Introduction
It has been established that Notch signaling plays
an important role in cell proliferation and apoptosis (1). A number of studies have confirmed
that Notch genes are abnormally activated in many human cancers,
including breast cancer (2). In
mammalian cells, activation of Notch signaling is induced through
the interaction of Notch ligands, Delta-like and Jagged, with their
receptors, Notch1, Notch2, Notch3 and Notch4 (3). Notch receptors are transmembrane
proteins that are comprised of a large extracellular domain and a
small intracellular domain (NCID). Overexpression of Notch1 or
Notch4 has been shown to promote tumorigenesis in murine mammary
glands (3,4). Furthermore, it has been reported that
the acquisition of epithelial-mesenchymal transition (EMT), coupled
to the cancer stem cell (CSC) phenotype, is induced by Notch1
overexpression in pancreatic cells (5). Finally, the generation of CSCs, which
is crucial for the genesis/maintenance of cancer and probably for
metastasis, is promoted by Notch signaling (6).
The process of EMT plays an important role in the
development of malignant tumors. It has been shown that EMT is a
pivotal step in cancer cell invasion and migration (7,8). EMT
is a multistep process in which epithelial cells lose their
polarity and adhesive properties and gain the migratory and
invasive properties of mesenchymal cells (9). The expression of epithelial marker
genes such as E-cadherin is downregulated, and the expression of
mesenchymal genes such as N-cadherin and vimentin is upregulated
during EMT (10,11). EMT not only facilitates tumor
metastasis, but it also promotes the occurrence of CSCs in breast
cancer. CSCs represent a subset of tumor cells that have a capacity
for selfrenewal, tumor propagation and differentiation into
multiple cell lineages (12).
Mammary epithelial tumor cells that have undergone EMT have been
shown to have CSC characteristics, such as the
CD44+/CD24− phenotype and an accelerated
selfrenewal ability (13).
Although in recent years the role of EMT in tumor development has
become a focus of many studies, the molecular mechanisms that
control EMT have not been completely elucidated.
Signal transducer and activator of transcription 3
(STAT3) is a transcription factor that is activated by
proinflammatory cytokines, growth factors and oncogenic proteins,
thus regulating a variety of biological processes (14,15).
The abnormal activation of STAT3 is associated with the progression
of numerous human carcinomas, including breast cancer (16). Furthermore, previous studies have
demonstrated that Notch activation induces EMT and the CSC
phenotype through STAT3 activation (17).
The transcription factor Twist, an important
regulator of EMT, is overexpressed in several cancers and regulates
tumorigenesis (18–20). It has been reported that Twist is
elevated through crosstalk with other pathways in breast cancer
(21,22). Upregulation of the Notch receptor
correlates with Twist expression in Drosophila. Moreover,
Drosophila mesoderm subdivision is regulated by the Notch
signaling pathway through controlling the expression Twist
(23,24). A previous study confirmed that
activation of the STAT3/Twist signaling pathway regulates cancer
progression through crosstalk with Notch1 signaling in gastric
cancer (25). Numerous studies
have demonstrated that inflammatory cytokines regulate EMT and the
CSC phenotype (26). However, at
present, the mechanism whereby Notch1 regulates EMT and the CSC
phenotype remains unclear. In this study, we investigated the
effect of Notch1 signaling on EMT and CSCs, with the focus on
crosstalk between Notch1, STAT3 and NF-κB signaling.
Materials and methods
Cell culture
The MCF10A and MCF7 cell lines were obtained from
The Cell Bank of Shanghai (Shanghai, China). MCF7 and MCF10A cells
were infected with lentiviral vector, where the HIV promoter
directs the expression of NICD. The lentiviral vector was obtained
from Genechem Co. Ltd. (Shanghai, China). Stable clones were
selected by the limiting dilution assay and the expression level of
NICD was confirmed by western blot analysis. MCF7-control and
MCF7-Notch1 cells were cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS); and
MCF10A-control and MCF10A-Notch1 cells were cultured in DMEM/F12
supplemented with 5% horse serum, 0.5 μg/ml hydrocortisone, 10
μg/ml insulin (Invitrogen, Carlsbad, CA, USA) and 100 ng/ml cholera
toxin (Biomal, Hamburg, Germany).
Cell proliferation assay
Cell proliferation assay was assayed by the
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich, St. Louis, MO, USA) assay. Tumor cells were seeded
in 96-well plates at a density of 1,000 cell/well. After 24, 48, 72
and 96 h, 20 μl of MTT solution (5 mg/ml) was added to each well.
Cells were incubated for 4 h at 37°C. The MTT was dissolved in 200
μl of dimethylsulfoxide (DMSO) and the absorbance was measured
using a microplate reader at 490 nm. Wells without cells were used
for the blank of the spectrophotometer. The assays were performed
in triplicate for each condition and the experiments were repeated
three times.
Western blot analysis
Total proteins were extracted from cells using RIPA
buffer containing proteinase inhibitors for 20 min on ice and then
cleared at 12,000 rpm for 20 min at 4°C. The protein concentration
was measured using the Bradford assay (Sigma Chemicals, Bangalore,
India). Equal amounts of total proteins were subjected to SDS-PAGE
under reducing conditions followed by transfer to polyvinylidene
difluoride membranes. After blocking, the membranes were incubated
at 4°C overnight with the primary antibody. Antibodies specific for
E-cadherin (Cell Signaling Technology, Inc.), occludin (Proteintech
Group Inc.), N-cadherin (Cell Signaling Technology, Inc.), vimentin
(Cell Signaling Technology, Inc.), p65 (Abcam, Cambridge, UK),
fibronectin (Cell Signaling Technology, Inc.), Notch1 (Abcam),
STAT3 (Proteintech Group Inc.), p-STAT3 (Epitomics, Inc.) and
interleukin (IL)-1β (Cell Signaling Technology, Inc.) were used in
this study. After a wash in Tris hydrochloride buffer (TBS)
containing 0.1% Tween-20 (TBS-T), the membranes were incubated with
an HRP-conjugated secondary antibody (Santa Cruz Biotechnology,
Inc.) for 2 h at room temperature. The membranes were treated with
ECL plus reagent (GE Healthcare) and exposed to X-ray film.
