Introduction
Rhabdomyosarcoma (RMS) is a soft-tissue malignancy
that is thought to arise from primitive mesenchymal cells of
skeletal muscle lineage. It is the most common soft tissue sarcoma
of childhood, accounting for ∼50% of all pediatric soft tissue
sarcomas, although it is infrequent in adults (1).
The aetiology of RMS is unknown, but has been
associated with growth factor pathways (2) and chromosomal translocations
(3). It is demonstrated that Mirk
is overexpressed in several solid tumors, including
rhabdomyosarcoma, colon carcinoma, prostate carcinoma, pancreatic
ductal adenocarcinoma and non-small cell lung carcinoma, where it
may play a role in prosurvival signaling (4). Minibrain-related kinase (Mirk) is a
member of the dual-specificity tyrosine-regulated kinase
(Dyrk)/Minibrain family of dual-specificity protein kinases
(5). Activation of the Dyrk family
kinases is accomplished by autophosphorylation mediated by a
transitional intermediate form of the nascent protein (6). The first primary function of Mirk to
be elucidated using myogenesis as a model system was the role of
Mirk as a G0 checkpoint kinase. Mirk is upregulated and activated
in myoblasts arrested in G0 when they initiate differentiation
(7). Since Mirk has limited
expression in normal tissue with highest expression seen in
skeletal muscle, heart, testes and brain (8), potential limitations of Mirk as a
pharmacological target may arise from the pathophysiological
consequences of inhibiting Mirk in normal tissues. To circumvent
this potential limitation, it may be possible to design RNA
interference(RNAi) tools that could specifically target the
survival functions of Mirk in tumorigenic cells according to the
differential functions and tissue distributions of the various
splice variants of Mirk.
RNAi is a sequence-specific gene silencing process
that occurs at the messenger RNA (mRNA) level. In mammalian cells
short dsRNAs (<30 bp) trigger the specific knockdown of mRNAs in
mammalian cells without interferon activation (9), based on the discovery above,
silencing of gene expression by RNA interference (RNAi) is
currently used as standard tool in cultured mammalian cells, which
has demonstrated great prospects for human gene function, signal
transduction research and gene therapy (10). Although chemically synthetic siRNAs
can be introduced into cultured cells and induce the transient
knockdown of target mRNAs, there is a clear problem in its use for
stable transcript knockdown and a high efficiency of RNAi delivery.
Alternatively, expression vectors driven by RNA polymerase III
enable the permanent production of small dsRNAs in mammalian cells
(11,12), furthermore the
doxycycline-controlled tet-on lentiviral system can be used to
reversibly induce gene silencing in a temporally and spatially
restricted manner (13).
Since there is little research on the relationship
between Mirk and tumor, and its function in tumor cells has not
been clearly studied so far. In the present study, a Tet-on
Lentivirus-mediated short hairpin RNA vector targeting human Mirk
gene was designed, such vector-derived transcripts were processed
by Dicer in a similar manner as siRNAs. Then we employed the
constructed lentivirus vector mediating RNAi targeting of Mirk gene
to study the influence of the knockdown of Mirk on rhabdomyosarcoma
RD cells growth in vitro.
Materials and methods
Cell culture
Rhabdomyosarcomas RD and 293T cell lines were
obtained from Chinese type culture collection (WuHan), they were
both cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco)
supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/ml
penicillin, and 100 mg/ml streptomycin (Invitrogen). Cells were
incubated at 37°C in 5% CO2 atmosphere, the medium was
refreshed every 2 or 3 days and passaged when confluent monolayers
were achieved using trypsin solution.
Design of human Mirk-shRNA
The mRNA sequence of human Mirk (GenBank acc. no.:
NM_004714) was placed in Ambion online RNAi Designer to design
siRNA. Since not every working siRNA sequence is equally effective
when incorporated into shRNA (14), we selected three optimal siRNAs in
different target sequences. One scrambled shRNA control was
specifically designed. All four sequences were aligned using the
GenBank BLAST program, no other homologous sequences matched except
the Mirk gene. To obtain the double-stranded RNA configuration
required for short hairpin RNA formation from single-stranded RNA,
we linked a 19-bp sense siRNA sequence to its complementary
antisense sequence via a 9-bp loop region and combine it with
additional two T nucleotides, which is flanked by restriction
enzyme recognition sequences and their protective bases (15). Two complementary single-stranded
DNA oligonucleotides of the four shRNAs were chemically synthesized
by Shanghai Genechem Co. Ltd. These oligonucleotides were annealed
to produce double-stranded oligonucleotides (Table I).
 | Table I.Sequences of four designed
Mirk-shRNA. |
Table I.
Sequences of four designed
Mirk-shRNA.
| Mirk-shRNA
name | Sequence |
|---|
| p1 |
5′-ccgCTCGAGGCAGCGCCATCAAGATTGTTTCAAGAGAACAATCTTGATGGCGCTGCTTACGCGTcg-3′
3′-ggcGAGCTCCGTCGCGGTAGTTCTAACAAAGTTCTCTTGTTAGAACTACCGCGACGAATGCGCAgc-5′ |
| p2 |
5′-ccgCTCGAGGGTGGTGAAAGCCTATGATTTCAAGAGAATCATAGGCTTTCACCACCTTACGCGTcg-3′
3′-ggcGAGCTCCCACCACTTTCGGATACTAAAGTTCTCTTAGTATCCGAAAGTGGTGGAATGCGCAgc-5′ |
| p3 |
5′-ccgCTCGAGGACCCTACGAAGGACGAAATTCAAGAGATTTCGTCCTTCGTAGGGTCTTACGCGTcg-3′
3′-ggcGAGCTCCTGGGATGCTTCCTGCTTTAAGTTCTCTAAAGCAGGAAGCATCCCAGAATGCGCAgc-5′ |
| p0 |
5′-ccgCTCGAGGGCCGGCCTTAAGCTAATATTCAAGAGATATTAGCTTAAGGCCGGCCTTACGCGTcg-3′
3′-ggcGAGCTCCCGGCCGGAATTCGATTATAAGTTCTCTATAATGCAATTCCGGCCGGAATGCGCAgc-5′ |
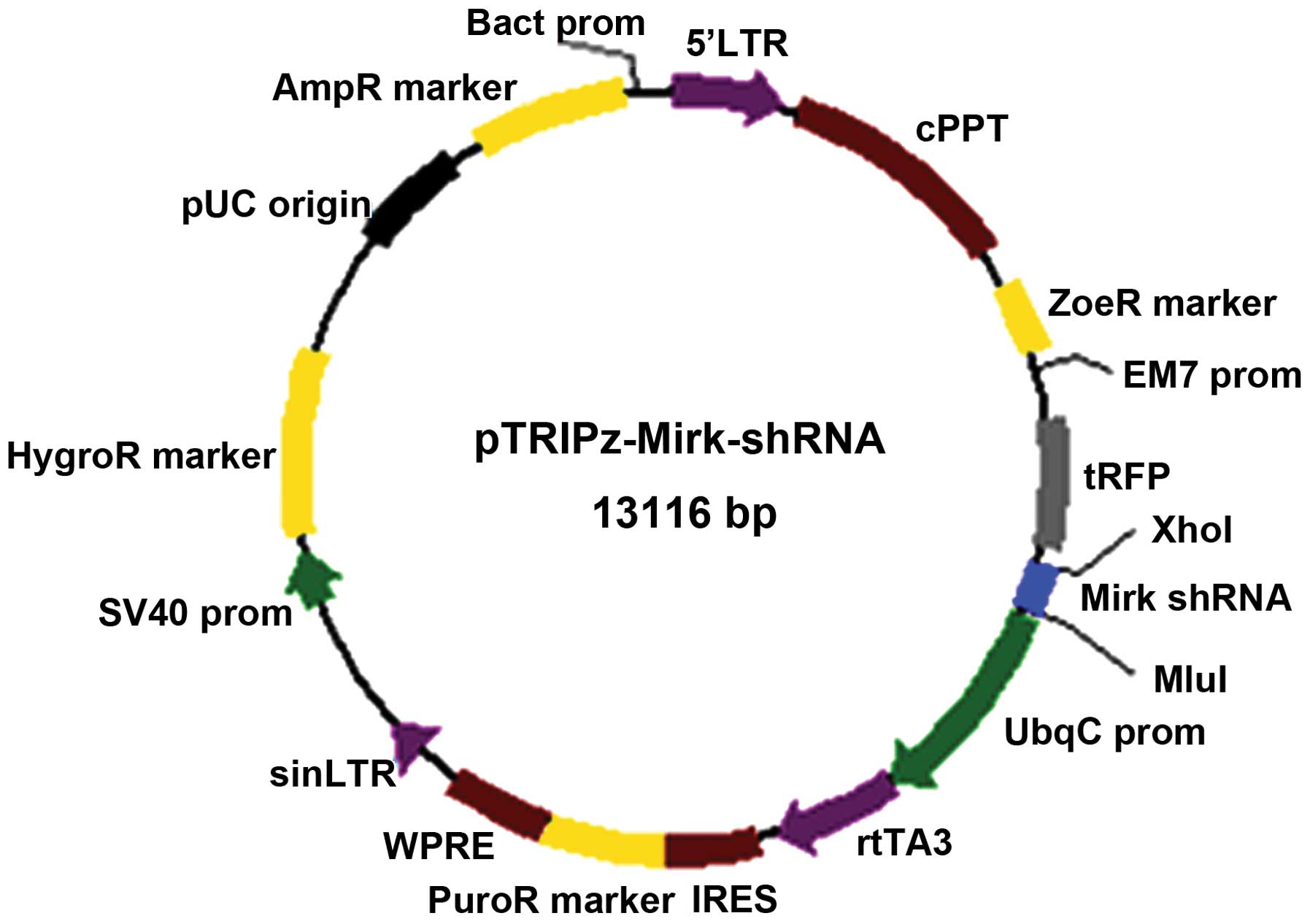
Construction of pTRIPz-Mirk-shRNA
vectors
The annealed Mirk-shRNA oligonucleotides were cloned
into linearized pTRIPZ empty vector (Open Biosystems catalog
#RHS4750) using the following steps: the pTRIPZ vectors and
double-stranded shRNA oligonucleotides were digested with both
XhoI and Mlul, respectively; recycling the big
fragment (13061 bp) of the former and the digested shRNA fragment
(55 bp); and then ligated by T4 DNA ligase according to the
manufacturer’s protocol. The ligated products were transformed into
competent DH5a cells using heat shock method. The transformed cells
were grown on an LB-agar plate containing ampicillin and
neomycin.
Identification of double digestion and
DNA sequence
To ensure that the shRNAs were inserted into the
vectors. We collected positive clones and grew them at 37°C in LB
broth (low salt) media plus 100 μg/ml ampicillin only, and
incubation at 37°C for 18 h with vigorous shaking. The plasmid DNA
was prepared using Plasmid mini kit (Qiagen) according to the
manufacturer’s instructions. and restriction digested with
XhoI and MluI using the plasmid DNA prepared and
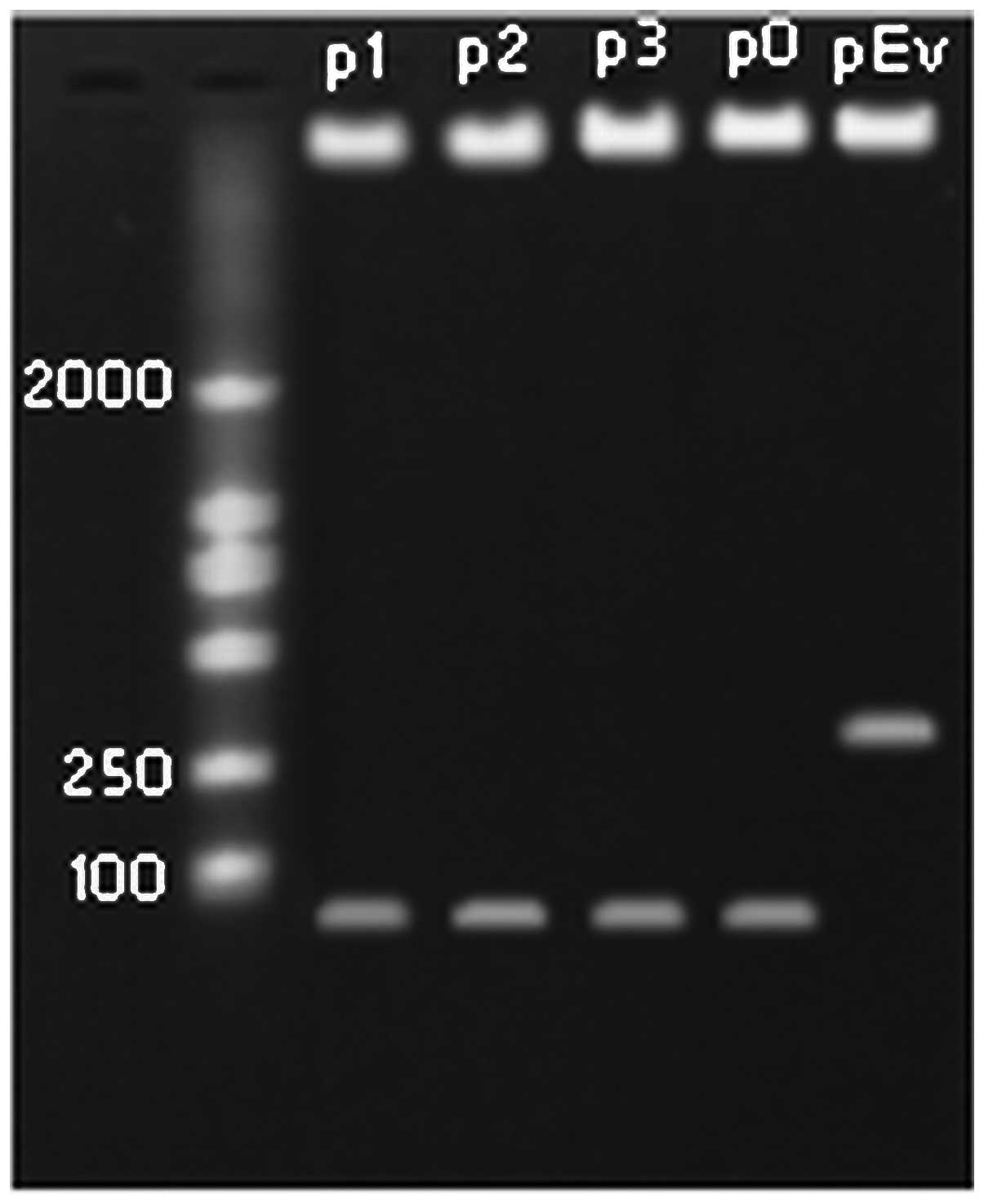
incubated at 37°C for 3 h, followed by digestion on a 1.5% agarose
gel. Two bands were seen (55 bp and a large band near 13 kp). The
extracted recombinant plasmids were used for DNA sequencing to
identify the inserted Mirk-shRNA fragments. The pTRIPZ sequencing
primer was: 5′-GGAAAGAATCAAGGAGG-3′.
