Introduction
Dose-dense docetaxel and cisplatin was previously
investigated as a first line treatment for patients with metastatic
non-small cell lung cancer (1).
The toxicities included significant neuropathy, nausea and
dehydration. The response rate achieved with this regimen was 53%
and the overall survival was 11 months with a 1-year survival rate
of 45%. Maintenance treatment was not given following completion of
dose-dense chemotherapy in this prior study.
Erlotinib improves the overall survival of patients
with metastatic non-small cell lung cancer in the maintenance,
second line and third line settings (2,3). The
improvement in overall survival is longer for patients who achieve
a rash after starting treatment with erlotinib (4,5).
Patients who continue to smoke cigarettes are less likely to
experience rash and are less likely to benefit from treatment.
These observations suggest that a failure to achieve adequate drug
levels may contribute to clinical erlotinib resistance in the
population of patients who continue to smoke cigarettes.
A phase I/II study investigated the maximal
tolerable dose (MTD) of erlotinib in patients who were smoking ≥10
cigarettes daily (6). This study
found that the dose of 300 mg daily in smokers achieved a similar
pharmacokinetic and side effect profile to non-smoking patients
treated with 150 mg daily. The increase in erlotinib metabolism was
attributed to induction of CYP1A1/1A2 enzymes by exposure to
tobacco smoke. However, this prior phase I/II study did not examine
the MTD of former smokers.
Molecular genetic properties of individual cancers
such as epidermal growth factor receptor (EGFR) mutations impact
the likelihood of achieving clinical benefit with treatment.
Progression-free survival is dramatically increased in patients
with certain EGFR activating mutations (3,7–9).
Patients with EGFR wild-type cancers treated with erlotinib also
experience improved progression-free survival and overall survival
in the maintenance setting (3) and
additional biomarkers are needed for clinical decision making.
Erlotinib functions by inducing cell cycle arrest at
the G1 checkpoint (10). Cell
cycle arrest is triggered by the transcriptional repression of the
cyclin D1 cell cycle regulatory protein (11). This effect has been documented in
both EGFR mutant and erlotinib-sensitive, EGFR wild-type lung
cancer cell lines (11,12). Erlotinib-resistant, EGFR wild-type
lung cancers do not exhibit this effect (11). The combination of bexarotene and
erlotinib also has been shown to reduce cyclin D1 expression
(13) and the BATTLE trial found
that high intratumoral cyclin D1 predicted favorable clinical
outcomes with this combination (14). In vitro studies have shown
that cyclin D3 is not repressed by erlotinib treatment and that
high cyclin D3 expression is associated with erlotinib resistance
(15). Based on this prior study,
we hypothesized that high cyclin D1 expression would predict
favorable outcomes and high cyclin D3 expression would predict
unfavorable outcomes with dose-intense erlotinib maintenance.
Materials and methods
Eligibility
Patients were required to have stage IV non-small
cell lung cancer. All patients were required to have a documented
histopathologic or cytopathologic diagnosis. Patients were allowed
to have either measurable disease or evaluable disease. ECOG
performance status (PS) of 0 or 1 was required. Patients were
ineligible if they had received prior chemotherapy, had inadequate
organ function, were pregnant or breast feeding, or were currently
receiving radiation therapy.
Treatment plan
This study was approved by the Institutional Review
Board of Wake Forest University and was registered with ClinicalTrials.gov (NCT00723138). After obtaining
written informed consent, patients were treated with cisplatin 75
mg/m2 and docetaxel 75 mg/m2 with both drugs
given intravenously on day 1 every two weeks. Prophylactic
anti-emetics were given based on investigator's preference and
generally included fosaprepitant or aprepitant as well as 5-HT3
antangonists. Prophylactic growth factor support with pegfilgrastim
was administered day 2 of every cycle. Treatment was repeated for
four cycles or until unacceptable toxicity or disease
progression.
