effects were mediated
through an overlapping domain that conferred ATGL repression and
altered G0S2 subcellular localization. Yet, this new G0S2 function
was not rescued by gain of ATGL expression or mimicked by
antagonizing ATGL activity. Thus, these findings revealed that
these G0S2 effects are independent of its previously recognized
role in regulating ATGL activity. The biological implications of
this G0S2 repression are discussed.
Materials and methods
Cell culture and reagents
Cell lines were cultured in their respective media
supplemented with penicillin (100 U/ml) and streptomycin (100
μg/ml) (Mediatech, Manassas, VA) in a humidified incubator
at 37°C with 5% CO2. The NB4 human APL cell line
(9) was cultured in advanced
RPMI-1640 media supplemented with 2% fetal bovine serum (FBS) and 4
mM L-glutamine. BEAS-2B immortalized human bronchial epithelial
cells were cultured in LHC-9 media, as before (23). The human 293T embryonic kidney cell
line (ATCC, Manassas, VA) was cultured in DMEM media supplemented
with 10% FBS. The multipotent NTERA-2 clone D1 (NT2/D1) human
embryonal carcinoma cells were cultured in DMEM media supplemented
with 10% FBS and 2 mM L-glutamine (24). Murine ED-1 (25) and the human A549 (ATCC) lung cancer
cell lines were each cultured in RPMI-1640 media supplemented with
10% FBS. To engineer cell lines with stable G0S2 expression, ED-1
cells were transduced with a G0S2 lentivirus (Addgene, Cambridge,
MA) (designated as ED-1-G0S2) and comparisons were made to an
insertless control lentivirus (ED-1-vector). Cells were then
selected in media supplemented with blasticidin S HCl (17.43
μM, Invitrogen, Grand Island, NY) in RPMI-1640 media
supplemented with 10% FBS.
Independent retinoid and rexinoid effects
on G0S2 expression
NB4 APL cells were individually treated for 2 days
with the RAR (RA, 1 μM) or RXR (LG268, 1 μM; Ligand
Pharmaceutical, La Jolla, CA) agonists. The proteasome inhibitors
MG132 (Calbiochem) and ALLN (Calbiochem), protease inhibitors PMSF
(Sigma, 1 mM) and EDTA (Sigma, 1 mM) and the lysosomal inhibitor
NH4Cl (Sigma, 2 mM) were each purchased. Five hours after
transfection, the original transfection medium was removed and
replenished with fresh media supplemented individually with each of
these inhibitors, except for the protea-some inhibitors (MG132, 10
μM and ALLN, 50 μM), which were each added 44 h after
transfection. Luciferase assays were performed 48 h after
transfection.
Plasmids and siRNAs
For luciferase assay experiments, pRL-TK (Promega,
Madison, WI), pGL3E (Promega), βRARE-TK-luc (26), pGL3-UBE1L-TK-luc (27) and pGL3-G0S2-FL-luc (9) were respectively used as reporter
constructs. For G0S2 gain of expression experiments with the
CMV-myc-G0S2 (myc-G0S2) vector (9), the results were compared to its empty
vector (CMV-myc ΔHD) as a control (9). The ΔHD G0S2 mutation of CMV-myc-G0S2
[myc-G0S2 was generated by polymerase chain reaction (PCR) assays
with deletions accomplished using primers that flanked the region
to be deleted in the full length CMV-myc-G0S2 vector. Primer
sequences were: forward primer
5′-GATGGTGAAGCTGATGGAGACTGTGTGCAGC-3′ and reverse primer
5′-CACAGTCTCCATCAGCTTCACCATCTTCCC-3′. For ATGL engineered
overexpression experiments, the pCMV-SPORT6-ATGL vector (Thermo
Fisher, Rockford, IL) was used to overexpress murine ATGL. An empty
vector pCMV-SPORT6 served as a control vector. Target sequence for
G0S2 siRNA (Thermo Fisher) was: 5′-AGATGGTGAAGCTGTACGT-3′. The
target sequences for ATGL siRNAs (Thermo Fisher) were: human ATGL
siRNA1: 5′-GTAAAGATCATCCGCAGTT-3′ and human ATGL siRNA2:
5′-GGGCGAGAGTGACATCTGT-3′; and for murine ATGL siRNA1: 5′-GAAATTGG
GTGACCATCTG-3′; and murine ATGL siRNA2: 5′-GGAGAGAACGTCATCATAT-3′.
A non-targeting RISC-free siRNA (Thermo Fisher) was used as a
control.
Transient transfection and luciferase
assays
NB4 APL cells were transiently co-transfected with
myc-G0S2 or a corresponding empty vector control with the indicated
luciferase construct using the AMAXA cell line Nucleofector kit V
(Lonza, Basel, Switzerland) according to the manufacturer’s
protocol. Following transfection, cells were plated at
2×106 cells/ml in individual wells of a 12-well tissue
culture plate and treated with RA (1 μM) or dimethyl
sulfoxide (DMSO) as vehicle control for 6 h. Cells were then
harvested in Passive Lysis Buffer as part of the Dual-Luciferase
Reporter Assay System kit (Promega). Analyses for luciferase
activities were performed according to the manufacturer’s
recommended protocol and luciferase activity was measured with a
TD-20/20 Luminometer (Promega). Renilla luciferase activity was
also measured. To normalize for total protein, total protein
concentrations within studied cell lysates were measured using the
BCA protein assay kit (Thermo Fisher). Luciferase activities were
normalized to the respective cellular protein concentrations and
activities were subsequently normalized to the vehicle-treated
insertless vector experimental arm. Similar transfection
efficiencies were confirmed by co-transfecting fluorescein-labeled
siRNA or GFP in desired cells and then by measuring fluorescein or
GFP-positive cells using flow cytometry (Becton Dickinson FACScan
cytometer, Franklin Lakes, NJ or MACSQuant VYB, Miltenyi Biotec,
Bergisch Gladbach, Germany). Cell lysates were harvested in
radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher)
supplemented with protease arrest (GBioscience, St. Louis, MO) for
immunoblot analysis to confirm that G0S2 knockdown or engineered
overexpression was achieved in the desired cells.
BEAS-2B, NT2/D1, ED-1, A549, ED-1-G0S2 and
ED-1-vector cells were individually plated at densities of
2×105, 2×105, 3.5×104 to
1×105, 6×105, 2×105 and
2×105 cells/well in 6-well tissue culture plates,
respectively. BEAS-2B cells were transiently transfected the next
day with indicated constructs using Fugene 6 (Roche, Indianapolis,
IN). ED-1, NT2/D1 and A549 cells were individually transfected with
Lipofectamine 2000 (Invitrogen). ED-1-G0S2 and ED-1-vector cells
were each transfected with TransIT-LT1 transfection reagent (Mirus,
Madison, WI) using the respective manufacturer’s protocol.