Transfection and lentivirus
production
Twenty-four hours before transfection, 293T
packaging cells were seeded in a 6-well plate and cultured in
growth medium without antibiotics to achieve ∼70% confluency in a
cell culture dish and passaging at a 1:2 ratio for at least two
consecutive days. For transfection, 6 μg pTRIPZ-Mirk-shRNA
vector and 4.3 μl Trans-Lentiviral packaging mix (Fisher
Scientific catalog no.: 14-959-1A) was used per well, which
co-transfect into cells by using the calcium phosphate reagent
according to the manufacturer’s instructions. Cells were incubated
at 37°C with 5% CO2 for 12 h. Calcium
phosphate-containing medium was removed from cells and replaced
with the indicated volume of reduced serum medium (High Glucose
DMEM 5% Fetal Bovine Serum 2 mM L-glutamine 1X
penicillin-streptomycin). Viral particle-containing supernatants
64-h post-transfection were harvested by removing medium to a 15 ml
sterile, capped, conical tube. Non-adherent cells and debris was
pelleted by centrifugation at 1600 × g and 4°C for 10 min. Each
viral titer was estimated as ×106 by counting the number
of RFP 293T cells by flow cytometry two days after transduction
with serial dilutions of the viral stocks. MOI of 0.3 was used.
Infection of target cells
In order to generate stable RD cell lines, we
determined that the minimum amount of puromycin required to kill
non-transduced cells is 1.1 μg/ml. We used the purified
viral particle to infect rhabdomyosarcomas RD cells, as previously
described (16). After 6 days
under puromycin selection, the fresh complete medium containing
doxycycline (0.5 μg/ml) was replaced in the cells,
doxycycline-free group, empty vector group and scrambled shRNA
vector group were used as controls. When the stable transgenic
cells reached confluence, some were collected for RNA extraction to
evaluate RNAi efficiency, and the rest were frozen in liquid
nitrogen for further experiments.
Quantitative RT-PCR
Total RNA was extracted from the Rhabdomyosarcomas
RD cells using the RNeasymini kit (Qiagen). RNA was digested by
DNaseI to remove the contamination of DNA, and single-stranded cDNA
was prepared from RNA using the High Capacity cDNA RT kit (Qiagen).
Quantitative RT-PCR primers were designed using Beacon Designer. A
reverse transcription PCR reaction was performed to screen the most
effective recombinant plasmids. Real-time PCR was performed using
LightCycler® 480 Gene Scanning software (Roche Applied
Science, USA), as previously described (17). A standard PCR program was used for
SYBR Green I: 95°C for 5 min; 45 cycles of 95°C for 10 sec, 55°C
for 40 sec; followed by 95°C for 10 sec, 65°C for 1 min, and 95°C
for 30 sec, melting curve analysis was performed to verify the
identities of PCR products. Each sample was tested three times to
obtain an average. Relative expression levels of Mirk gene were
normalized to GAPDH expression levels. Primer sequences for
amplification of Mirk and GAPDH are listed in Table II.
| Table II.Primers of Mirk and GAPDH. |
Table II.
Primers of Mirk and GAPDH.
| Name | Primer | Product total
length (bp) |
|---|
| Human Mirk | | |
| Sense |
5′-ATTCACTGCGACCTCAAG | 126 |
| Antisense |
5′-GCGGCTCTGGATATACTG | |
| Human GAPDH | | |
| Sense |
5′-agaaggctggggctcatttg | 258 |
| Antisense |
5′-aggggccatccacagtcttc | |
Western blot analysis
RD cells were lysed in 40 μl of 1X Radio
Immunoprecipitation Assay Lysis Buffer (Sigma) on ice for 30 min.
The lysates were cleared by centrifugation. Proteins were separated
on SDS-PAGE (4% stacking gel, 12% separating gel) and transferred
to nitrocellulose membranes, as previously described (18). The membranes were blocked with 5%
skimmed milk in TS buffer (10 mm Tris-HCl, 150 mm NaCl, pH 7.4),
then probed with anti-Mirk (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) and anti-GAPDH (Sigma, USA) at room temperature, followed
by incubation with horseradish peroxidase-conjugated goat
anti-mouse secondary antibody (Amersham Pharmacia Biotech). After
several washes, the membranes were incubated with an enhanced
chemiluminescence system (Invitrogen, USA) and exposed to Kodak
Biomax light film. GAPDH protein levels were used as a control to
verify equal protein loading.