Erlotinib was started immediately after completion
or discontinuation of chemotherapy for all patients regardless of
response or progression on chemotherapy. The starting doses of
erlotinib were 300 mg daily for patients who were smoking at ≥10
cigarettes per day and 150 mg daily for all other patients. Doses
of erlotinib were increased in 75 mg increments every two weeks
until patients developed either grade 2 or 3 toxicities (according
to the National Cancer Institute Common Terminology Criteria for
Adverse Events version 3.0). In the event of grade 3 toxicities,
erlotinib was held until resolution to grade 1 and then the dose
was reduced by 75 mg daily. If the reduced dose was tolerated with
grade 2 or less toxicity, that was determined to be the MTD for
that patient. In the event of grade 2 toxicities, medical
interventions were added and erlotinib was continued at that dose
which was determined to be the MTD for that patient.
Study procedures
At the time of enrollment, all patients completed a
detailed smoking history questionnaire. Patients were categorized
as never smokers (<100 lifetime cigarettes), distant former
(>1 year since cessation), former (1 year - 1 month since
cessation), recent former (<1 month since cessation), and
current smokers. Physical examination and standard laboratory tests
were performed prior to each cycle of chemotherapy. Complete blood
counts were performed weekly during dose-dense chemotherapy. After
initiation of erlotinib, physical examination and laboratory tests
were performed every two weeks until the patient's erlotinib MTD
was established and then every four weeks. Tumor measurements were
performed prior to initiation of treatment, after completion of
dose-dense chemotherapy and then every eight weeks until disease
progression or unacceptable toxicity. Tumor response was assessed
using the Response Evaluation Criteria in Solid Tumors (RECIST)
(16).
Immunohistochemistry procedure
Formalin-fixed paraffin-embedded biopsy specimens
that had been obtained prior to enrollment were analyzed after the
completion of the study by a pathologist who was unaware of
clinical outcomes (Jennifer Laudadio). Antigen retrieval was
performed using the Leica antigen retrieval system according to the
manufacturer's protocol for 20 min prior to applying antibodies
(Leica Microsystems, Wetzlar, Germany). Cyclin D1 primary antibody
(Clone SP4, ThermoScientific, Waltham, MA, USA) at a dilution of
1:50 and cyclin D3 primary antibody (DCS-22, Leica Microsystems) at
a dilution of 1:20 were independently applied to prepared slides.
The percent of cancer cells staining positive for each cyclin was
determined. Tumors were then categorized as having high or low
expression based on whether the expression was above or below the
median percent staining result for each marker.
EGFR mutation analyses
Genomic DNA was extracted from paraffin-embedded
biopsy tissues using the DNEasy Tissue kit (Qiagen, Valencia, CA,
USA) according to the manufacturer's protocol. DNA concentrations
were measured using spectroscopy. Polymerase chain reaction (PCR)
assays were performed using the EGFR PCR Kit Using Scorpions and
Amplification Refractory Mutation System (Qiagen) to assess for 28
activating mutations in the EGFR gene. Activating mutations
identified with this method were confirmed and tested for the T790M
resistance mutation by a second independent analysis using Rotor
Gene analysis with the EGFR RGQ PCR kit (Qiagen).
Statistical methods
A sample size of 45 evaluable patients was selected
to provide 85% power to detect an improvement in time to
progression (TTP) by 2.5 months over the historical control of 4
months (17) using a two-sided
test, assuming exponential distribution of times and a Type I error
rate = 5%. Overall survival (OS), TTP and toxicity statistics were
performed on an intent-to-treat basis. Comparisons of patient
characteristics were performed using χ2 test except for
age which was compared using t-test. OS and TTP were assessed using
the Kaplan-Meier method. Comparisons of survival curves were
performed using the log-rank test. Cox proportional hazards models
were used to assess differences between biomarker expression groups
after controlling for chemotherapy response, age and PS. All
P-values shown are two-sided.
Results
Patients
Forty-five patients were enrolled from August, 2007
to February, 2011. The patient characteristics are displayed in
Table I. All patients were
eligible and received at least one cycle of chemotherapy. Five
patients did not receive erlotinib for the following reasons: one
died from a pulmonary embolism during dose-dense chemotherapy, two
initiated treatments other than erlotinib after dose-dense
chemotherapy and two discontinued all treatment. Two patients
initiated erlotinib but progressed prior to achieving MTD.
 | Table I.Baseline characteristics. |
Table I.