Twenty-four hours after transient transfection, the medium was
replaced with fresh medium supplemented respectively with RA or
DMSO as a vehicle for NT2/D1 and BEAS-2B cells, and with fresh
media for ED-1 and A549 cells. For oleic acid treatment
experiments, varying concentrations of oleic acid (Sigma, St.
Louis, MO) were added 24 h after transfection; cell lysates for the
NT2/D1, BEAS-2B, ED-1 and A549 cell lines were individually
harvested 48 h after transfection for luciferase activity analyses.
Cell lysates for stably transfected ED-1-G0S2 and ED-1-vector cells
were harvested 24 h after transfection to measure luciferase
activity and also placed in RIPA buffer for immunoblot
analyses.
Real-time PCR assays
To evaluate effects of G0S2 transient transfection
on endogenous gene expression, ED-1 cells were transfected with the
myc-G0S2 vector. RA or vehicle (DMSO) was added 24 h after
transfection. Total RNA was isolated 48 h after transfection using
TRIzol reagent (Invitrogen). Reverse transcription was performed
using the High Capacity cDNA Reverse Transcription Kit (Life
Technologies, Carlsbad, CA) with a Peltier Thermal Cycler (GMI,
Ramsey, MN). Real-time PCR assays were performed using SYBR-Green
PCR master mix (Life Technology) with the 7500 fast Real-time PCR
system (Life Technology). Primer sequences were as follows: murine
RARβ forward primer: 5′-CAGTGAGCTGGCCACCAAGT-3′; reverse primer:
5′-GCGATGGTCAGACCTGTGAA-3′; murine UBE1L forward primer:
5′-CTACGAGCGACTCCATATACCT-3′; reverse primer:
5′-TACACACAGGGTAGGGAGCAT-3′; murine G0S2 forward primer:
5′-AGTGCTGCCTCTCTTCCCAC-3′; reverse primer:
5′-TTTCCATCTGAGCTCTGGGC-3′; murine GAPDH forward primer:
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse primer:
5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Subcellular localization and immunoblot
analysis
For subcellular localization of endogenous G0S2, NB4
cells were plated at 105/ml and treated with RA (1 μM) for
48 h. Cells were then harvested and fractionated using the
Subcellular Protein Fractionation Kit according to manufacturer’s
protocol (Thermo Fisher, Rockford, IL). To confirm that the ΔHD
G0S2 mutant protein was of the expected size and to establish that
respective gain or loss of G0S2 or ATGL expression was achieved,
myc-G0S2 and myc-G0S2 ΔHD plasmids were individually transfected
into 293T cells. Human ATGL siRNAs were individually transfected
into A549 cells, and murine ATGL siRNA and the pCMV-SPORT6-ATGL
vector were each transfected into ED-1 cells. Cell lysates were
harvested 48 h after transfection in RIPA buffer (Thermo Fisher)
with protease arrest (GBioscience) added. Protein concentrations
were determined using the BCA Protein Assay Kit (Thermo Fisher).
Samples were run on SDS-PAGE gels and transferred to nitrocellulose
membrane, as before (9). Membranes
were individually probed with antibody recognizing G0S2 (9) to detect endogenous or stably
overexpressed G0S2 proteins with antibody recognizing myc (Covance,
Princeton, NJ) to individually detect myc-G0S2 and myc-G0S2 ΔHD,
with antibody recognizing ATGL (Cell Signaling, Danvers, MA) to
detect both human and mouse ATGL species, with antibody recognizing
transglutaminase II (TGase II) (Thermo Fisher), or with respective
antibodies that recognized UBE1L (9) or RARβ (Santa Cruz Biotechnology,
Santa Cruz, CA) proteins. Antibodies that recognized actin, COX-4
or nucleoporin (all from Santa Cruz Biotechnology) were used to
confirm similar protein loadings were achieved for the desired
subcellular immunoblot analyses.
Confocal microscopy
ED-1 cells were plated at a density of
3×104 cells/well on a poly-D-lysine (Sigma)-coated cover
slip in individual wells of a 12-well tissue culture plate. Cells
were transfected on the following day with the desired myc-tagged
G0S2 expression or control constructs using Lipofectamine 2000
(Invitrogen) according to the manufacturer’s protocol. Twenty-four
hours later, cells were fixed in 4% paraformaldehyde for 15 min,
washed with phosphate-buffered saline (PBS), incubated in 0.1%
Triton in PBS (PBT) for 10 min and blocked with 7.5% bovine serum
albumin (BSA) in PBS overnight at 4°C. Cells were subsequently
washed in 0.1% PBT solution and stained with an anti-myc antibody
(Convance, at 1:100 dilution) in 1% BSA in PBT for 45 min at room
temperature and with goat anti-mouse secondary antibody conjugated
to Alexa Fluor 647 fluorophor (Invitrogen, at 1:1,000 dilution) in
1% BSA in PBT for 30 min at room temperature. F-actin was stained
using Alexa Fluor 568 Phalloidin (Invitrogen, 1:40 dilution) in 1%
BSA in PBT for 20 min at room temperature in the dark. Coverslips
were gently washed with PBS three times before mounting onto slides
using Prolong Gold with DAPI (Invitrogen) staining. The slides were
viewed using a Zeiss LSM 510 confocal microscope (Zeiss,
Oberkochem, Germany) and representative images were obtained.
Statistical analysis
Two-sample t-tests were used for statistical
analyses using Microsoft Excel software, with significance defined
as a two-sided P<0.05.