Detection of apoptosis by flow cytometry
and fluorescence microscopy
After transduction with lentivirus, stable RD cell
lines were seeded in 6-well plates at a density of 2×104
cells per well. After 24 h Dox (0.5 μg/ml) was added, RD
cells were harvested and washed in cold phosphate-buffered saline
(PBS). Cell suspension with density of 1×106/ml was
preparing for each assay. Cells were stained with fluorescein
isothiocyanate (FITC) labeled Annexin V, and simultaneously with
propidium iodide (PI) stain, as previously described (19), to discriminate intact cells
(Annexin−/PI−) from apoptotic cells
(Annexin+/PI−), and necrotic cells
(Annexin+/PI+). FACS analysis for Annexin V
and PI staining was performed by flow cytometery. All experiments
were performed in triplicate.
Flow cytometry analysis of the cell
cycle
The cells were harvested by trypsinization, fixed
with cold 70% ethanol, and stored at 4°C until analyzed. The cells
were resuspended in phosphate buffered saline (PBS) containing 10
μg/ml RNaseA and 20 μg/ml propidium iodide (PI) for
30 min at room temperature, and DNA content was detected by flow
cytometry (BD, USA). The relative proportions of cells in the
G1/G0, S and G2/M phases of the cell cycle were determined from the
flow cytometry data.
Statistical analysis
The software of SPSS version 17.0 (SPSS Inc., IL,
USA) was used for statistical analysis. Values shown are
representative of triplicate determinations in no less than three
experiments. Data are expressed as mean ± SD. Results were
considered significant, if P<0.05 was obtained by a two-sided
Student’s t-test.
Results
Sequencing profile of the recombinant
Mirk-shRNA expression vectors
Schematic diagram for the construction of the
pTRIPz-shRNA expression vector to knockdown Mirk is shown as shown
in Fig. 1. Recombinant plasmids
were digested with XhoI and MluI, and the fragments
were identified on 1.5% agarose gel (Fig. 2). The results of DNA sequencing
provided further confirmation of the presence of the recombinant
plasmids, indicating that all the shRNA expression plasmids carried
the correct sequence.
Generation of stable transgenic RD
cells
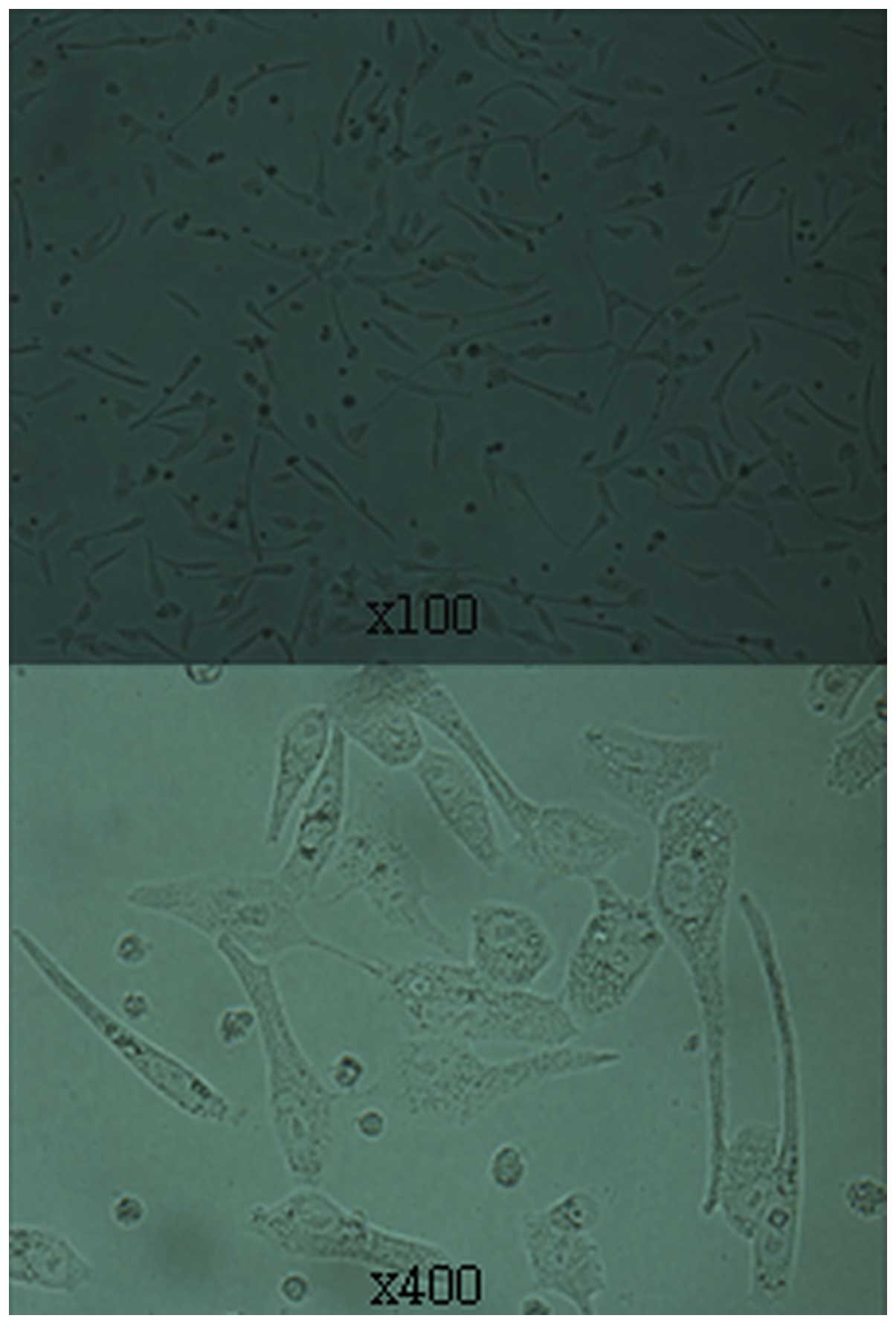
Puromycin selection was used to obtain stable
transfected RD cell lines from rhabdomyosarcomas. We noted that a
few cells aged and lost their growing ability after puromycin
selection. Well-grown colonies were passaged after puromycin
selection, and then transferred to 60-mm culture dishes to
facilitate expansion (Fig. 3).
Expression of the reporter gene (RFP) was assessed by visualization
of fluorescence (Fig. 4).