Baseline characteristics.
| Characteristic | No. (N=45) | % |
|---|
| Sex | | |
| Female | 20 | 44 |
| Male | 25 | 56 |
| Age (years) | | |
| Median | 60 | |
| Range | 33–80 | |
| Performance
status | | |
| 0 | 8 | 18 |
| 1 | 37 | 82 |
| Race/ethnicity | | |
| White | 35 | 78 |
| Black or African
American | 9 | 20 |
| Hispanic or
Latino | 1 | 2 |
| Pathologic
subtype | | |
| Adenocarcinoma | 27 | 60 |
| Squamous cell
carcinoma | 7 | 16 |
| Other | 11 | 24 |
| High-risk metastatic
sites | | |
| Brain
metastases | 16 | 36 |
| Subcutaneous tissue
metastases | 4 | 9 |
| Smoking status | | |
| Never | 7 | 16 |
| Distant former
(>1 year since cessation) | 16 | 36 |
| Former (1 year - 1
month since cessation) | 6 | 13 |
| Recent former (<1
month since cessation) | 8 | 18 |
| Current | 8 | 18 |
Toxicity
The toxicities during dose-dense chemotherapy and
dose-intense erlotinib are displayed in Table II. Toxicities during dose-dense
chemotherapy were primarily non-hematologic. Only one case of
febrile neutropenia occurred. Significant fatigue, anorexia and
dehydration were common, and 69% of patients received all four
cycles of chemotherapy.
| Table II.Adverse events. |
Table II.
Adverse events.
| Adverse event | Dose-dense
chemotherapy | Maintenance
Dose-intense erlotinib |
|---|
|
|
|---|
| All Grades | Grade 3 | Grade 4 | Grade 2 | Grade 3 | Grade 4 |
|---|
|
|
|
|
|
|
|---|
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | % |
|---|
| Nausea | 28 | 62 | 1 | 2 | | | | | | | | |
| Diarrhea | 14 | 31 | 3 | 7 | | | 19 | 42 | 4 | 9 | | |
| Constipation | 3 | 7 | 1 | 2 | | | | | | | | |
| Anorexia | 11 | 24 | | | | | 2 | 4 | 2 | 4 | | |
| Dehydration | 8 | 18 | 4 | 9 | 2 | 4 | 1 | 2 | 1 | 2 | | |
| Rash | 9 | 20 | 6 | 13 | | | 6 | 13 | 11 | 24 | | |
| Fatigue | 27 | 60 | 7 | 16 | | | | | 1 | 2 | | |
| Paronychia | | | | | | | 2 | 4 | | | | |
| Conjunctivis | 4 | 9 | 2 | 4 | | | | | | | | |
| Mucositis | 5 | 11 | 1 | 2 | | | | | 1 | 2 | | |
| Ototoxicity | 6 | 13 | 3 | 7 | | | | | | | | |
| Peripheral
neuropathy | 8 | 18 | 1 | 2 | | | | | | | | |
| Allergic
reaction | 3 | 7 | 3 | 7 | | | | | | | | |
| Neutropenia | 3 | 7 | 1 | 2 | 1 | 2 | | | | | | |
|
Hyperbilirubinemia | | | | | | | 1 | 2 | | | | |
| Transaminitis | | | | | | | | | | | 1 | 2 |
Toxicities during dose-intense erlotinib were
primarily rash and diarrhea. However, other toxicities including
anorexia, dehydration and fatigue were dose limiting in 14% of
patients. No patient on protocol developed grade 5 toxicity.