Results
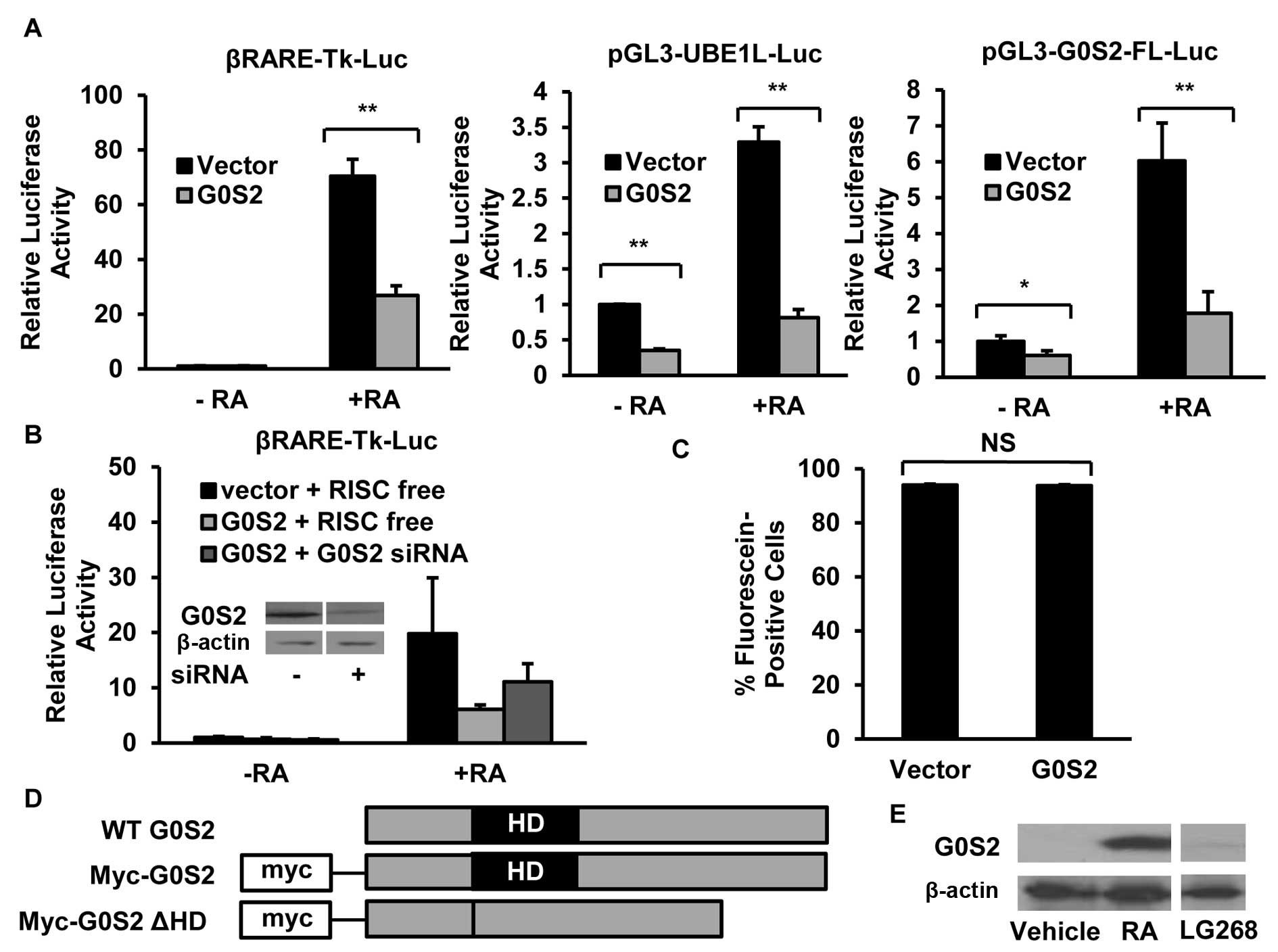
G0S2 inhibits exogenous reporter activity
of several retinoid-regulated genes
Our prior study identified G0S2 as a direct retinoid
target gene that was markedly induced after in vitro
RA-treatment of cultured NB4 APL cells and leukemic cells from APL
patients as well as after in vivo RA-treatment of transgenic
APL mice (9). The current study
confirmed and extended that prior work by showing that RAR but not
RXR agonists induced G0S2 expression in NB4 APL cells (Fig. 1E).
To investigate the role of G0S2 in
retinoid-dependent pathways, myc-tagged G0S2 was transiently
transfected into NB4 APL cells along with the respective luciferase
reporter constructs of the individual retinoid-regulated species:
RARβ, UBE1L or G0S2 itself. Notably, exogenous G0S2 expression
significantly (P<0.05) reduced individual reporter activity in
NB4 APL cells for each of the respective reporter constructs
(Fig. 1A). This repression was
observed in the presence and absence of RA-treatment (Fig. 1A). As expected, G0S2 knockdown by
an siRNA that targeted G0S2 for repression partially reversed these
inhibitory effects versus a control siRNA (Fig. 1B). This decline in luciferase
activity in NB4 cells was not due to changes in transfection
efficiency, as shown in Fig. 1C.
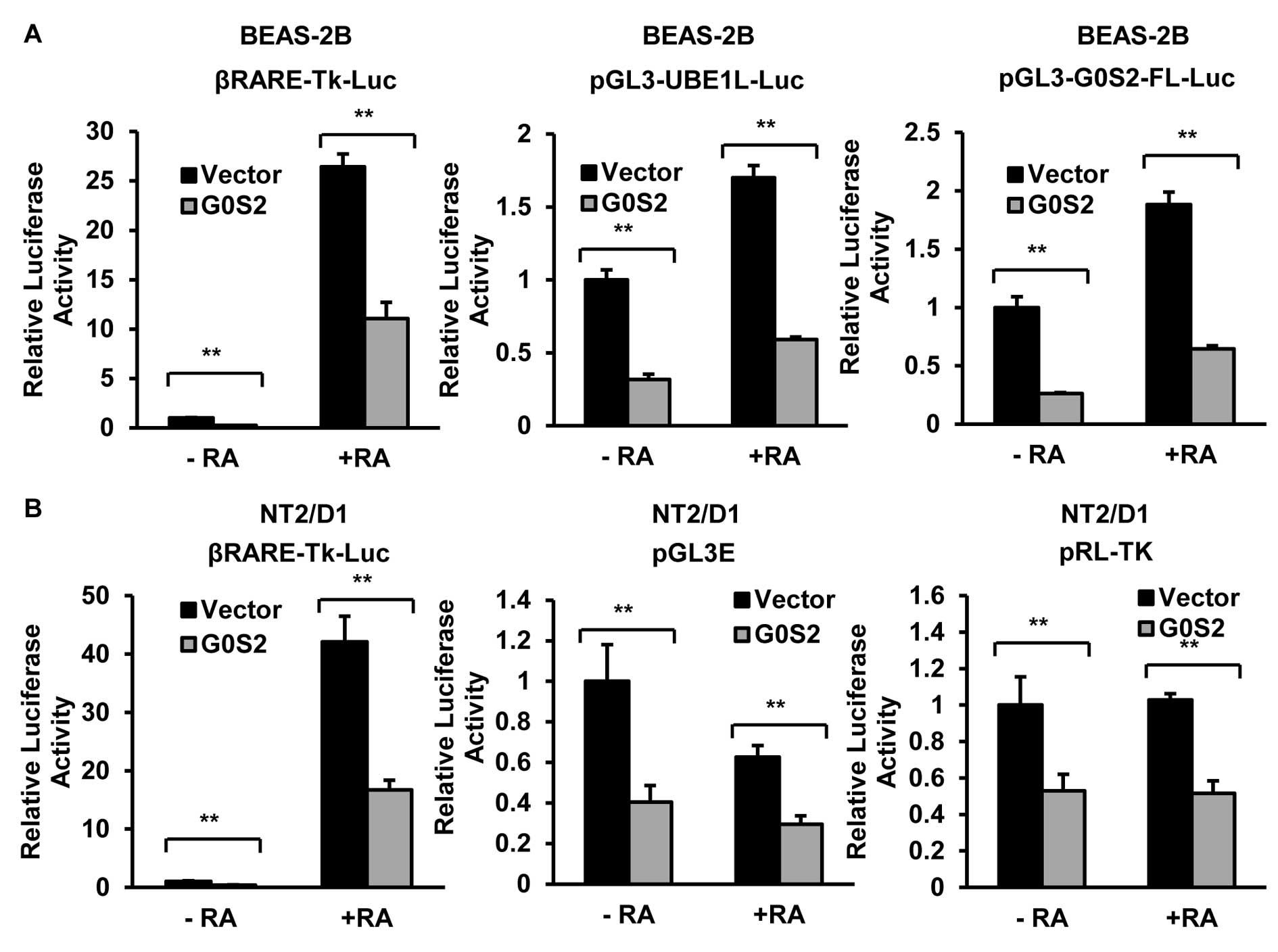
Similar inhibitory effects of G0S2 were observed in other
retinoid-responsive cell lines including BEAS-2B immortalized human
bronchial epithelial (23) and
NT2/D1 multipotent human embryonal carcinoma (24) cell lines (Fig. 2). This inhibition by G0S2 was
extended to include other reporter constructs that did not contain
retinoid responsive elements such as pGL3E (firefly), which was the
control vector for the indicated luciferase reporter constructs,
and pRL-TK (renilla), as shown in Fig.