Autoradiogram signals were documented by a gel densitometric
scanning program.
Cell migration and invasion assays
For the migration assays, 8×104 cells
were seeded on 8-μm transwell filters (Millipore) in 200 μl of
complete medium without FBS. In the lower chamber, 500 μl of
complete medium supplemented with 10% FBS was added. Cells were
incubated for 12 or 24 h. For the invasion assays, the membranes
were coated with Matrigel (1.5 mg/ml) and the cells were cultured
for 48 h. Non-intruding cells were removed with a cotton swab. The
cells that adhered to the underside of the membrane were fixed and
stained with crystal violet solution. Ten random fields were
counted under a bright-field microscope.
Flow cytometry
The identification of
CD44+/CD24− was assayed using anti-CD44-FITC
(Abcam) and anti-CD24-PE antibodies (Abcam). Cells were cultured in
DMEM/F12 supplemented with 20 ng/ml EGF and 20 ng/ml FGF-β. After 2
weeks, cells were centrifuged at 1,000 rpm for 5 min and
re-suspended in 100 μl of FACS buffer containing 20 μl of CD44 and
CD24 antibodies for 30 min on ice. Labeled cells were analyzed by
flow cytometer (BD Biosciences, San Diego, CA, USA).
Immunofluorescence assay
A total of 2×104 cells were plated on
poly-L-lysine-coated coverslips in 24-well plates. After 48 h, the
cells were washed with phosphate-buffered saline (PBS) and fixed
with 4% paraformaldehyde for 30 min. Next, the cells were
permeabilized with 0.3% Triton X-100 for 5 min. After blocking with
goat serum for 30 min, the cells were incubated with a rabbit
polyclonal CD24 antibody (Abcam) and a mouse polyclonal CD44
antibody (Abcam) overnight at 4°C. The slides were washed with PBS,
stained with Cy3-conjugated secondary antibody for 2 h at room
temperature and imaged under a fluorescence microscope (Ex and Em
wavelengths were set at 550 and 620 nm, respectively, with exposure
at 400 msec).
Tumor formation in vivo
Athymic nude mice (Silaike Laboratory Animal Co.,
Ltd., Shanghai, China) were used to assess the effect of Notch1 on
tumor growth and metastasis in vivo. The animal protocol was
authorized by the Animal Care and Use Committee of Xi’an Jiaotong
University. Approximately 8×106 of MCF7 or MCF7-notch1
cells resuspended in 0.3 ml of serum-free medium were implanted
into the fat pads of mice. Tumor growth was monitored every 2 days
for 28 days. The tumor volume was calculated using the following
formula: length × width2 × 0.5. The tumor tissues was
removed for immunohistochemistry.
Immunohistochemistry
Fresh tissue was fixed in 4% formaldehyde and