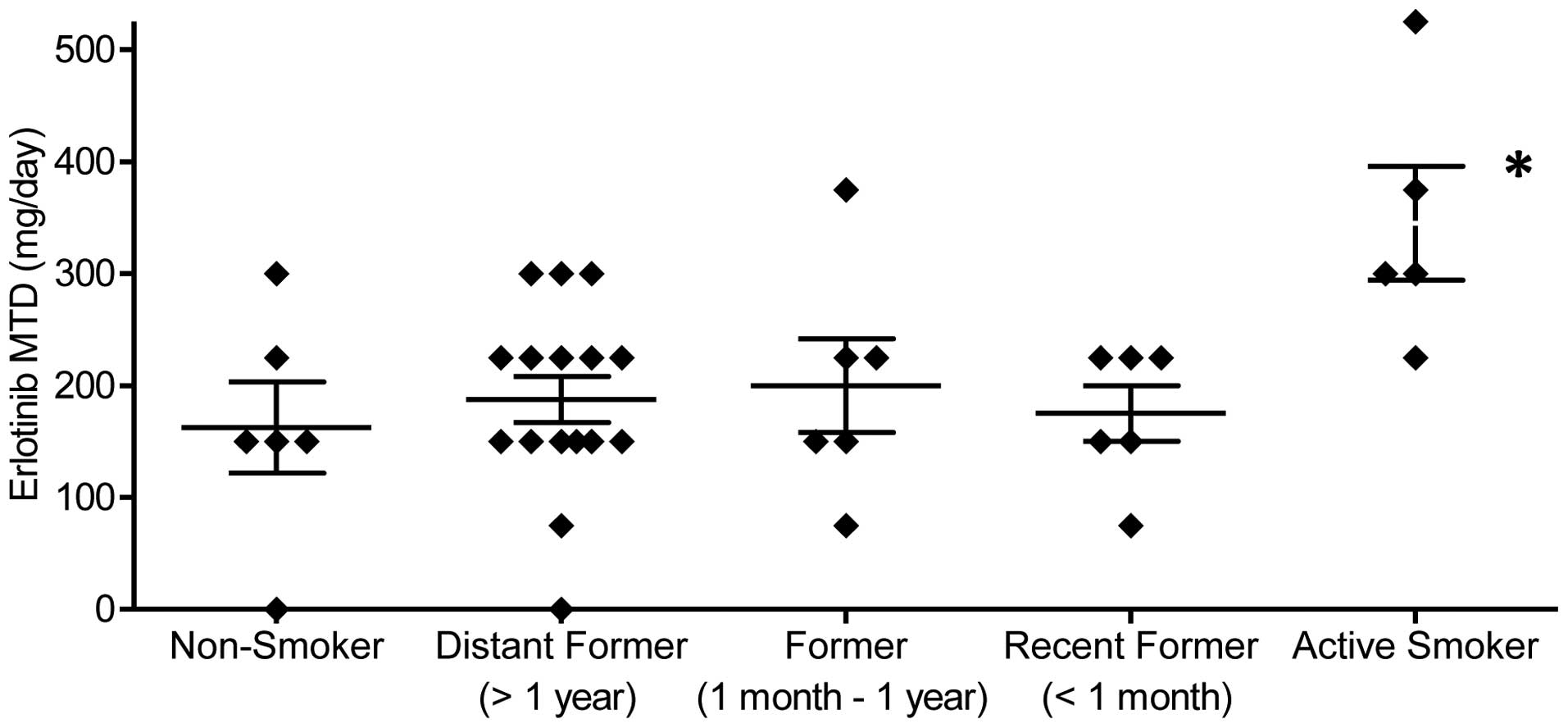
Impact of smoking on erlotinib MTD
The median erlotinib MTDs for lifelong non-smokers,
former smokers, and current smokers were 150, 187.5 and 300 mg
daily. The MTD for current smokers was significantly higher as
compared to lifelong non-smokers (P=0.019) and former smokers
(P<0.001). The MTD for former smokers was not significantly
different as compared to lifelong non-smokers (P=0.51). Fig. 1 depicts the MTD grouped by smoking
status. Within the group of former smokers, recent (quit <1
month prior) and distant (quit >1 year prior) former smokers
exhibited a similar median MTD.
Biomarker analyses
Thirty-four patients had adequate tissue for
immunohistochemical biomarker and EGFR mutation studies. Cyclin D1
expression ranged from 1 to 95% with a median of 33%. Cyclin D3
expression ranged from 5 to 85% with a median of 28%.
Samples from three patients were initially
identified as potentially harboring EGFR mutations. A subsequent
analysis confirmed that two of these three patients harbored EGFR
activating mutations (one del 19 and one L858R). Neither of these
co-expressed the T790M resistance mutation. The biomarker and
clinical outcomes for these patients are shown in Table III.
| Table III.EGFR mutation positive cases. |
Table III.