2B.
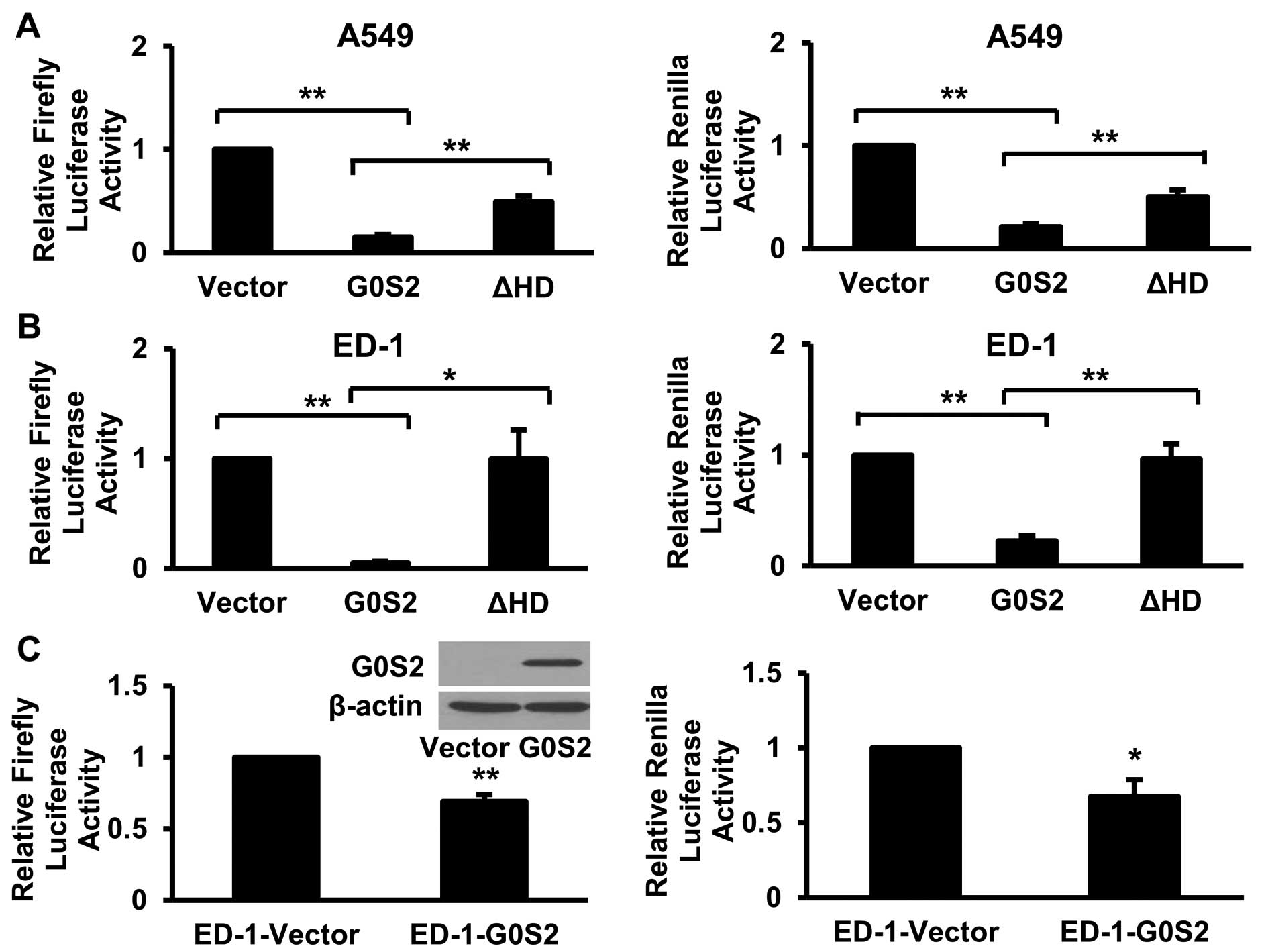
The G0S2 hydrophobic domain (HD) mediates
the G0S2 inhibitory effects
G0S2 was previously shown to require its HD domain
(Fig. 1D) to inhibit ATGL activity
(13). To explore whether this
domain was also important for G0S2 inhibitory effects, myc-G0S2 ΔHD
was individually co-transfected into ED-1 and A549 cells with
firefly or renilla luciferase constructs. The G0S2 mutation
designated as ΔHD in which the HD domain was removed had
significantly (P<0.05) less ability to repress either firefly or
renilla luciferase reporter activities than the full-length G0S2
species (Fig. 3A and B). To
exclude the possibility that removal of the HD domain destabilized
G0S2, immunoblot analysis was performed. The stability of this
mutant G0S2 protein was similar to that of wild-type G0S2 (data not
shown). Thus, the HD domain of G0S2 exerted this inhibitory effect
of G0S2.
Stable G0S2 expression inhibits reporter
plasmid activity
To exclude the possibility that the G0S2 inhibitory
effects were due to transient G0S2 co-transfection, G0S2 was stably
expressed in ED-1 cells (ED-1-G0S2) in Fig. 3C. The consequences of this on
luciferase reporter activities were next examined. As compared with
ED-1 cells that were stably transfected with an insertless vector
(designated as ED-1-vector cells), the G0S2 transductants
significantly (P<0.05) inhibited firefly or renilla luciferase
reporter activities (Fig. 3C). To
exclude the possibility that G0S2 transfection activated protein
degradation programs that would destabilize genetically-introduced
proteins, transient transfections of luciferase reporter plasmids
were repeated in 293T cells in the presence of transfected G0S2 and
co-treatment with individual proteasomal, protease, or lysosomal
inhibitors. These inhibitors did not abrogate the repressive
effects of G0S2 expression on transcriptional activity (data not
shown). Thus, induced protein destabilization did not appear to
confer these inhibitory effects of expressed G0S2.