embedded into paraffin with 4-μm thickness. The slides were
de-paraffinized with xylene, dehydrated through a graded alcohol
series and subsequently incubated with 0.3% hydrogen peroxide for
10 min. Antigens were retrieved via heating in citrate buffer for 1
h at 95°C. The slides were incubated with hydrogen peroxide for 5
min and blocked with 10% of normal goat serum for 15 min at room
temperature and then incubated with anti-E-cadherin antibody
(1:100; Beijing Biosynthesis Biotechnology), anti-N-cadherin
antibody (1:50; Beijing Biosynthesis Biotechnology) and
anti-vimentin antibody (1:100; Beijing Biosynthesis Biotechnology)
in humidified chamber overnight at 4°C, washed in PBS and incubated
with secondary antibody for 15 min at 37°C, followed by incubation
with streptavidin-peroxidase (Dako) for 15 min at 37°C. The slides
were counterstained with hematoxylin.
Statistical methods
The data were presented as means ± standard
deviation using GraphPad Prism software (version 5.0). Comparisons
between the groups were analyzed using the Student’s t-test.
P-values <0.05 were considered statistically significant.
Results
Notch1 overexpression promotes cell
proliferation in MCF7 and MCF10A cells
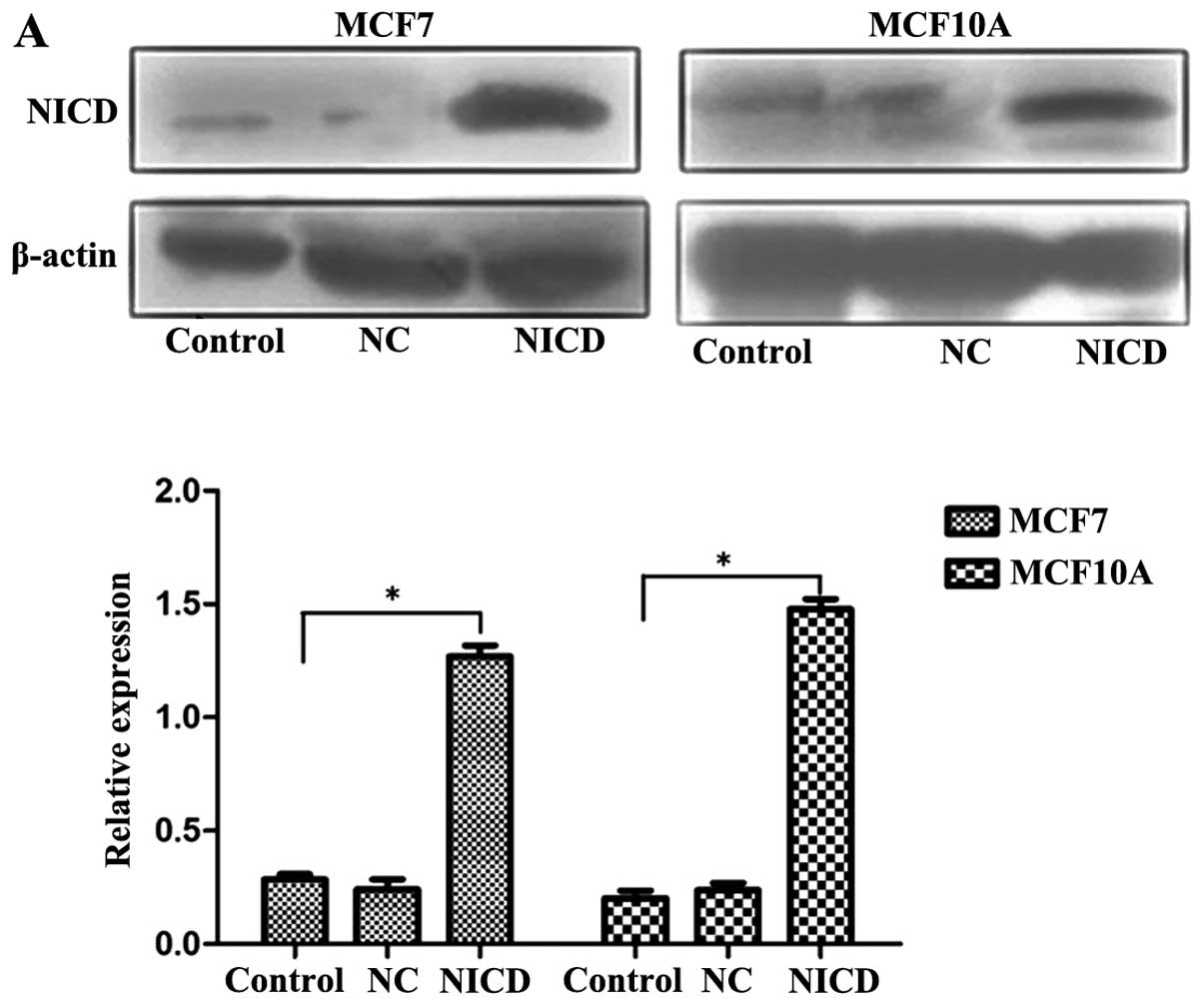
Transfected cells were analyzed by western blot
analysis using an anti-Notch1 antibody. We demonstrated that the
expression of Notch1 was significantly increased in MCF7 and MCF10A
cells (Fig. 1A). The effect of
Notch1 on the growth of breast cancer cells is shown in Fig. 1B. We demonstrated that
proliferation of MCF7-notch1 cells was increased at 24, 48, 72 and
96 h compared to control MCF7 cells (P<0.05). Similarly, Notch1
increased proliferation of MCF10A cells (P<0.05).
Notch1 overexpression promotes EMT
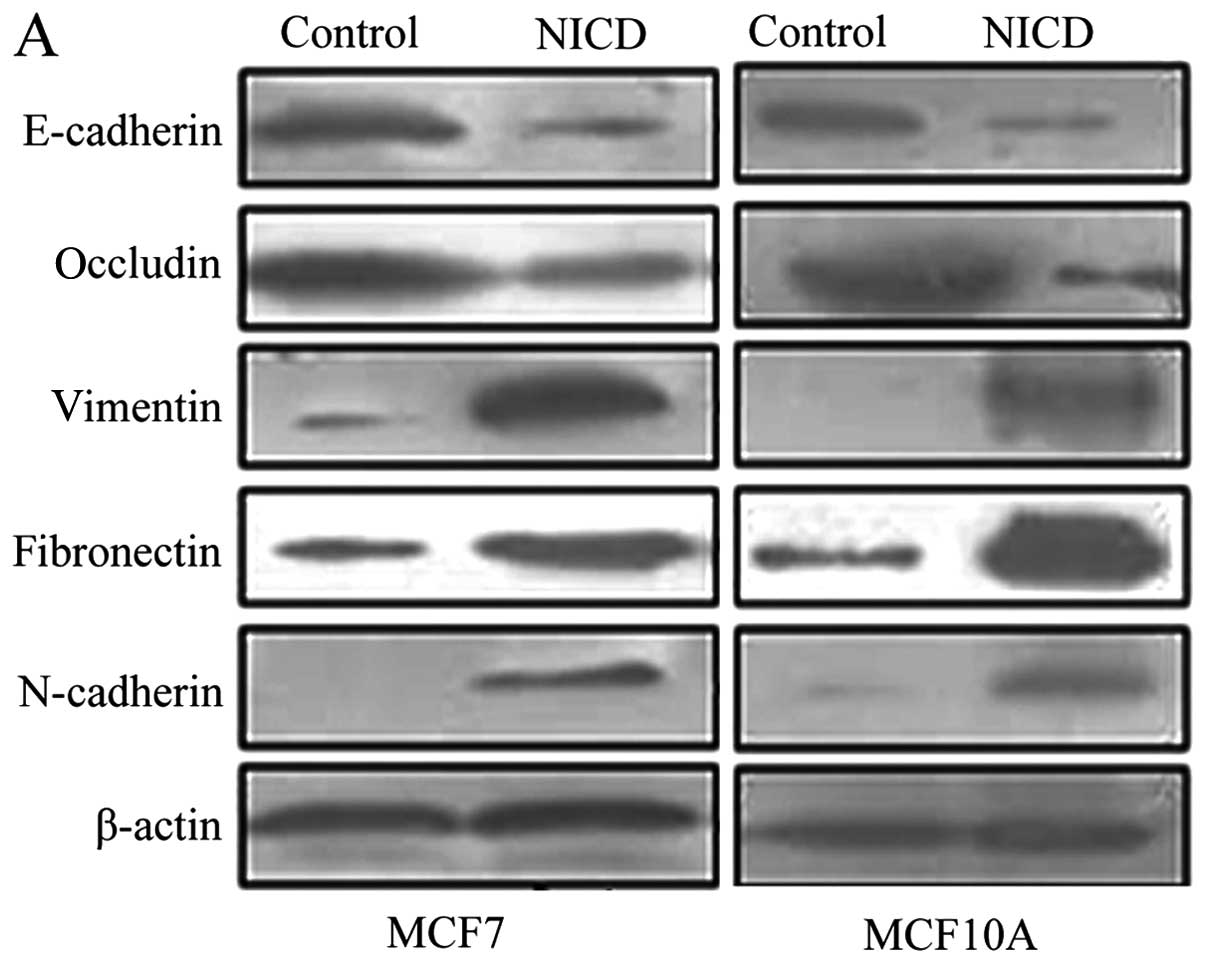
To assess the effect of Notch1 overexpression on
EMT, we compared the expression of E-cadherin, occludin,
N-cadherin, vimentin and fibronectin in MCF7 and MCF10A cells as
well as MCF7 and MCF10A cells transfected with Notch1 by western
blot analysis. We showed that Notch1 overexpression led to
increased expression of N-cadherin, vimentin, and fibronectin,
while the expression of E-cadherin and occludin was decreased
(Fig. 2A) (P<0.05). We also
found morphological changes in MCF7-notch1 and MCF10A-notch1 cells
that were consistent with EMT. The cell-cell junctions were
disrupted and the cells regained a fibroblast-like appearance
(Fig. 2B), characteristics of the
EMT phenotype. These findings suggested that overexpression of
Notch1 promotes acquisition of the EMT phenotype.