EGFR mutation positive cases.
| EGFR mutation
testing
| | | | |
|---|
| Case | Initial
testing | Confirmatory
testing | T790M testing | Cyclin D1 (%) | Cyclin D3 (%) | TTP | OS |
|---|
| 1 | G719X | wt | Negative | Low (25) | High (35) | 3.7 | 10.4 |
| 2 | Exon 19 del | Exon 19 del | Negative | Low (1) | High (40) | 6.3 | 8.1 |
| 3 | L858R | L858R | Negative | High (40) | High (40) | 49.7 | 55+ |
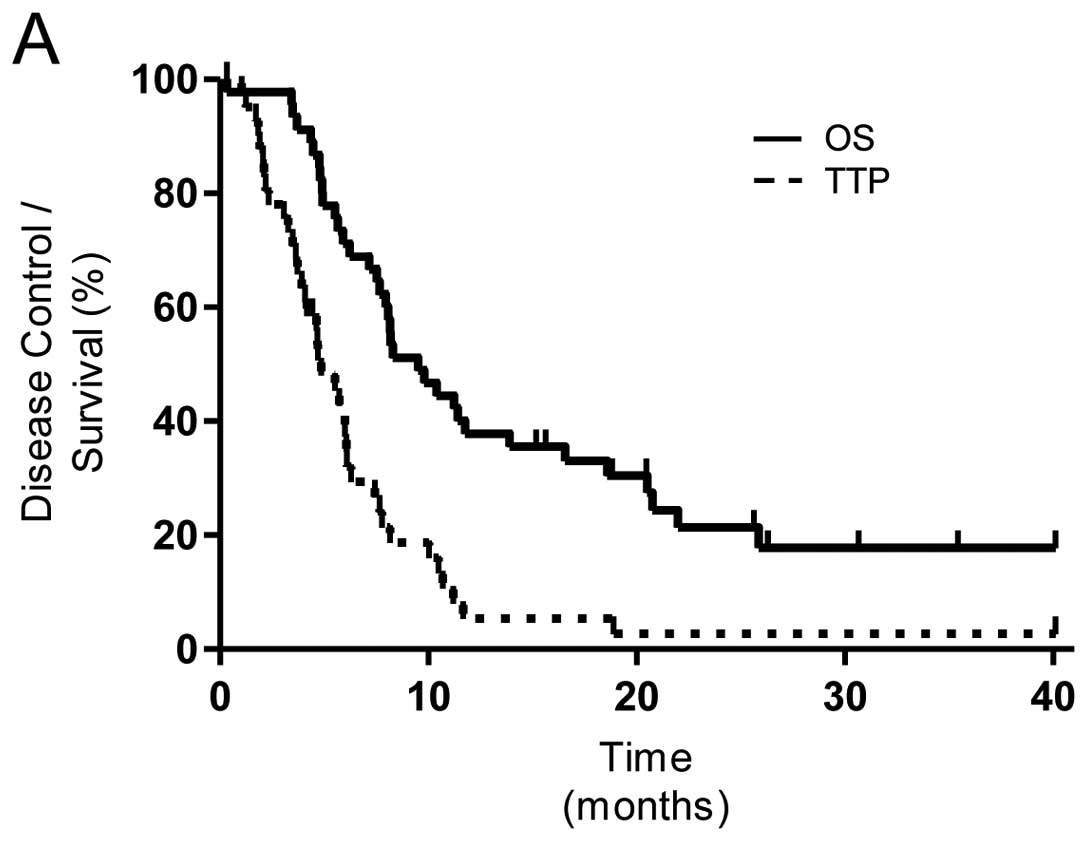
Response and survival
Radiographic responses following dose-dense
chemotherapy were partial response (20%), stable disease (63%) and
progressive disease (18%). For the entire study population, the
median time to progression was 4.6 months (95% CI, 3.7–6.1 months)
which was not significantly improved compared to the historical
control of 4 months. The median overall survival for the entire
study population was 9.5 months (Fig.
2).
As compared to low cyclin D1 expression, high cyclin
D1 expression was associated with longer TTP on erlotinib (8.2 vs.