G0S2 does not affect endogenous gene
expression
To investigate G0S2 effects on endogenous expression
of retinoid regulated species, G0S2 was transiently transfected
into ED-1 murine lung cancer cells. The expression of endogenous
RA-induced species was next examined after G0S2 transfection. In
contrast to the marked ability of G0S2 to inhibit the activity of
the individually co-transfected retinoid regulated RARβ, UBE1L and
G0S2 reporter plasmids, transiently transfected G0S2 did not
appreciably affect endogenous mRNA levels of these respective
species, as displayed in Fig. 4A.
Similarly, stable G0S2 overexpression in ED-1-G0S2 cells did not
affect endogenous RARβ or UBE1L protein expression as compared to
its control ED-1-vector transfected cells (Fig. 4B). In addition, siRNA-mediated
knockdown of the endogenous G0S2 expression (induced by
RA-treatment) in NB4 APL cells did not affect endogenous protein
expression of the retinoid regulated genes UBE1L or TGase II
(Fig. 4C). Together, these studies
extended prior findings that G0S2 inhibited exogenously introduced
reporter activities by showing that engineered gain or loss of G0S2
also did not affect endogenous expression of the examined
retinoid-augmented species.
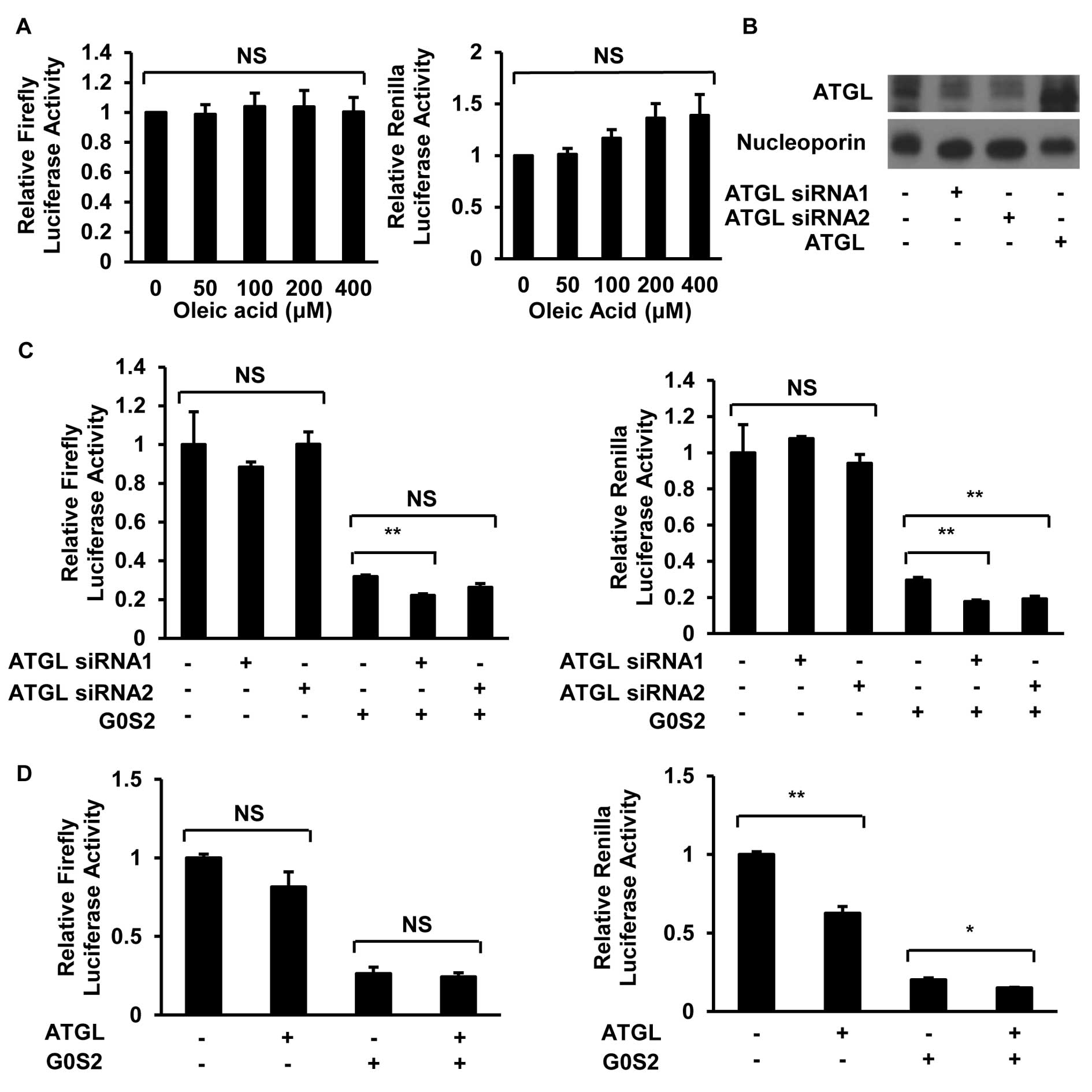
Changes in lipid droplet size or ATGL
levels do not affect G0S2 inhibitory effects
G0S2 interacts with and inhibits ATGL and this can
decrease lipolysis and increase cellular lipid droplet size
(13). Since G0S2 interacts with
ATGL via its HD domain and because the HD domain conferred G0S2
inhibitory effects, the role of ATGL-regulated lipolysis in this
repression was examined. ED-1 cells were treated with oleic acid to
increase lipid droplet size (13).
This experiment was designed to investigate whether mimicking
inhibition of lipolysis reproduced G0S2 repressive effects. Oleic
acid treatment of ED-1 cells markedly increased lipid droplet size
even at the lowest concentration tested (50 μM), as
visualized by BODIPY staining (data not shown). Yet, the reporter
activity of the indicated transiently transfected luciferase
constructs did not exhibit inhibition after oleic acid treatment
(Fig. 5A).
To learn whether ATGL plays a role in the
suppressive effect of G0S2, ATGL levels were reduced by siRNA
knockdown (Fig. 5B and C) and
independently increased by gain of ATGL expression (Fig. 5D). G0S2 inhibitory effects were
minimally affected by ATGL knockdown or gain of ATGL expression as
shown in Fig. 5C and D. Luciferase
reporter constructs were transiently co-transfected into A549 cells
(in the presence or absence of G0S2 transfection) with individual
ATGL-targeting versus control siRNAs. In A549 cells that
overexpressed the empty vector pCMV-myc, co-transfection of
individual ATGL-targeting siRNAs did not appreciably decrease the
respective luciferase activity as compared to cells co-transfected
with control siRNA (Fig. 5C). In
A549 cells overexpressing G0S2, there was a minor decline in
luciferase activity between ATGL siRNA- and control
siRNA-co-transfected cells, but this was not as large as the
inhibitory effect caused by the transfection of G0S2 alone versus
its empty vector control. In addition, co-transfection of ATGL and
G0S2 in A549 cells did not reverse the inhibitory effect of G0S2 on
luciferase activity (Fig. 5D).