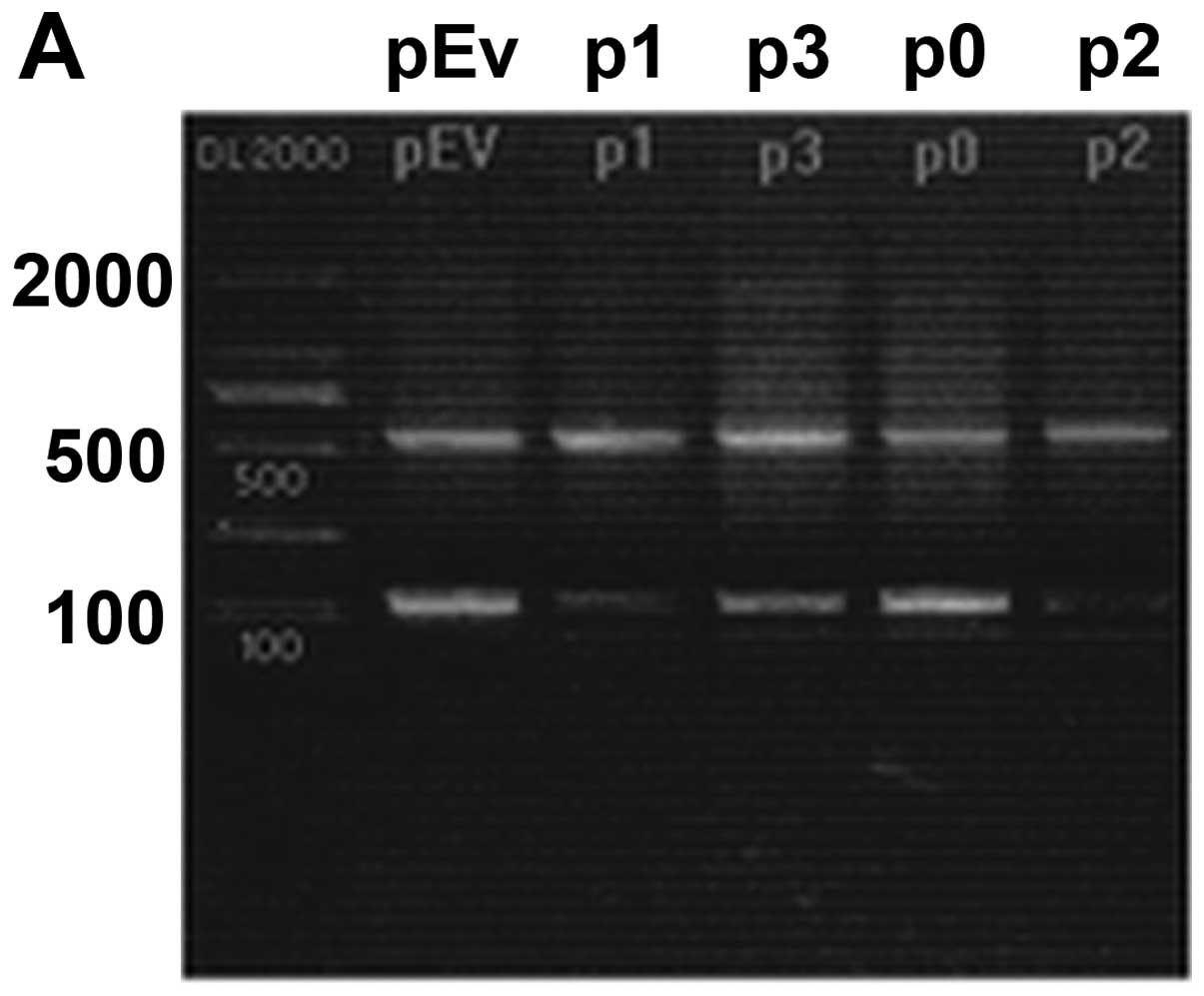
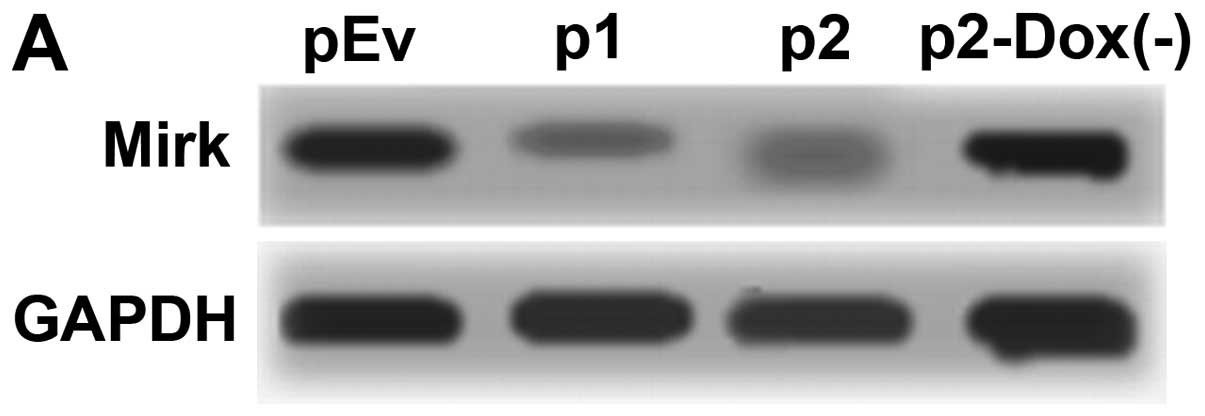
Silencing of Mirk mRNA and protein
expression after lentivirus transduction
To exclude off-target silencing effect mediated by
specific-shRNA, we designed three different Mirk-shRNAs (p1, p2 and
p3) to silence the expression of Mirk gene in the RD cell line,
with scrambled shRNA (p0) and empty vector (pEv) as controls. The
mRNA levels of Mirk gene expression were determined by reverse
transcription PCR (Fig. 5A) and
real-time quantitative PCR (Fig.
5B). The screened p1 and p2 were the most effective groups.
Then p1 and p2 were transduced into stable RD cells again, controls
were pEv and p2-Dox(−). Protein levels of Mirk gene expression were
determined by western blotting. As shown in Fig. 6A, compared with both pEv and
p2-Dox(−) control groups, the protein levels of Mirk in p1 and p2
groups were significantly reduced respectively (P<0.05), but
there was no obvious difference between the two controls (Fig. 6B).