4.7 months; hazard ratio, 4.1; 95% CI, 1.6–10.6; P=0.003) and
improved OS (20.5 vs. 8.0 months; hazard ratio 2.8; 95% CI 1.2–6.3;
P=0.016) as shown in Fig. 2.
Cyclin D3 has been proposed as a marker of erlotinib resistance.
However, no significant difference between high cyclin D3 and low
cyclin D3 was observed for TTP on erlotinib (6.1 vs. 5.7 months;
hazard ratio 1.0; 95% CI, 0.43–2.6; P=0.98) or OS (11.8 vs. 8.3
months; hazard ratio 1.3; 95% CI, 0.63–2.8; P=0.46).
Cox proportional hazards models were used to control
for the potential confounding effects of chemotherapy response, age
and PS on clinical outcomes. After controlling for these variables,
high cyclin D1 continued to predict improvements in TTP on
erlotinib (hazard ratio 3.0; 95% CI, 1.32–6.78; P=0.009) and OS
(hazard ratio 3.38; 95% CI, 1.28–8.94; P=0.014). After controlling
for these variables, the effect of cyclin D3 continued to be
non-significant for TTP on erlotinib (P=0.71) and OS (P=0.94).
Clinical characteristics of patients with high
cyclin D1 and low cyclin D1 expressing cancers are compared in
Table IV. Radiographic responses
to dose-dense chemotherapy were similar between these groups of
patients. High cyclin D1 expressing cancers were more like to be
adenocarcinomas and were more likely to present with brain or
soft-tissue metastases.
| Table IV.Characteristics of low versus high
cyclin D1 expressing cancers. |
Table IV.
Characteristics of low versus high
cyclin D1 expressing cancers.
| Characteristic | Low cyclin D1 | High cyclin D1 | P-value |
|---|
|
|
|---|
| No. | % | No. | % |
|---|
| Sex | | | | | 0.05 |
| Female | 5 | 29 | 9 | 53 | |
| Male | 12 | 71 | 8 | 47 | |
| Age (years) | | | | | 0.98 |
| Median | 61 | | 60 | | |
| Range | 38–72 | | 45–80 | | |
| Performance
status | | | | | 0.11 |
| 0 | 2 | 12 | 5 | 29 | |
| 1 | 15 | 88 | 12 | 71 | |
| Race/ethnicity | | | | | 0.22 |
| White | 16 | 94 | 13 | 76 | |
| Black or African
American | 1 | 6 | 3 | 18 | |
| Hispanic or
Latino | | | 1 | 6 | |
| Pathologic
subtype | | | | | 0.03 |
|
Adenocarcinoma | 9 | 53 | 12 | 71 | |
| Squamous cell
carcinoma | 5 | 29 | 0 | 0 | |
| Other | 3 | 18 | 5 | 29 | |
| High-risk
metastatic sites | | | | | 0.02 |
| Brain
metastases | 4 | 24 | 8 | 47 | |
| Subcutaneous
tissue metastases | 0 | 0 | 3 | 18 | |
| No high-risk
sites | 13 | 76 | 8 | 47 | |
| Smoking status | | | | | 0.27 |
| Never | 1 | 6 | 4 | 24 | |
| Distant former
(>1 year since cessation) | 6 | 35 | 5 | 29 | |
| Former (1 year - 1
month since cessation) | 3 | 18 | 2 | 12 | |
| Recent former
(<1 month since cessation) | 4 | 24 | 2 | 12 | |
| Current | 3 | 18 | 4 | 24 | |
| Chemotherapy
response (N=16 and N=15) | | | | | 0.51 |
| Progressive
disease | 3 | 19 | 4 | 27 | |
| Stable
disease | 9 | 56 | 9 | 60 | |
| Partial
response | 4 | 25 | 2 | 13 | |
Discussion
In this study, dose-dense chemotherapy was
associated with high degree of treatment related toxicities and a
response rate lower than that observed in a prior
multi-institutional study (1).