Notch1 promotes the migration and
invasion of MCF7 and MCF10A cells
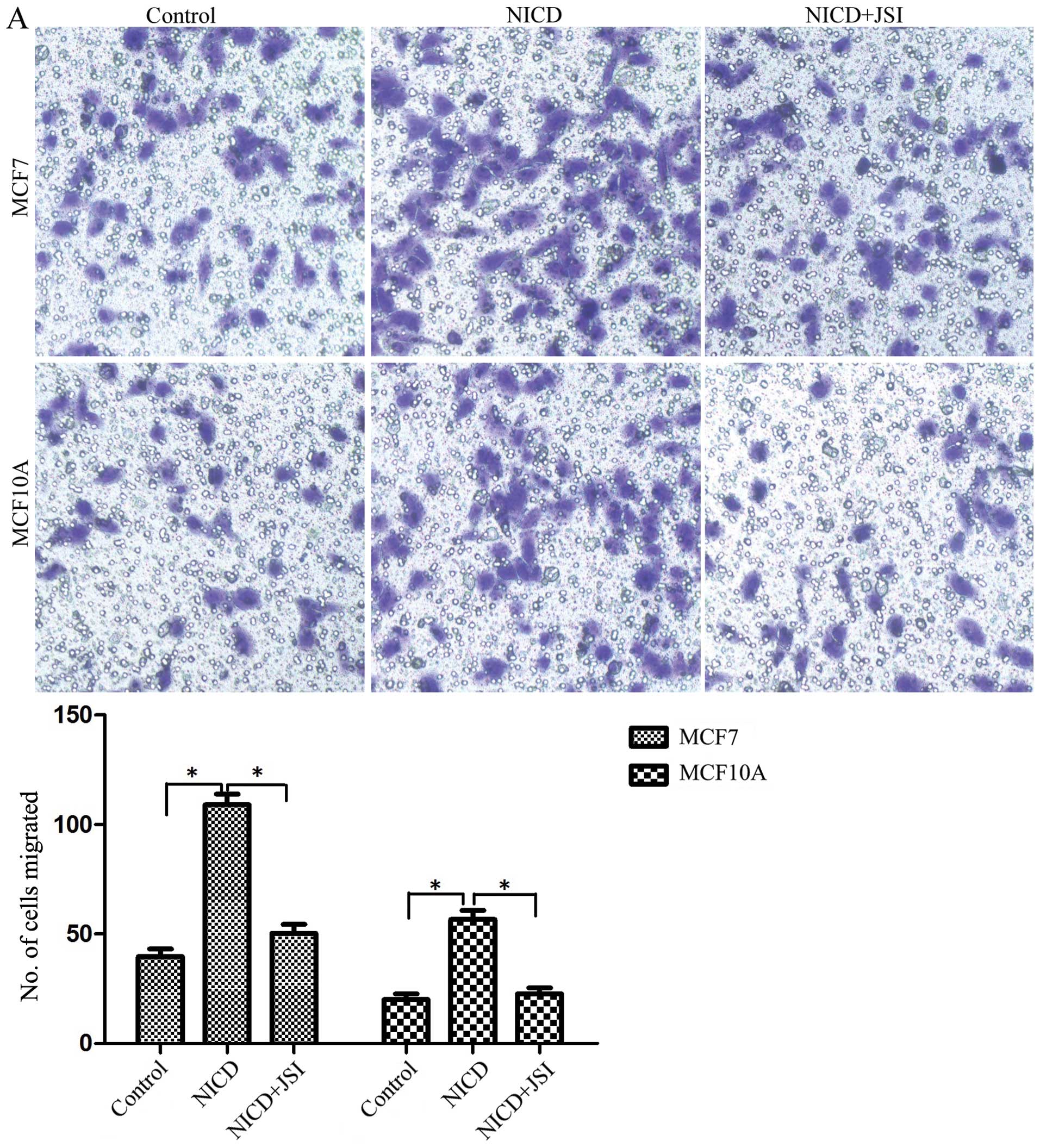
Next, we tested the effect of Notch1 on the
migration and invasion of MCF7 and MCF10A cells. As shown in
Fig. 3A, Notch1 overexpression led
to a 3.2-fold increase in migration of MCF7 cells (P<0.05) and a
2-fold increase in MCF10A-notch1 cells (P<0.05). In addition, we
also assessed the capability of these cells to invade using a
Matrigel-coated transwell assay. The expression of Notch1 increased
the invasion of MCF7 cells by 2.8- (P<0.05) and 1.9-fold in
MCF10A cells (P<0.05) (Fig.
3B). These results established that Notch1 promotes the
migration and invasiveness of breast cancer cells. A selective
JAK/STAT3 inhibitor, JSI124, decreased the migration of MCF7-notch1
cells by 2.6- (P<0.05) and by 1.7-fold in MCF10A-notch1 cells
(P<0.05) (Fig. 3A). The
invasion of MCF7-notch1 cells was reduced by JSI124 3.2-
(P<0.05) and 1.5-fold in MCF10A-notch1 cells (P<0.05)
(Fig. 3B). These results suggested
that Notch1 stimulates the invasion and migration of cancer cells
via STAT3.
