Introduction
Pancreatic cancer (PanC) is a devastating disease
with an extremely poor prognosis, and ranks as the fifth leading
cause of cancer-related death in Western countries (1). The main reason for poor prognosis of
PanC is due to high resistance to currently available
chemotherapeutic agents (2). The
other curative treatment of PanC is surgical resection that is
possible only in 10–15% of the cases and it only slightly-improves
the overall survival rate of 5% after 5 years (3). Although gemcitabine is the first-line
chemotherapy for PanC patients, the response rate remains low
(4). One of the major mechanisms
of drug resistance in these cells is an increased energy-dependent
drug efflux, resulting in decreased intracellular drug accumulation
(5). Drug efflux and metabolism
consume large amounts of ATP that is mainly generated via
glycolysis; thereby high glycolytic rate protects cancer cells from
the toxic effects of drugs by providing constant energy supply
required for drug efflux and metabolism (6). Thus, the bioenergetic pathways in
cancer cells could be targeted to overcome the chemoresistance and
to inhibit cell proliferation and long-term survival (7). The survival signaling pathways such
as PI3K/Akt and ERK that play important role in cellular functions
such as proliferation, survival and metabolism, are also
responsible for chemoresistance in cancer cells (8–10).
Specifically, Akt activation has been directly correlated with
increased rates of glucose metabolism in cancer cells (11). Akt activation stimulates anabolic
metabolism, and enhances survival and suppresses apoptosis in
cancer cells (12–14). Importantly, the enhanced Akt
phosphorylation also confers resistance to chemotherapy (15). Duxbury et al have reported
that Akt knockdown enhances gemcitabine chemosensitivity in PanC
cells (16). All together, these
studies suggest that altered metabolism and bioenergetic functions
together with activated signaling pathways such as PI3K/Akt and
ERK1/2 might be the major contributors to gemcitabine resistance in
PanC cells, and that the agents which target them could be
effective in treating gemcitabine-resistant (GR) PanC.
Bitter melon (Momordica charantia, Family:
Cucurbitaceae) is a well-consumed vegetable in Asian countries, and
is widely used for medicinal purposes; specifically, it has the
ability to enhance insulin sensitivity in the body (17). There is a growing interest in
bitter melon because of its beneficial effects against several
diseases such as diabetes, obesity and hyperlipidemia. In addition,
several studies have demonstrated that the leaf or fruit extract of
bitter melon exerts antineoplastic effects against various cancers
(18–21). The methanolic extract of bitter
melon inhibited the colon cancer stem cell proliferation by
altering energy homeostasis and inducing autophagy (22,23).
Several cucurbitane-type triterpene glycosides from bitter melon
have also shown strong antiproliferative activity against human
breast adenocarcinoma MCF-7 cells, human colon adenocarcinoma WiDr
cells, human laryngeal carcinoma HEp-2 cells, and human
medulloblastoma Daoy cells (24).
Importantly, bitter melon leaf extract is shown to inhibit
P-glycoprotein-mediated drug efflux and to increase the efficacy of
chemotherapeutic drugs in multidrug-resistant human cervical KBV1
carcinoma cells (25). Recently,
we reported that bitter melon juice (BMJ) inhibits the growth of
human pancreatic carcinoma cells both in vitro and in
vivo through activating cellular metabolic energy sensor AMPK
(26). However, BMJ efficacy
against GR PanC cells has not yet been studied. Accordingly, in the
present study, we investigated the mechanisms (metabolic,
bioenergetic and signaling) underlying gemcitabine resistance in
PanC cells, and BMJ efficacy and associated mechanism in these
cells.
Materials and methods
Chemicals and reagents
Primary antibodies for phosphorylated and total
PI3K, Akt, ERK1/2, and PTEN as well as hexokinase I and II, hypoxia
inducible factor (HIF)-1α, and E-cadherin; and anti-rabbit
peroxidase-conjugated secondary antibody were purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). Anti-LC3B and
anti-Atg5 were from Novus Biologicals LLC (Littleton, CO, USA);
anti-Beclin 1 was from BD Biosciences (San Jose, CA, USA).
Anti-GLUT1 and 4 were from Abcam (Cambridge, MA, USA). β-actin
antibody, gemcitabine, oligomycin, antimycin A, 2-deoxyglucose
(2-DG) and carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
(FCCP) were from Sigma-Aldrich (St. Louis, MO, USA). MK-2206 was
from Selleck Chemicals (Houston, TX, USA); PD98059 from EMD
Millipore (Billerica, MA, USA), and LY-294002 from Adipogen Corp.
(San Diego, CA, USA). ECL detection system and anti-mouse
HRP-conjugated secondary antibody were from GE Healthcare
(Buckinghamshire, UK). BMJ was prepared and stored as detailed
recently (26). As needed, 1–4%
(v/v in medium) of pure BMJ was used for cell culture studies.