This argued against ATGL playing a driving role in the observed
G0S2 repression.
G0S2 localization
Subcellular localization of G0S2 was next examined.
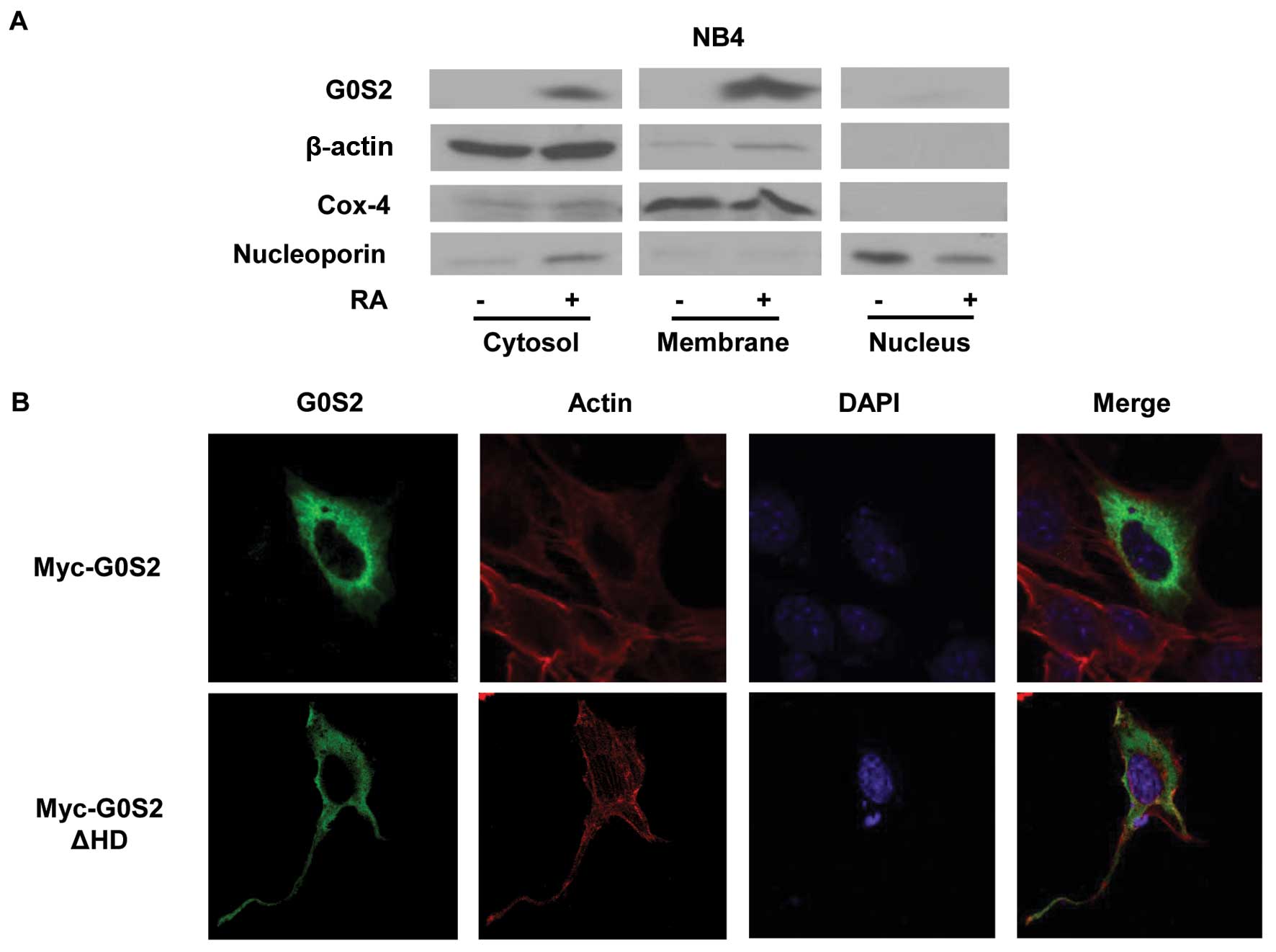
To investigate the subcellular localization of G0S2 protein, NB4
cells were treated with RA (1 μM) for 2 days to induce
endogenous G0S2 expression. In the absence of RA-treatment G0S2 was
not appreciably expressed. Subcellular fractionation and immunoblot
analysis after RA-treatment of NB4 cells revealed that endogenous
G0S2 was predominantly expressed in the cytosolic and membrane
fractions of these cells (Fig.
6A). This subcellular localization of G0S2 was independently
determined in ED-1 cells, where ED-1 cells were transfected with
myc-G0S2 and probed with an anti-myc antibody to detect G0S2
protein, and also with Phalloidin to detect microfilaments. As
visualized by confocal microscopy, G0S2 was distributed in the
cytoplasm, especially in the peri-nuclear region, but with
relatively diffuse cytoplasmic staining (Fig. 6B). Interestingly, after its
transfection into ED-1 cells, the myc-G0S2 ΔHD protein was
distributed diffusely throughout the cytoplasm and G0S2 expression
was extended to cellular processes and projections, with reduced
perinuclear staining as compared to transfected wild-type G0S2
(Fig. 6B).
Discussion
G0S2 is an RA target gene (9) and its functions are under active
study. Studies performed here in APL cells found that G0S2 was
induced after treatment with RAR, but not RXR agonists (Fig. 1). It is previously reported that
G0S2, via its HD domain, complexes with ATGL and inhibits ATGL
lipolytic activity (13). The
current study advanced prior work by identifying a previously
unrecognized G0S2 function. This is its ability to inhibit
exogenous reporter activities, as shown in Figs. 1, 2 and 3.
Yet, G0S2 did not appear to affect endogenous gene expression, as
displayed in Fig. 4. G0S2
repressed transcriptional activities of several retinoid responsive
reporter plasmids including those containing individual regulatory
elements for RARβ, UBE1L or G0S2 itself; this occurred in diverse
cell lines including those that were retinoid responsive or not, as
found in Figs. 1, 2 and 3.
This inhibitory effect was antagonized by siRNA-mediated knockdown
of G0S2 (Fig. 1). The inhibitory
effects of G0S2 were not restricted to retinoid responsive reporter
plasmid activities. Both firefly luciferase and renilla luciferase
activities as well as GFP fluorescent intensities driven by
constitutively active promoters had decreased activity or
expression in the presence of G0S2 co-transfection (Fig. 3 and data not shown). Notably, this
decline could not be explained by a substantial change in
transfection efficiency as noted in Fig. 1 (and data not shown). Stable G0S2
expression also inhibited exogenous reporter activities (Fig. 3), which indicated that these
inhibitory effects depended on expression of G0S2 protein.