Notch1 interacts with the STAT3 and NF-κB
signaling pathways in mammary cells
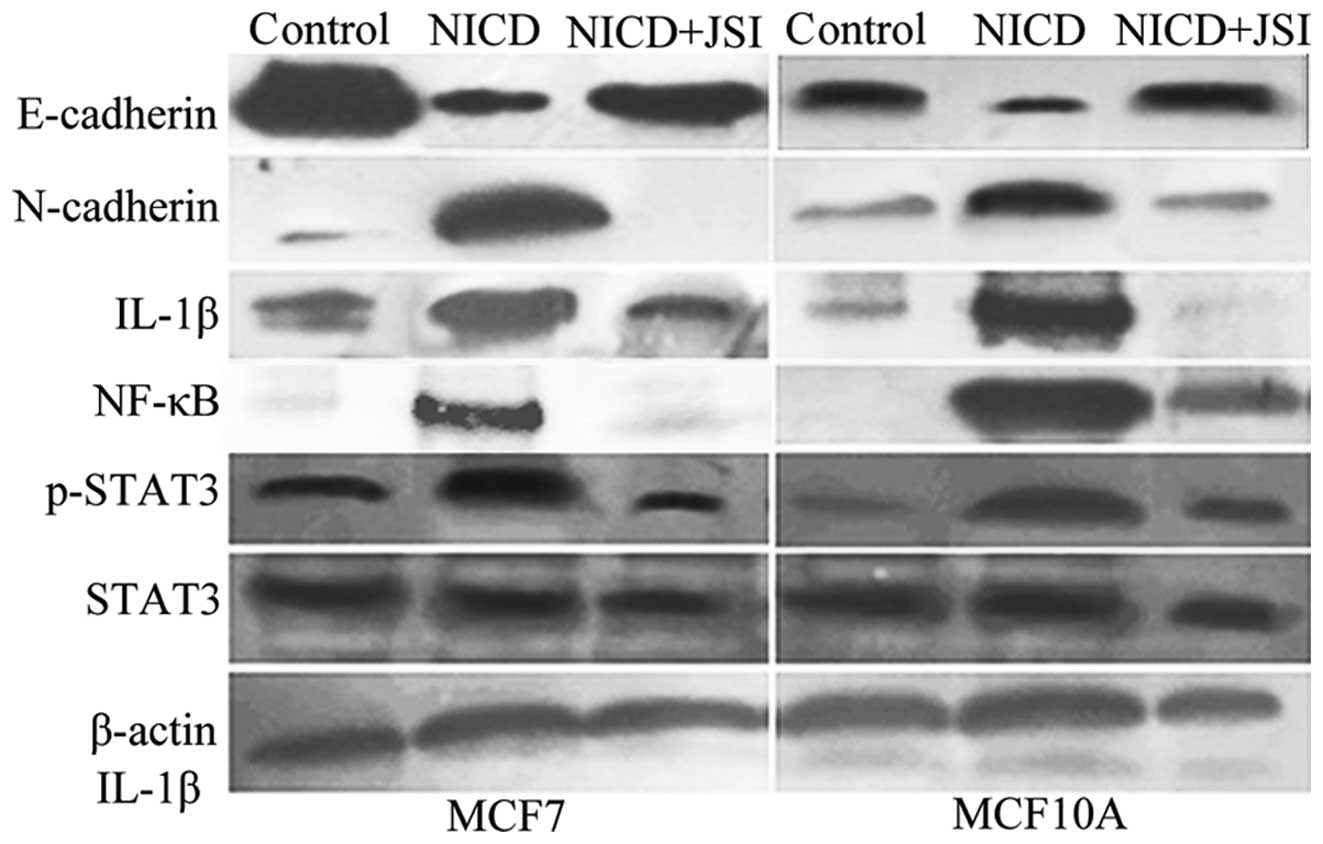
In order to test our hypothesis that Notch1 induces
EMT through interaction with other signaling pathways, such as
STAT3, we monitored the expression of STAT3 and p-STAT3 (phospho
S727) in breast cancer cells by western blot analysis. We showed
that Notch1 increased the expression of p-STAT3 in MCF7 and MCF10A
cells; the expression of total STAT3 was not affected by Notch1.
Several studies have suggested that NF-κB signaling also influences
the Notch signaling pathway (27).
We demonstrated that Notch1 increased the expression of p65 and the
NF-κB target gene, IL-1. As shown in previous studies, NF-κB plays
a key role in the regulation of apoptosis (27). To investigate the role of STAT3 in
Notch1 signaling, we inhibited STAT3 activity by the specific
inhibitor JSI-124. We demonstrated that JSI124 downregulated the
levels of p65 and IL-1β in MCF7-notch1 and MCF10A-notch1 cells
(P<0.05). We showed that JSI124 increased the expression of
E-cadherin and reduced the expression the EMT marker N-cadherin in
MCF7 and MCF10A cells expressing Notch1 (P<0.05) (Fig. 4).