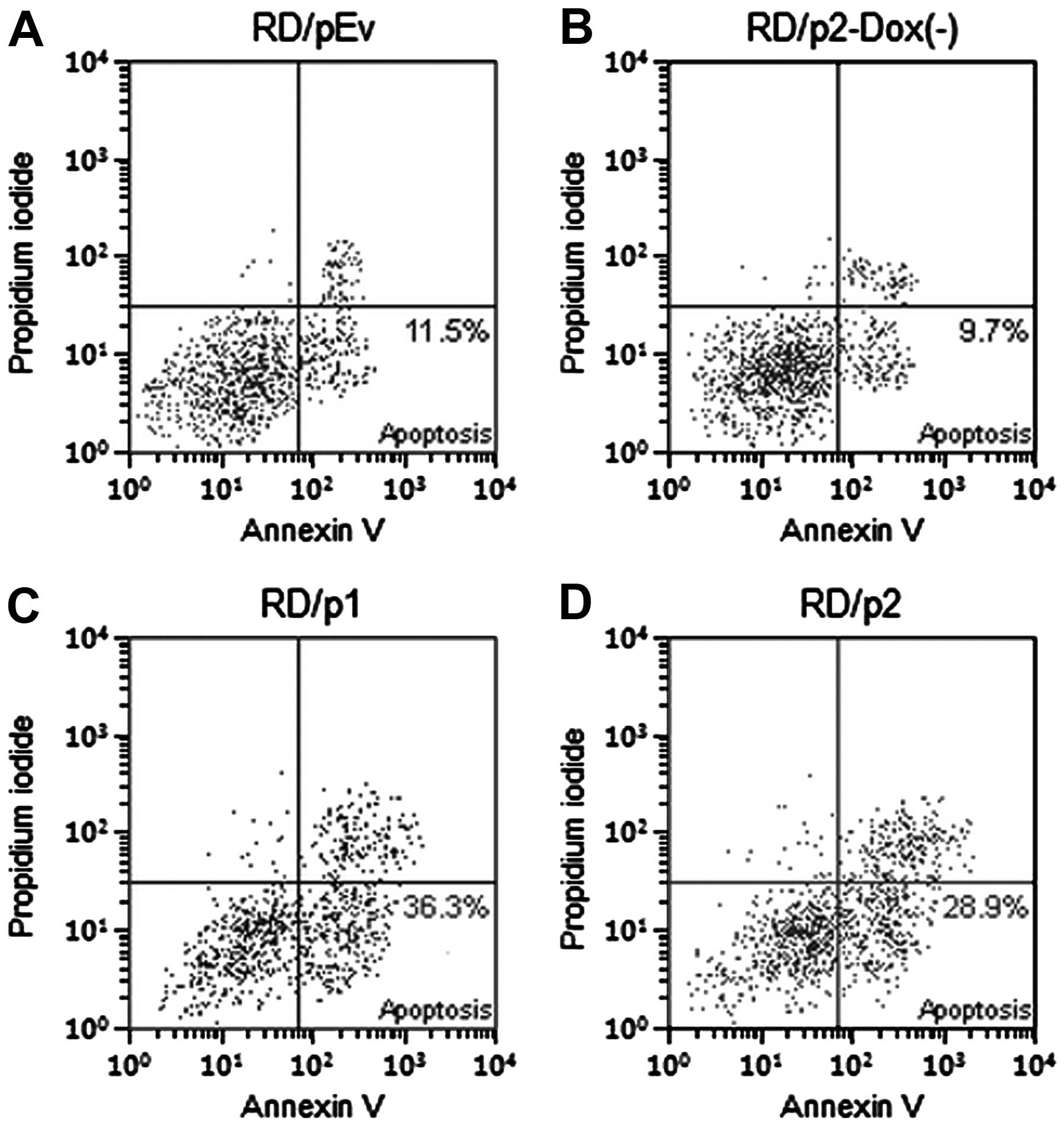
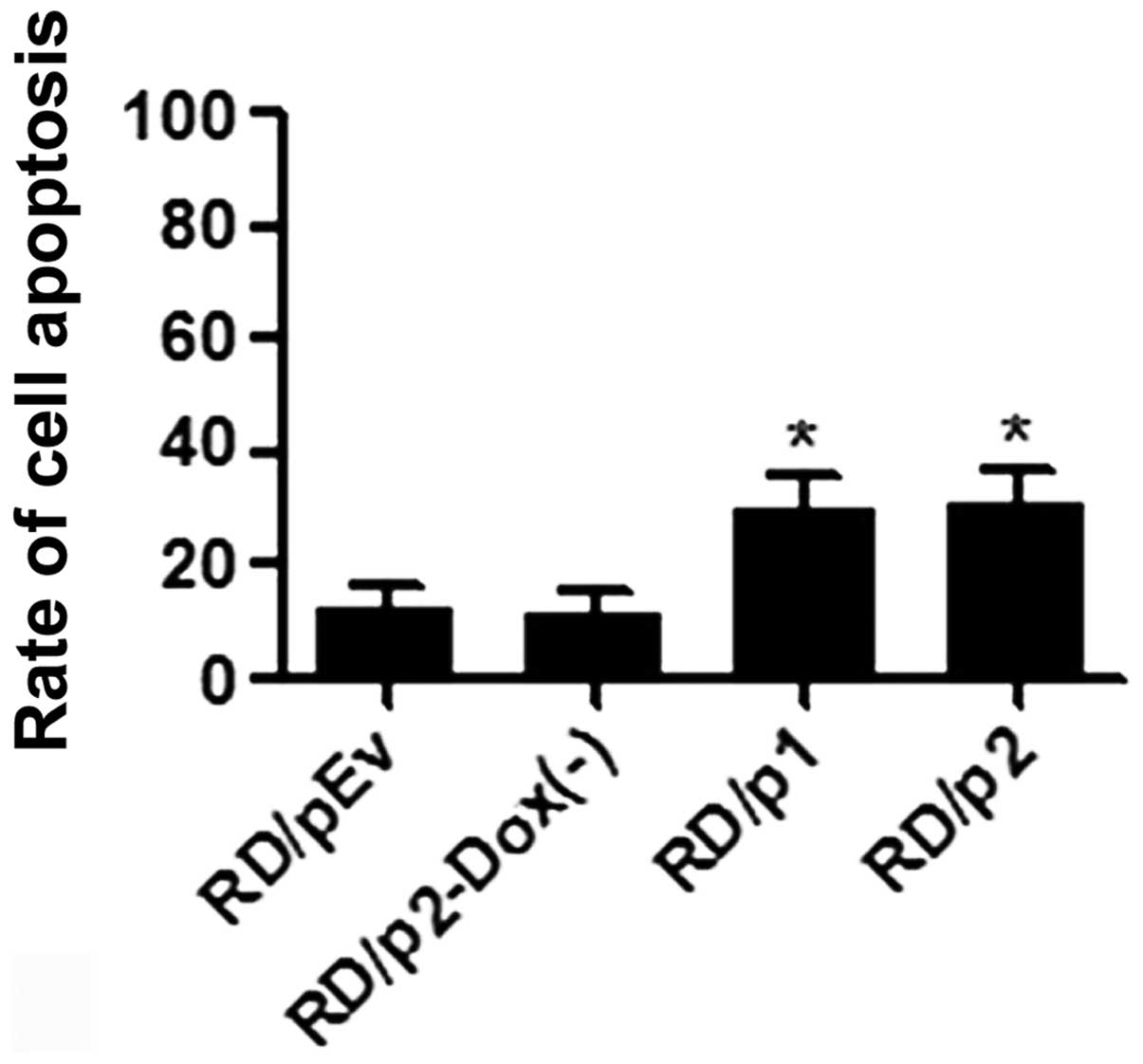
Apoptosis was increased in RD cells by
knockdown of endogenous Mirk
To ascertain whether apoptosis was increased in RD
cells by the knockdown of Mirk, we used Annexin V-FITC and
propidium iodide staining. Flow cytometric analysis was performed
to evaluate apoptotic cells (Fig.