This limits interest in future studies utilizing the dose-dense
cisplatin and docetaxel regimen for unselected patients. Rapid
dose-escalation of erlotinib following completion of chemotherapy
was safe and well-tolerated. Increasing erlotinib dose by 75 mg
every two weeks effectively achieved MTD with only two patients
progressing prior to reaching MTD. While TTP and OS outcomes were
lower than those obtained in the SATURN study, a direct comparison
cannot be made between these studies since the SATURN population
was restricted to patients with clinical benefit from initial
chemotherapy treatment (stable disease, partial response, or
complete response) (3).
In some situations, erlotinib resistance may be
related to achieving inadequate drug levels. Intensification of
erlotinib dose in the maintenance setting is an attractive approach
to prevent this form of resistance. Our findings confirm prior
reports indicating that the MTD of erlotinib is higher in patients
who continue to smoke. A randomized phase III trial is ongoing to
test whether high dose erlotinib treatment will improve the
clinical outcomes over standard dose erlotinib for patients who
continue to smoke (18).
In the present study, the MTD for former smokers was
also examined. The median MTD for former smokers was significantly
lower than for current smokers and was not elevated compared to
lifelong non-smokers. These findings indicate that the increase in
erlotinib metabolism triggered by tobacco exposure is reversible
following smoking cessation. Providing smoking cessation
interventions to patients after diagnosis of lung cancer improves
clinical outcomes and reducing the risk of inadequate drug levels
may help to explain this effect (19,20).
EGFR mutation analysis is a valuable test for
identifying highly sensitive tumors in patients who will benefit
from first line erlotinib instead of chemotherapy. Consistent with
this, the patient in the present study with the longest TTP and OS
demonstrated an EGFR activating mutation as well as high cyclin D1
(Table III). Some patients with
EGFR wild-type cancers also benefit from erlotinib treatment
(3). Several emerging biomarkers
that could identify the subset of erlotinib-sensitive EGFR
wild-type cancers are undergoing clinical testing. These include
Ras mutations, TGF-α, E-cadherin and cyclin D1 among others
(14,21–23).
The present study supports high cyclin D1 expression as a marker of
erlotinib sensitivity. High cyclin D3 expression failed to predict
erlotinib resistance in this study.
Cyclin D1 was identified as a biomarker by studying
the mechanism of action of erlotinib using in vitro models
as well as pre- and post-treatment cancer biopsies (11). Cyclin D1 has been proposed as a
nodal point for EGFR signaling with multiple pathways leading from
EGFR activation to induction of this cell cycle regulator. The
present study found no significant difference in chemotherapy
response for high cyclin D1 expressing cancers but did show
significant improvements in TTP on erlotinib and OS. In light of
these findings and the results of the BATTLE trial, cyclin D1
immunohistochemical staining appears to be a promising biomarker
for predicting erlotinib sensitivity and additional clinical
testing is warranted.
Acknowledgements
W.J.P. received funding from Astellas
Pharmaceuticals to conduct this study. Additional funding was
provided by Cancer Center Support Grant P30 CA12197 from the
National Cancer Institute.
References
|
1.
|
Miller AA, Wang XF, Gu L, et al: Phase II
randomized study of dose-dense docetaxel and cisplatin every 2
weeks with pegfilgrastim and darbepoetin alfa with and without the
chemo-protector BNP7787 in patients with advanced non-small cell
lung cancer (CALGB 30303). J Thorac Oncol. 3:1159–1165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Shepherd FA, Rodrigues Pereira J, et al:
Erlotinib in previously treated non-small-cell lung cancer. N Engl
J Med. 353:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Cappuzzo F, Ciuleanu T, Stelmakh L, et al:
Erlotinib as maintenance treatment in advanced non-small-cell lung
cancer: a multicentre, randomised, placebo-controlled phase 3
study. Lancet Oncol. 11:521–529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Petrelli F, Borgonovo K, Cabiddu M, et al:
Relationship between skin rash and outcome in non-small-cell lung
cancer patients treated with anti-EGFR tyrosine kinase inhibitors:
a literature-based meta-analysis of 24 trials. Lung Cancer.
78:8–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lee SM, Khan I, Upadhyay S, et al:
First-line erlotinib in patients with advanced non-small-cell lung
cancer unsuitable for chemotherapy (TOPICAL): a double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 13:1161–1170.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hughes AN, O'Brien ME, Petty WJ, et al:
Overcoming CYP1A1/1A2 mediated induction of metabolism by
escalating erlotinib dose in current smokers. J Clin Oncol.