Notch1 overexpression promotes the
expression of CSC surface markers in breast cancer cells
It has been suggested that the CSC phenotype is
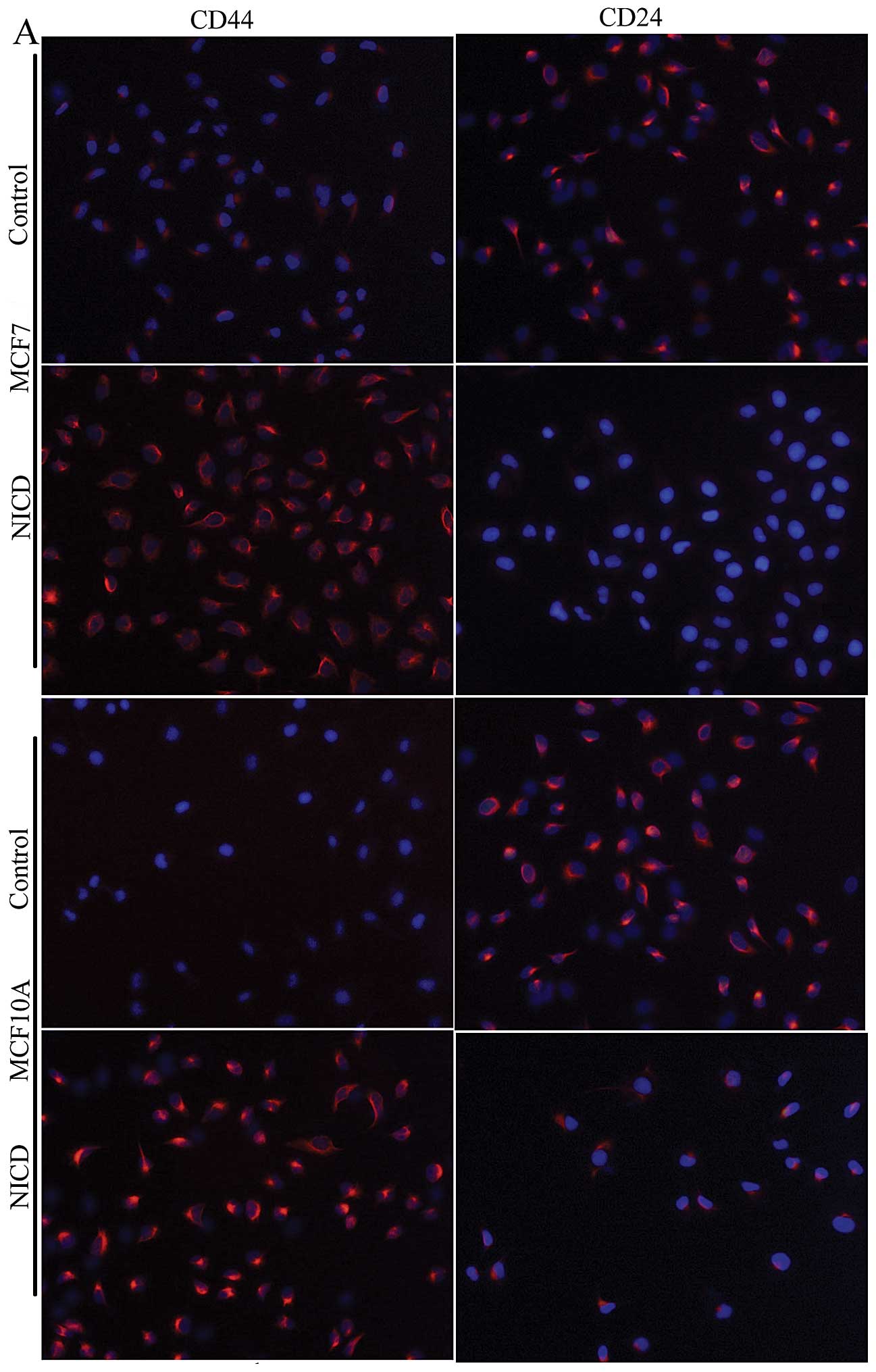
induced by Notch1 overexpression (6). We tested the CSC surface markers
using anti-CD44 and anti-CD24 antibodies by an immunofluorescence
assay. Notch1 increased the expression of CD44 and decreased the
expression of CD24 (Fig. 5A).
Accordingly, we demonstrated that the proportion of
CD44+/CD24low cancer stem cells was increased
in MCF7 and MCF10 cells upon the expression of Notch1 (Fig. 5B). Notch 1 increased the proportion
of CD44+/CD24low cells from 12.41 to 28.26%
in MCF7 cells and from 9.40 in to 19.21% in MCF10A cells. Thus, our
data suggested that Notch1 overexpression promotes the acquisition
of the breast CSC phenotype in MCF7 and MCF10A cells.
Notch1 overexpression promotes tumor
growth in vivo
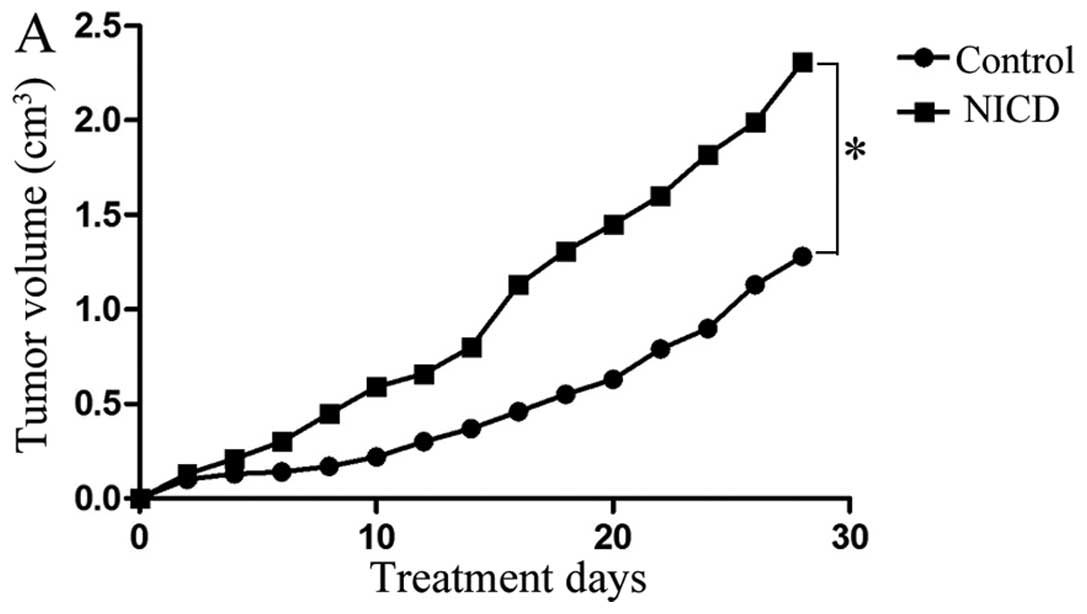
Next, we explored the effect of Notch1
overexpression on tumor growth and metastatic potential in
vivo. Consistent with our in vitro data, we showed that
MCF7 cells that overexpressed Notch1 grew significantly faster than
control cells (P<0.05) (Fig.
6A), confirming that Notch1 has a positive effect on tumor
growth in vivo. Immunohistochemical analysis showed that
tumors derived from MCF7-notch1 cells display elevated levels of
N-cadherin and vimentin, but reduced levels of E-cadherin (Fig. 6B).