Interestingly, neither transient nor stable
expression of G0S2 (Fig. 4)
affected endogenous levels of retinoid regulated species. This
indicated that the G0S2 inhibitory effect appears restricted to the
regulation of exogenous reporter activities. The deletion of the
G0S2 HD domain at least partially rescued G0S2 inhibitory effects,
indicating that this domain played a direct role in conferring
G0S2-mediated transcriptional repression (Fig. 3). Yet, this activity was likely
independent of the ATGL inhibitory function of G0S2 because
respective engineered gain or loss of ATGL expression as well as
induced changes in lipid droplet size after oleic acid treatment
each did not appreciably affect reporter construct activities
(Fig. 5). G0S2 exerted these
actions via its predominant localization to the cytosol and
membrane rather than to the nuclear compartment, as established in
Fig. 6. Yet, deletion of the G0S2
HD domain altered both the repressive effects of G0S2 and its
subcellular localization, as shown in Figs. 3 and 6. In contrast to the localization of
wild-type G0S2, the ΔHD mutation of G0S2 was localized to the
cytoplasm in a more prominent pattern within cellular processes and
projections (Fig. 6). This could
contribute to the reduced transcriptional repression exerted by
this G0S2 mutation.
A repressive effect on exogenously expressed
species, but not on endogenous gene expression was previously
reported. It was found that spermidine/spermine
N1-acetyltransferase 1 (SSAT1) exerted similar effects as reported
here for G0S2 (28). SSAT1 is an
enzyme involved in polyamine catabolism and it catalyzes the
N1-acetylation of spermidine and spermine to form acetyl
derivatives (28). Like G0S2,
SSAT1 inhibited expression of exogenously expressed proteins
including GFP and GFP-elF5A (28).
The precise mechanism responsible for this inhibition by SSAT1 was
not found (28). Yet, similar to
that described here for G0S2, the repressive effect of SSAT1 was
also independent of increased protein degradation since proteasome,
protease, lysosome or autophagy inhibitors did not antagonize SSAT1
repressive activity (28).
SSAT1-dependent repression was limited to
transiently transfected SSAT1 species (28). Neither the induction of endogenous
SSAT1 by a potent SSAT1 inducer such as BENSpm nor stable
expression of SSAT1 repressed exogenously examined proteins
(28). These data indicated that
the inhibitory effects of SSAT1 were likely due to its transient
transfection. In contrast to SSAT1, stable expression of G0S2 also
repressed activities of exogenously expressed reporter plasmids as
shown in Fig. 3B, implicating a
direct role for G0S2 protein in this observed inhibition. Other
factors could still play a role in this process since the extent of
inhibition by stable G0S2 protein expression was less than observed
after transient G0S2 transfection. Even so, changes in transfection
efficiency conferred by introduction of a G0S2 expression vector
played at most a minor role in this effect, as displayed in
Fig. 1C (and in data not
shown).
G0S2 was previously reported to complex with and
inhibit ATGL, which reduced lipolysis and increased lipid droplet
size in cells (13). Because the
G0S2 HD domain was responsible for this interaction with ATGL and
was the domain that antagonized the repressive effects of G0S2
described in this study, it was hypothesized that transcriptional
repression by G0S2 was also mediated by ATGL. Intriguingly, this
was not found to be the case since mimicking the consequences of
increased G0S2 expression by increasing lipid droplet size via
oleic acid treatment did not reproduce this G0S2 inhibition
(Fig. 5). In addition, ATGL
overexpression did not reverse G0S2 repression (Fig. 5). Consistent with this observation,
the siRNA-mediated knockdown of ATGL did not mimic G0S2 repressive
effects (Fig. 5). Hence,
ATGL-regulation of lipolysis does not likely play a major role in
this G0S2 inhibitory effect. When ATGL targeting siRNA was
transfected with a G0S2 overexpression vector, it slightly
increased the G0S2 repressive effect, but the degree of this
inhibition was not as great as observed with G0S2 alone (Fig. 5C). This could be due to a reduction
of the G0S2-ATGL complex after ATGL knockdown, leading to the
release of free G0S2 that can then exert an inhibitory effect.
It was previously reported that G0S2 was localized
to the endoplasmic reticulum (ER) and this conclusion was based on
the subcellular localization of GFP-tagged G0S2 (12). The subcellular localization
findings presented here likely diverge from this prior work because
the GFP-tagged G0S2 used was much larger than the native G0S2
protein. This GFP-tag could lead to non-physiologic localization of
G0S2. Consistent with this interpretation was the observation that
introduction of GFP-tagged G0S2 in studied cells did not repress
reporter activities to the same extent as myc-tagged G0S2 (data not
shown). Unlike GFP-tagged G0S2, myc-tagging leads to a slight
change in the size of G0S2 protein and this did not appear to
affect subcellular localization versus endogenously induced G0S2
protein, as shown in Fig. 6. In
the current study, a prominent localization of G0S2 to the ER
compartment was not appreciated.
The biological basis for the inhibitory effect of
G0S2 is not well understood. Yet, there are several plausible
explanations. G0S2 expression is enhanced in autoimmune and
inflammatory diseases (15–17).
Perhaps the G0S2 inhibitory effect is related to a G0S2 role in
immunity. A class of small proteins known as antimicrobial peptides
(AMPs) is implicated in innate immunity (29). LL-37, one of the most studied AMPs,
has an ability to bind DNA (30).
It is conceivable that G0S2 exerts its inhibitory effect on the
diverse reporter plasmids shown in the studies presented here
through this recognized function of antimicrobial peptides. Future
study will explore this possibility. If this provides a mechanistic
basis for the observed inhibitory actions of G0S2, this finding
would establish a previously unrecognized role for G0S2 beyond what
has recently been found (31).
Of course, to discern a potential role for G0S2 in
immunity, it is necessary to explore the in vivo activities
of G0S2. In this regard, it is interesting that G0S2 transgenic
mice exist (17). Although they
did not exhibit an obvious immune system abnormality, these mice
did have evidence for this because increased autoimmunity-related
antibodies were detected in their serum as compared to wild-type
mice (17). To build on this prior
study, it would be useful to engineer a G0S2 knockout mouse model
in the future. This would be a new tool to discern the precise
biological role for G0S2 beyond its known role in regulating
metabolism (31). Until such a
model is at hand, our data extend the prior study by highlighting a
previously unrecognized G0S2 activity. This is the ability of the
retinoic acid target gene G0S2 to repress both exogenous gene
expression and reporter activity.
Acknowledgements
We thank Dr William Lamph (Ligand
Pharmaceutical) for providing us LG268. The authors also thank the
staff of the Flow Cytometry-Dartlab and Cell Imaging facility at
Dartmouth College for their technical assistance. This study was
supported by the National Institutes of Health and National Cancer
Institute grants R01-CA087546 (E.D.) and R01-CA062275 (E.D.), by a
Samuel Waxman Cancer Research Foundation award (E.D.), and by an
American Cancer Society Clinical Research Professorship (E.D.)
supported by a generous gift from the FM Kirby Foundation.