27:1220–1226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Jänne PA, Wang X, Socinski MA, et al:
Randomized phase II trial of erlotinib alone or with carboplatin
and paclitaxel in patients who were never or light former smokers
with advanced lung adenocarcinoma: CALGB 30406 trial. J Clin Oncol.
30:2063–2069. 2012.
|
|
8.
|
Fukuoka M, Wu YL, Thongprasert S, et al:
Biomarker analyses and final overall survival results from a phase
III, randomized, open-label, first-line study of gefitinib versus
carboplatin/paclitaxel in clinically selected patients with
advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol.
29:2866–2874. 2011. View Article : Google Scholar
|
|
9.
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ling YH, Li T, Yuan Z, et al: Erlotinib,
an effective epidermal growth factor receptor tyrosine kinase
inhibitor, induces p27KIP1 up-regulation and nuclear translocation
in association with cell growth inhibition and G1/S phase arrest in
human non-small-cell lung cancer cell lines. Mol Pharmacol.
72:248–258. 2007. View Article : Google Scholar
|
|
11.
|
Petty WJ, Dragnev KH, Memoli VA, et al:
Epidermal growth factor receptor tyrosine kinase inhibition
represses cyclin D1 in aerodigestive tract cancers. Clin Cancer
Res. 10:7547–7554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kobayashi S, Shimamura T, Monti S, et al:
Transcriptional profiling identifies cyclin D1 as a critical
downstream effector of mutant epidermal growth factor receptor
signaling. Cancer Res. 66:11389–11398. 2006. View Article : Google Scholar
|
|
13.
|
Dragnev KH, Petty WJ, Shah S, et al:
Bexarotene and erlotinib for aerodigestive tract cancer. J Clin
Oncol. 23:8757–8764. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Kim ES, Herbst RS, Wistuba II, et al: The
BATTLE trial: personalizing therapy for lung cancer. Cancer Discov.
1:44–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Petty WJ, Voelzke WR, Urbanic JJ, et al:
High cyclin D3 expression confers erlotinib resistance in
aerodigestive tract cancer. Lung Cancer. 74:384–391. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Therasse P, Arbuck SG, Eisenhauer EA, et
al: New guidelines to evaluate the response to treatment in solid
tumors. J Natl Cancer Inst. 92:205–216. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Schiller JH, Harrington D, Belani CP, et
al: Comparison of four chemotherapy regimens for advanced non-small
cell lung cancer. N Engl J Med. 346:92–98. 2002. View Article : Google Scholar
|
|
18.
|
Roche trials database: A study of Tarceva
(erlotinib) to compare two different doses in currently smoking
patients with advanced or metastatic non-small cell lung cancer
(CURRENTS). <http://www.roche-trials.com/trialDetailsGet.action?studyNumber=M022162>.
|
|
19.
|
Parsons A, Daley A, Begh R, et al:
Influence of smoking cessation after diagnosis of early stage lung
cancer on prognosis: systematic review of observational studies
with meta-analysis. BMJ. 340:b55692010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Waller LL, Weaver KE, Petty WJ, et al:
Effects of continued tobacco use during treatment of lung cancer.
Expert Rev Anticancer Ther. 10:1569–1575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Brugger W, Triller N, Blasinska-Morawiec
M, et al: Prospective molecular marker analyses of EGFR and KRAS
from a randomized, placebo-controlled study of erlotinib
maintenance therapy in advanced non-small-cell lung cancer. J Clin
Oncol. 29:4113–4120. 2011. View Article : Google Scholar
|
|
22.
|
Addison CL, Ding K and Zhao H: Plasma
transforming growth factor alpha and amphiregulin protein levels in
NCIC Clinical Trials Group BR.21. J Clin Oncol. 28:5247–5256. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Witta SE, Jotte RM, Konduri K, et al:
Randomized phase II trial of erlotinib with and without
entinostatin patients with advanced non-small-cell lung cancer who
progressed on prior chemotherapy. J Clin Oncol. 30:2248–2255. 2012.
View Article : Google Scholar : PubMed/NCBI
|