Discussion
Several studies have suggested that the Notch
signaling pathway regulates the progression of solid tumors
(28). However, the molecular
mechanism linking the Notch signaling pathway and tumorigenesis has
not been completely elucidated. It has been demonstrated that the
upregulation of Notch promotes the growth of lung and breast cancer
cells (29,30). In this study, we showed that Notch1
overexpression enhanced the proliferation of MCF7 and MCF10A breast
cancer cells. The invasion and migration of cancer cells were also
promoted by Notch1 overexpression. In addition, we showed that
Notch1 overexpression advanced the growth of tumor cells in
vivo. All of these results are consistent with the notion that
the Notch signaling pathway plays an important role in the
progression of breast cancer.
Recent data have suggested that EMT is a key process
in tumor progression and metastasis related to abnormal Notch
signaling (31). Xie et al
confirmed that the upregulation of Notch1 signaling reinforces the
process of EMT in lung cancer (32). Our results showed that EMT markers,
such as N-cadherin and vimentin, were upregulated, but epithelial
markers (e.g., E-cadherin and occludin) were downregulated in MCF7
and MCF10A cells overexpressing Notch1. The extracellular matrix
(ECM) plays an important role in mammary gland progression and
breast cancer formation (33) and
rearrangement of ECM proteins is a hallmark of EMT (34). Our results support this viewpoint,
as we showed that fibronectin was increased in breast cancer cells
undergoing EMT.
Previous reports have shown that CSCs are present in
acute myeloid leukemia, breast cancer, pancreatic cancer, colon
carcinoma and melanoma (35–37).
EMT has been shown to generate cells with stem cell-like properties
(38–40). In addition, recent studies have
demonstrated that Notch signaling is involved in EMT and the
generation of CSCs (41). McGowan
et al have confirmed that the repression of Notch1 leads to
the generation of CD44+/CD24− cells and
reduces the formation of brain metastasis in breast cancer
(42). Notch1 also induced EMT
consistent with the CSC phenotype in pancreatic cancer cells
(6). In this study, we confirmed
that Notch1 regulates EMT and CSCs. The proportion of
CD44+/CD24− cells was higher in MCF7 and
MCF10A cells overexpressing Notch1. Furthermore, mammary gland
development is regulated by various signaling pathways that
regulate cell fate and cell differentiation. Notch4 and Notch3
genes have been shown to regulate normal mammary development.
Indeed, Notch family members play a significant role in mammosphere
formation and their ligands may affect mammary epithelial cell
development (43–45). In this study, we only explored the
gene function of Notch1; Notch2, Notch3 and Notch4 were not
studied.
The effect of Notch signaling on CSCs is enhanced by
crosstalk with other signaling pathways, such as Hedgehog and Wnt
(46). Hsu et al have
confirmed that activation of the Notch1/STAT3/Twist pathways is
involved in the development of human gastric cancer (25). Several studies have shown that
inflammatory cytokines such as IL-1 and IL-6 are important factors
in Notch signaling (47,48) and that there is crosstalk between
Notch and NF-κB signaling (44).
Liu et al have investigated the effect of crosstalk of the
Notch1, STAT3 and NF-κB signaling pathways on human epidermal
tumors (27). We showed that the
expression of p65 and IL-1β were increased in Notch1-overexpressing
cells. To establish the role of STAT3 signaling, we used a
STAT3-specific inhibitor. Strikingly, we found that the expression
levels of p65 and IL-1β were downregulated upon inhibition of
STAT3. Inhibition of STAT3 activity by JSI124 inhibited EMT, as
shown by increased levels of E-cadherin and reduced levels of
N-cadherin. In addition, inhibition of STAT3 activity also
suppressed cell migration and invasion. These results suggest that
STAT3 phosphorylation is a key downstream target of Notch signaling
and that Notch1 induces the acquisition of EMT and the CSC
phenotype in a STAT3-dependent manner.
In conclusion, our findings suggest that Notch1
plays a crucial role in breast cancer and may be a key target for
cancer therapy. However, the precise mechanism of the crosstalk
between Notch and other signaling pathways requires further
investigations.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81102027).
References
|
1
|
Wang Z, Li Y, Banerjee S and Sarkar FH:
Exploitation of the Notch signaling pathway as a novel target for
cancer therapy. Anticancer Res. 28:3621–3630. 2008.
|
|
2
|
Rizzo P, Osipo C, Foreman K, Golde T,
Osborne B and Miele L: Rational targeting of Notch signaling in
cancer. Oncogene. 27:5124–5131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gallahan D, Jhappan C, Robinson G, et al:
Expression of a truncated Int3 gene in developing secretory mammary
epithelium specifically retards lobular differentiation resulting
in tumorigenesis. Cancer Res. 56:1775–1785. 1996.PubMed/NCBI
|
|
4
|
Kiaris H, Politi K, Grimm LM, et al:
Modulation of notch signaling elicits signature tumors and inhibits
hras1-induced oncogenesis in the mouse mammary epithelium. Am J
Pathol. 165:695–705. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bao B, Wang Z, Ali S, et al: Notch-1
induces epithelial-mesenchymal transition consistent with cancer
stem cell phenotype in pancreatic cancer cells. Cancer Lett.
307:26–36. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shook D and Keller R: Mechanisms,
mechanics and function of epithelial-mesenchymal transitions in
early development. Mech Dev. 120:1351–1383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levayer R and Lecuit T: Breaking down EMT.
Nat Cell Biol. 10:757–759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duband JL, Blavet C, Jarov A and
Fournier-Thibault C: Spatio-temporal control of neural epithelial
cell migration and epithelium-to-mesenchyme transition during avian
neural tube development. Dev Growth Differ. 51:25–44. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Clevers H: The cancer stem cell: premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baker SJ, Rane SG and Reddy EP:
Hematopoietic cytokine receptor signaling. Oncogene. 26:6724–6737.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Groner B, Lucks P and Borghouts C: The
function of Stat3 in tumor cells and their microenvironment. Semin
Cell Dev Biol. 19:341–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berishaj M, Gao SP, Ahmed S, et al: Stat3
is tyrosine-phosphorylated through the interleukin-6/glycoprotein
130/Janus kinase pathway in breast cancer. Breast Cancer Res.