7). After silencing of Mirk was induced, the percentage of
apoptotic cells in p1 and p2 groups is much greater than that in
pEv and p2-Dox(−) groups (Fig.
8).
Mirk RNAi induces the changes of the cell
cycle
In the current study, the effect of Mirk
downregulation on the cell cycle of RD cells was determined and
each assay was performed in triplicate. The flow cytometry analysis
revealed that more RD cells in p1 and p2 groups were arrested in
the G0/G1 phase of the cell cycle (65.8±2.3 and 67.8±1.8%,
respectively), and the proliferation index (PI) of RD cells
(44.4±4.1 and 41.4±3.4%, respectively) was decreased
correspondingly (P<0.05), while there was no statistical
difference between pEv and p2-Dox(−) groups, PI =
(S+G2/M)/(G0/G1+G2/M) ×100% (Table
III).
| Table III.Cell cycle phases detected by flow
cytometry. |
Table III.
Cell cycle phases detected by flow
cytometry.
| Cells | Cell cycle phases
(mean ± SD, %)
|
|---|
| G0/G1 | S | G2/M | PI |
|---|
| pEv | 54.5±2.7 | 31.8±1.8 | 13.7±0.9 | 66.8±5.7 |
| p2-Dox(−) | 54.3±3.0 | 32.4±2.9 | 14.2±1.1 | 69.1±7.2 |
| p1 | 65.8±2.3a | 23.0±2.0a | 11.1±0.3 | 44.4±4.1a |
| p2 | 67.8±1.8a | 22.1±2.4a | 10.1±1.3 | 41.4±3.4a |
Discussion
Rhabdomyosarcoma (RMS) is generally diagnosed in
younger children, 60% of cases are diagnosed in children younger
than 5 years of age, nearly one-third of all RMS cases occur in the
head and neck area (20). Current
approaches of treatment include surgical removal, radiation and
chemotherapy (21). However, both
chemotherapy and radiotherapy sometimes are not sensitive to
patients (22) and may result in
facial growth retardation, neuroendocrine dysfunction of oral
tissues, such as visual problems, hearing loss, delayed eruption of
the teeth, hypodontia and velopharyngeal insufficiency (23,24).
In addition, surgical resection of rhabdomyosarcoma is not always
possible because of extent of disease and involvement of
surrounding critical organs (25),
which is challenging and can result in large maxillofacial defects
with loss of function and esthetics of the surrounding tissues. In
recent years, with the development of RNA interference (RNAi)
technology, RNAi shows enormous potential in therapeutics for
tumors (26).
RNAi constructs can be designed to target any known
gene. As they make use of a conserved biological silencing pathway,
they are a particularly effective method to inhibit aberrant gene
expression that results in pathogenesis. The sequence specificity
of the RNAi mechanism provides a high specificity required for
targeted therapies, overcoming the side effects of several
traditional therapies. Specific gene silencing can be achieved in
many sorts of cell systems using chemically synthesized small
interference RNA (siRNA) or DNA vector-based shRNA. Functional
genomics using RNAi has facilitated the rate in which genes are
assigned function and thereby expedited the identification of
potential therapeutic targets for some gene related diseases
(27–29). Importantly, several RNAi-based
therapies are on the way to being developed (30). Doxycycline-controlled tet-on
lentiviral system enables the reversible and body-wide expression
of shRNAs (13,31) and offers the opportunity to reverse
the induced gene knockdown at a given time. This property of the
shRNA system offers unique applications to study gene function in
animal experiments that cannot be achieved with knockout
technologies (32,33). Although lentivirus vectors has been
used for several years, the use of Tet-on lentiviral vector
expressing shRNA as a therapeutic tool for rhabdomyosarcoma has not
been clearly explored.
In this study, we used lentivirus-mediated RNAi to
silence the endogenous Mirk expression and explored the effects of
Mirk downregulation on the phenotypes of RD cells. We designed
three shRNAs targeted at Mirk gene and successfully transfected
them into rhabdomyosarcoma RD cell line by lentivirus. Since the
inducible pTRIPz vector include a puromycin resistance gene,
positive cell lines can be conveniently selected using puromycin.
After stable integration, vectors expressing Mirk-shRNAs
constitutively will directly induce downregulating of target gene
with the control of doxycycline. Permanent Mirk gene silencing is
achieved in RD cells by integrating shRNA transgenes into the
genome. Mirk-shRNAs (p1, p2) have been identified as the most
effective ones, because the stable transfectants show significantly
decreased levels of Mirk mRNA and protein, which subsequently
indicated that our constructional lentivirus-mediated Mirk-specific
shRNA was able to silence the expression of Mirk effectively and
specifically in RD cells. The efficiency of gene silencing can be
as high as 60%.
In addition, we observed that the depletion of Mirk
resulted in apoptosis in RD cells in vitro. Further research
showed that most of the cells were arrested in G0/G1 phase.
However, the mechanism has not been elucidated in detail. Mirk has
been previously shown to mediate cell survival by binding to a
variety of proapoptotic molecules, including procaspase-3 and
apoptosis signaling kinase 1 (34,35)
in colon carcinoma cells (8). A
previous study has demonstrated the importance of kinases such as
Mirk/Dyrk1B and Dyrk3 which were found to participate in
prosurvival signaling (36). Mirk
is likely to mediate tumor survival during periods when tumorigenic
cells temporarily outgrow their nutrient support, and the
Mirk-induced survival pathway may provide a strong selective
pressure to maintain expression of Mirk in tumors and complement
other survival pathways activated by growth factors such as
fibroblast growth factors (4).
In conclusion, we established a conditional and
effective method for screening the most effective shRNA for
suppressing Mirk gene overexpression in rhabdomyosarcoma RD cell
line and generated stable transgenic RD cell line for further
study. This method is likely to be useful in exploring biochemical
mechanisms of RNA interference pathways and it has the potential to
provide more rational strategies for efficient targeting
suppression of any desired gene. We also found that depletion of
Mirk inhibited RD cells proliferation due to G0/G1 arrest and
apoptosis. There is an increasing awareness that survival pathways
in tumors are much more critical compared with normal progenitors.