References
|
1.
|
Massaro D and Massaro GD: Lung
development, lung function, and retinoids. New Eng J Med.
362:1829–1831. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Palczewski K: Chemistry and biology of
vision. J Biol Chem. 287:1612–1619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Pino-Lagos K, Benson MJ and Noelle RJ:
Retinoic acid in the immune system. Ann NY Acad Sci. 1143:170–187.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Freemantle SJ, Spinella MJ and Dmitrovsky
E: Retinoids in cancer therapy and chemoprevention: promise meets
resistance. Oncogene. 22:7305–7315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Warrell RP Jr, Frankel SR, Miller WH Jr,
et al: Differentiation therapy of acute promyelocytic leukemia with
tretinoin (all-trans-retinoic acid). New Eng J Med. 324:1385–1393.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kakizuka A, Miller WH Jr, Umesono K, et
al: Chromosomal translocation t(15;17) in human acute promyelocytic
leukemia fuses RARα with a novel putative transcription factor,
PML. Cell. 66:663–674. 1991.PubMed/NCBI
|
|
7.
|
Melnick A and Licht JD: Deconstructing a
disease: RARalpha, its fusion partners, and their roles in the
pathogenesis of acute promyelocytic leukemia. Blood. 93:3167–3215.
1999.PubMed/NCBI
|
|
8.
|
Pitha-Rowe I, Petty WJ, Kitareewan S and
Dmitrovsky E: Retinoid target genes in acute promyelocytic
leukemia. Leukemia. 17:1723–1730. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kitareewan S, Blumen S, Sekula D, et al:
G0S2 is an all-transretinoic acid target gene. Int J Oncol.
33:397–404. 2008.PubMed/NCBI
|
|
10.
|
Tamayo P, Slonim D, Mesirov J, et al:
Interpreting patterns of gene expression with self-organizing maps:
methods and application to hematopoietic differentiation. Proc Natl
Acad Sci USA. 96:2907–2912. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Russell L and Forsdyke D: A human putative
lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a
small basic protein with the potential to be phosphorylated. DNA
Cell Biol. 10:581–591. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zandbergen F, Mandard S, Escher P, et al:
The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem J.
392:313–324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Yang X, Lu X, Lombès M, et al: The
G(0)/G(1) switch gene 2 regulates adipose lipolysis through
association with adipose triglyceride lipase. Cell Metab.
11:194–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Cristillo AD, Heximer SP, Russell L and
Forsdyke DR: Cyclosporin A inhibits early mRNA expression of G0/G1
switch (G0S2) in cultured human blood mononuclear cells. DNA Cell
Biol. 16:1449–1458. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Koczan D, Guthke R, Thiesen HJ, et al:
Gene expression profiling of peripheral blood mononuclear
leukocytes from psoriasis patients identifies new immune regulatory
molecules. Euro J Dermatol. 15:251–257. 2005.
|
|
16.
|
Nakamura N, Shimaoka Y, Tougan T, et al:
Isolation and expression profiling of genes upregulated in bone
marrow-derived mononuclear cells of rheumatoid arthritis patients.
DNA Res. 13:169–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kobayashi S, Ito A, Okuzaki D, et al:
Expression profiling of PBMC-based diagnostic gene markers isolated
from vasculitis patients. DNA Res. 15:253–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Tokumaru Y, Yamashita K, Osada M, et al:
Inverse correlation between cyclin A1 hypermethylation and p53
mutation in head and neck cancer identified by reversal of
epigenetic silencing. Cancer Res. 64:5982–5987. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Chang X, Monitto CL, Demokan S, et al:
Identification of hypermethylated genes associated with cisplatin
resistance in human cancers. Cancer Res. 70:2870–2879. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kusakabe M, Kutomi T, Watanabe K, et al:
Identification of G0S2 as a gene frequently methylated in squamous
lung cancer by combination of in silico and experimental
approaches. Int J Cancer. 126:1895–1902. 2010.PubMed/NCBI
|
|
21.
|
Kusakabe M, Watanabe K, Emoto N, et al:
Impact of DNA demethylation of the G0S2 gene on the transcription
of G0S2 in squamous lung cancer cell lines with or without nuclear
receptor agonists. Biochem Biophys Res Commun. 390:1283–1287. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Welch C, Santra MK, El-Assaad W, et al:
Identification of a protein, G0S2, that lacks Bcl-2 homology
domains and interacts with and antagonizes Bcl-2. Cancer Res.
69:6782–6789. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Boyle JO, Langenfeld J, Lonardo F, et al:
Cyclin D1 proteolysis: a retinoid chemoprevention signal in normal,
immortalized, and transformed human bronchial epithelial cells. J
Natl Cancer Inst. 91:373–379. 1999. View Article : Google Scholar
|
|
24.
|
Spinella MJ, Freemantle SJ, Sekula D,
Chang JH, Christie AJ and Dmitrovsky E: Retinoic acid promotes
ubiquitination and proteolysis of cyclin D1 during induced tumor
cell differentiation. J Biol Chem. 274:22013–22018. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Liu X, Sempere LF, Galimberti F, et al:
2009. Uncovering growth-suppressive microRNAs in lung cancer. Clin
Cancer Res. 15:1177–1183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
White KA, Yore MM, Deng D and Spinella MJ:
Limiting effects of RIP140 in estrogen signaling: potential
mediation of anti-estrogenic effects of retinoic acid. J Biol Chem.
280:7829–7835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Kitareewan S, Pitha-Rowe I, Sekula D, et
al: UBE1L is a retinoid target that triggers PML/RARalpha
degradation and apoptosis in acute promyelocytic leukemia. Proc
Natl Acad Sci USA. 99:3806–3811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lee SB, Park JH, Woster PM, Casero RA Jr
and Park MH: Suppression of exogenous gene expression by
spermidine/spermine N1-acetyltransferase 1 (SSAT1) cotransfection.
J Biol Chem. 285:15548–15556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Zaiou M: Multifunctional antimicrobial
peptides: therapeutic targets in several human diseases. J Mol Med.
85:317–329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Sandgren S, Wittrup A, Cheng F, Jönsson M,
Eklund E, Busch S and Belting M: The human antimicrobial peptide
LL-37 transfers extracellular DNA plasmid to the nuclear
compartment of mammalian cells via lipid rafts and
proteoglycan-dependent endocytosis. J Biol Chem. 279:17951–17956.
2004. View Article : Google Scholar
|
|
31.
|
Heckmann BL, Zhang X, Xie X and Liu J: The
G0/G1 switch gene 2 (G0S2): regulating metabolism and beyond.
Biochim Biophys Acta. 1831:276–281. 2013. View Article : Google Scholar : PubMed/NCBI
|