9:R322007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marotta LL, Almendro V, Marusyk A, et al:
The JAK2/STAT3 signaling pathway is required for growth of CD44(+)
CD24(−) stem cell-like breast cancer cells in human tumors. J Clin
Invest. 121:2723–2735. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barnes RM and Firulli AB: A twist of
insight - the role of Twist-family bHLH factors in development. Int
J Dev Biol. 53:909–924. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Puisieux A, Valsesia-Wittmann S and
Ansieau S: A twist for survival and cancer progression. Br J
Cancer. 94:13–17. 2006. View Article : Google Scholar
|
|
20
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watanabe O, Imamura H, Shimizu T, et al:
Expression of twist and wnt in human breast cancer. Anticancer Res.
24:3851–3856. 2004.
|
|
22
|
Vesuna F, Lisok A, Kimble B and Raman V:
Twist modulates breast cancer stem cells by transcriptional
regulation of CD24 expression. Neoplasia. 11:1318–1328.
2009.PubMed/NCBI
|
|
23
|
Anant S, Roy S and VijayRaghavan K: Twist
and Notch negatively regulate adult muscle differentiation in
Drosophila. Development. 125:1361–1369. 1998.PubMed/NCBI
|
|
24
|
Tapanes-Castillo A and Baylies MK: Notch
signaling patterns Drosophila mesodermal segments by regulating the
bHLH transcription factor twist. Development. 131:2359–2372. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hsu KW, Hsieh RH, Huang KH, et al:
Activation of the Notch1/STAT3/Twist signaling axis promotes
gastric cancer progression. Carcinogenesis. 33:1459–1467. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Wang L, Pappan L, Galliher-Beckley A
and Shi J: IL-1beta promotes stemness and invasiveness of colon
cancer cells through Zeb1 activation. Mol Cancer. 11:872012.
View Article : Google Scholar
|
|
27
|
Liu ZL, Li Y, Kong QY, et al:
Immunohistochemical profiling of Wnt, NF-kappaB, Stat3 and Notch
signaling in human epidermal tumors. J Dermatol Sci. 52:133–136.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koch U, Lehal R and Radtke F: Stem cells
living with a Notch. Development. 140:689–704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Osanyingbemi-Obidi J, Dobromilskaya I,
Illei PB, Hann CL and Rudin CM: Notch signaling contributes to lung
cancer clonogenic capacity in vitro but may be circumvented in
tumorigenesis in vivo. Mol Cancer Res. 9:1746–1754. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bolos V, Mira E, Martinez-Poveda B, et al:
Notch activation stimulates migration of breast cancer cells and
promotes tumor growth. Breast Cancer Res. 15:R542013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Espinoza I and Miele L: Deadly crosstalk:
Notch signaling at the intersection of EMT and cancer stem cells.
Cancer Lett. 341:41–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie M, Zhang L, He CS, et al: Activation
of Notch-1 enhances epithelial-mesenchymal transition in
gefitinib-acquired resistant lung cancer cells. J Cell Biochem.
113:1501–1513. 2012.
|
|
33
|
Ghajar CM and Bissell MJ: Extracellular
matrix control of mammary gland morphogenesis and tumorigenesis:
insights from imaging. Histochem Cell Biol. 130:1105–1118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park J and Schwarzbauer JE: Mammary
epithelial cell interactions with fibronectin stimulate
epithelial-mesenchymal transition. Oncogene. 33:1649–1657. 2014.
View Article : Google Scholar :
|
|
35
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li C, Heidt DG, Dalerba P, et al:
Identification of pancreatic cancer stem cells. Cancer Res.
67:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mallini P, Lennard T, Kirby J and Meeson
A: Epithelial-to-mesenchymal transition: what is the impact on
breast cancer stem cells and drug resistance. Cancer Treat Rev.
40:341–348. 2014. View Article : Google Scholar
|
|
39
|
Castellanos JA, Merchant NB and
Nagathihalli NS: Emerging targets in pancreatic cancer:
epithelial-mesenchymal transition and cancer stem cells. Onco
Targets Ther. 6:1261–1267. 2013.PubMed/NCBI
|
|
40
|
Takebe N, Warren RQ and Ivy SP: Breast
cancer growth and metastasis: interplay between cancer stem cells,
embryonic signaling pathways and epithelial-to-mesenchymal
transition. Breast Cancer Res. 13:2112011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Espinoza I, Pochampally R, Xing F, Watabe
K and Miele L: Notch signaling: targeting cancer stem cells and
epithelial-to-mesenchymal transition. Onco Targets Ther.
6:1249–1259. 2013.PubMed/NCBI
|
|
42
|
McGowan PM, Simedrea C, Ribot EJ, et al:
Notch1 inhibition alters the CD44hi/CD24lo population and reduces
the formation of brain metastases from breast cancer. Mol Cancer
Res. 9:834–844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Soriano JV, Uyttendaele H, Kitajewski J
and Montesano R: Expression of an activated Notch4(int-3)
oncoprotein disrupts morphogenesis and induces an invasive
phenotype in mammary epithelial cells in vitro. Int J Cancer.
86:652–659. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dontu G, Jackson KW, McNicholas E,
Kawamura MJ, Abdallah WM and Wicha MS: Role of Notch signaling in
cell-fate determination of human mammary stem/progenitor cells.
Breast Cancer Res. 6:R605–R615. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bellavia D, Checquolo S, Campese AF, Felli
MP, Gulino A and Screpanti I: Notch3: from subtle structural
differences to functional diversity. Oncogene. 27:5092–5098. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo S, Liu M and Gonzalez-Perez RR: Role
of Notch and its oncogenic signaling crosstalk in breast cancer.
Biochim Biophys Acta. 1815:197–213. 2011.PubMed/NCBI
|
|
47
|
Lin JT, Wang JY, Chen MK, et al: Colon
cancer mesenchymal stem cells modulate the tumorigenicity of colon
cancer through interleukin 6. Exp Cell Res. 319:2216–2229. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang H, Tian Y, Wang J, et al:
Inflammatory cytokines induce NOTCH signaling in nucleus pulposus
cells: implications in intervertebral disc degeneration. J Biol
Chem. 288:16761–16774. 2013. View Article : Google Scholar : PubMed/NCBI
|