The evidence of Mirk to mediating cell survival in RMS suggests
that it may have some potential as a pharmacological target. These
results have paved the way for the study of the function of Mirk in
tumor cells, and could be of great benefit for gene therapy in the
future.
Acknowledgements
This work was supported by the
National Natural Science Fund of China (no. 81072187). We are
indebted to Professor Y. Liu for providing experimental supplies
and technical assistance.
Reference
|
1.
|
Ulutin C, Bakkal H and Kuzhan O: A cohort
study of adult rhabdomyosarcoma: a single institution experience.
World J Med Sci. 3:54–59. 2008.
|
|
2.
|
Merlino G and Helman LJ: Rhabdmyosarcoma -
working out the pathways. Oncogene. 18:5340–5348. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Barr FG: Molecular genetics and
pathogenesis of rhabdomyosarcoma. J Paediatr Hematol Oncol.
19:483–491. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Mercer SE and Friedman E: Mirk/Dyrk1B-A
multifunctional dual-specificity kinase involved in growth arrest,
differentiation, and cell survival. Cell Biochem Biophys.
45:303–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Deng X, Ewton D, Pawlikowski B, et al:
Mirk/Dyrk1B is a rho-induced kinase active in skeletal muscle
differentiation. J Biol Chem. 278:41347–413003
|
|
6.
|
Lochhead PA, Sibbet G, Morrice N and
Cleghon V: Activation-loop autophosphorylation is mediated by a
novel transitional intermediate form of DYRKs. Cell. 121:925–936.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Becker W, Weber Y, Wetzel K, et al:
Sequence characteristics, subcellular localization and substrate
specificity of Dyrk-related kinases, a novel family of dual
specificity protein kinases. J Biol Chem. 273:25893–25902. 1998.
View Article : Google Scholar
|
|
8.
|
Lee K, Deng X and Friedman E: Mirk protein
kinase is a mitogen-activated protein kinase substrate that
mediates survival of colon cancer cells. Cancer Res. 60:3631–3637.
2000.PubMed/NCBI
|
|
9.
|
Elbashir SM, Harborth J, Lendeckel W, et
al: Duplexes of 21-nucleotide RNAs mediate RNA interference in
cultured mammalian cells. Nature. 411:494–498. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Karagiannis TC and El-Osta A: siRNAs:
mechanism of RNA interference, in vivo and potential clinical
applications. Cancer Biol Ther. 3:1069–1074. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Paddison PJ, Caudy AA, Bernstein E, et al:
Short hairpin RNAs (shRNAs) induce sequence-specific silencing in
mammalian cells. Genes Dev. 16:948–958. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Seibler J, Kleinridders A, Küter-Luks B,
et al: Reversible gene knockdown in mice using a tight, inducible
shRNA expression system. Nucleic Acids Res. 35:e542007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Reynolds A, Leake D, Boese Q, et al:
Rational siRNA design for RNA interference. Nat Biotechnol.
22:326–330. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Barøy T, Sørensen K, Lindeberg MM and
Frengen E: shRNA expression constructs designed directly from siRNA
oligonucleotide sequences. Mol Biotechnol. 45:116–120.
2010.PubMed/NCBI
|
|
16.
|
Das AT and Zhou X: Viral evolution as a
tool to improve the tetracycline-regulated gene expression system.
J Biol Chem. 279:18776–18782. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nolan T, Hands RE and Bustin SA:
Quantification of mRNA using real-time RT-PCR. Nat Protoc.
1:1559–1582. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ghaemmaghami S, Huh WK, Bower K, et al:
Global analysis of protein expression in yeast. Nature.
425:737–741. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flowcytometry. Nat
Protoc. 1:1458–1461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Arndt CA, Rose PS, Folpe AL and Laack NN:
Common musculo-skeletal tumors of childhood and adolescence. Mayo
Clin Proc. 87:475–487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fyrmpas G, Wurm J, Athanassiadou F, et al:
Management of paediatric sinonasal rhabdomyosarcoma. J Laryngol
Otol. 123:990–996. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Fatusi OA, Ajike SO, Olateju SO, et al:
Clinicoepidemiological analysis of orofacial rhabdomyosarcoma in a
Nigerian population. Int J Oral Maxillofac Surg. 38:256–260. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kaste SC, Goodman P, Leisenring W, et al:
Impact of radiation and chemotherapy on risk of dental
abnormalities: a report from the Childhood Cancer Survivo Study.
Cancer. 115:5817–5827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Estilo CL, Huryn JM, Kraus DH, et al:
Effects of therapy on dentofacial development in long-term
survivors of head and neck rhabdomyosarcoma: the memorial
sloan-kettering cancer center experience. J Pediatr Hematol Oncol.
25:215–222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Paulino AC and Okcu MF: Rhabdomyosarcoma.
Curr Probl Cancer. 32:7–34. 2008. View Article : Google Scholar
|
|
26.
|
Stevenson M: Therapeutic potential of RNA
interference. N Engl J Med. 351:1772–1777. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hannon GJ and Rossi JJ: Unlocking the
potential of the human genome with RNA interference. Nature.
431:371–378. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ito M, Kawano K, Miyagishi M and Taira K:
Genome-wide application of RNAi to the discovery of potential drug
targets. FEBS Lett. 579:5988–5995. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Silva J, Chang K, Hannon GJ and Rivas FV:
RNA-interference-based functional genomics in mammalian cells:
reverse genetics coming of age. Oncogene. 23:8401–8409. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Samakoglu S, Lisowski L, Budak-Alpdogan T,
et al: A genetic strategy to treat sickle cell anemia by
coregulating globin transgene expression and RNA interference. Nat
Biotechnol. 24:89–94. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Kotnik K, Popova E, Todiras M, et al:
Inducible transgenic rat model for diabetes mellitus based on
shRNA-mediated gene knockdown. PLoS One. 4:e51242009. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Christoph T, Bahrenberg G, De Vry J, et
al: Investigation of TRPV1 loss-of-function phenotypes in
transgenic shRNA expressing and knockout mice. Mol Cell Neurosci.
37:579–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Sacca R, Engle SJ, Qin W, et al:
Genetically engineered mouse models in drug discovery research.
Methods Mol Biol. 602:37–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Asada M, Yamada T, Ichijo H, et al:
Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in
monocytic differentiation. EMBO J. 18:1223–1234. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Blagosklonny MV: Are p27 and p21
cytoplasmic oncoproteins? Cell Cycle. 1:391–393. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Mackeigan JP, Murphy LO and Blenis J:
Sensitized RNAi screen of human kinases and phosphatases identifies
new regulators of apoptosis and chemoresistance. Nat Cell Biol.
7:591–600. 2005. View
Article : Google Scholar : PubMed/NCBI
|