Cell culture and generation of GR PanC
cells
Human pancreatic adenocarcinoma AsPC-1 and MiaPaCa-2
cells were obtained from ATCC (Manassas, VA, USA). AsPC-1 cells
were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10%
FBS with essential amino acids; and MiaPaCa-2 cells were cultured
in DMEM with 10% FBS and 2.5% horse serum under standard culture
conditions (37°C, 95% humidified air and 5% CO2). To
generate GR cell lines, at first, AsPC-1 cells were exposed to 0.1
μM concentration of gemcitabine for 3–4 days, the dead cells were
removed by washing with media, and the viable cells were further
exposed with 2-fold concentration of gemcitabine. The same
gemcitabine treatment cycle was repeated for 3 months with
increasing concentration of gemcitabine in every cycle up to 200
μM. GR MiaPaCa-2 cells were also generated by exposing to 0.1 μM
gemcitabine at first and gradually increasing it up to 5 μM. Dead
cells were removed regularly following each gemcitabine exposer.
Both GR AsPC-1 and MiaPaCa-2 cells were grown under 5 μM
gemcitabine for all the experiments.
Cell viability assays
GR AsPC-1 cells (3×104 cells/well) were
seeded in complete media in 6-well plates with 5 μM gemcitabine.
Next day, cells were treated with different doses of Akt and/or MEK
inhibitor or BMJ for 24, 48 and 72 h. Thereafter, total cells were
collected by brief trypsinization and counted using a
haemocytometer. Trypan blue dye was used for assessing the number
of dead cells. For apoptosis analyses, cells were stained with
Annexin V/propidium iodide (PI) using Apoptosis Assay kit 2
(Molecular probes, Eugene, OR, USA) following the manufacturer’s
instructions. The extent of apoptosis was determined by flow
cytometry analysis of Annexin V/PI-stained cells using the
fluorescence-activated cell sorting (FACS) core facility of the
University of Colorado Cancer Center (Aurora, CO, USA). In another
experiment, GR AsPC-1 cells were treated with 1–4% BMJ 24 and 48 h
without or with pre-treatment with autophagy inhibitor
3-methyladenine (3-MA) or bafilomycin A1 (BafA1) for 2 h, and cell
viability was analyzed by trypan blue assay.
Western blotting
For western blotting, following desired treatment,
total cell lysates were prepared, protein concentration estimated,
and samples were subjected to SDS-PAGE on 8–16% tris-glycine gels
and blotted onto nitrocellulose membrane as detailed earlier
(27). Membranes were probed with
specific primary antibodies overnight at 4°C followed by
peroxidase-conjugated appropriate secondary antibody for 1 h at
room temperature, and visualized by ECL detection system from GE
Healthcare. For certain proteins, membranes were also probed with
appropriate secondary IRDye-tagged antibodies and visualized using
Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE, USA).
Membranes were also stripped and re-probed again for the protein of
interest or β-actin antibody to check protein loading; however,
only representative β-actin blots are shown.
Bioenergetics analysis
XF24 Extracellular Flux Analyzer from Seahorse
Bioscience, Inc. (North Billerica, MA, USA) was utilized to detect
oxygen consumption rate (OCR) and extracellular acidification rate
(ECAR), representing oxidative phosphorylation (OXPHOS) and
glycolysis, respectively, in AsPC-1 cells (both sensitive and
resistant). Briefly, cells were plated in 24-well XF cell culture
microplates at 3.2×104 cells/well using regular growth
medium and then incubated at 37°C/5% CO2 for 24 h. After
incubation, cells were washed twice with XF24 running medium (DMEM
unbuffered assay medium adjusted to pH 7.4) and run on the XF24
analyzer to obtain real-time OCR and ECAR. As indicated four
injections of compounds that modulate mitochondrial respiration and
glycolysis, namely oligomycin (injection A: 1 μg/ml), FCCP
(injection B: 1 μM), 2-DG (injection C: 10 mM), and antimycin A
(injection D: 3 μM) were injected sequentially, in each well.
Inhibitors used in the study included oligomycin that blocks ATP
synthase required to determine ATP turnover rates, FCCP that
uncouples mitochondria and stimulates maximal respiration and
glycolysis, 2-DG that inhibits hexokinase, the first enzyme in the
glycolytic pathway, and antimycin A that inhibits electron
transport chain and indicates non-mitochondrial respiration
(28–30). Real-time OCR and ECAR were recorded
during specified programmed time periods (three readings each) as
the average numbers between the injections of inhibitors mentioned
above. In general, baseline OCR was calculated as respiration
before injection of any compounds minus OCR after antimycin
injection, and respiratory reserve capacity (RRC) was calculated
using FCCP minus the basal OCR. The final data calculation was
performed after the readings had been normalized with protein
concentration of each well. Similarly, baseline ECAR was calculated
as the recorded acidification rate during the respiratory
conditions explained earlier in this section. OCR and ECAR are
expressed as pmol/min/μg of protein and mpH unit change/min/μg of
protein, respectively.
Statistical analysis
All statistical analyses were performed with
SigmaStat software version 2.03 (Jandel Scientific, San Rafael, CA,
USA). One-way ANOVA followed by Tukey’s test was used for multiple
comparisons and statistically significant difference was considered
at p≤0.05.
Results
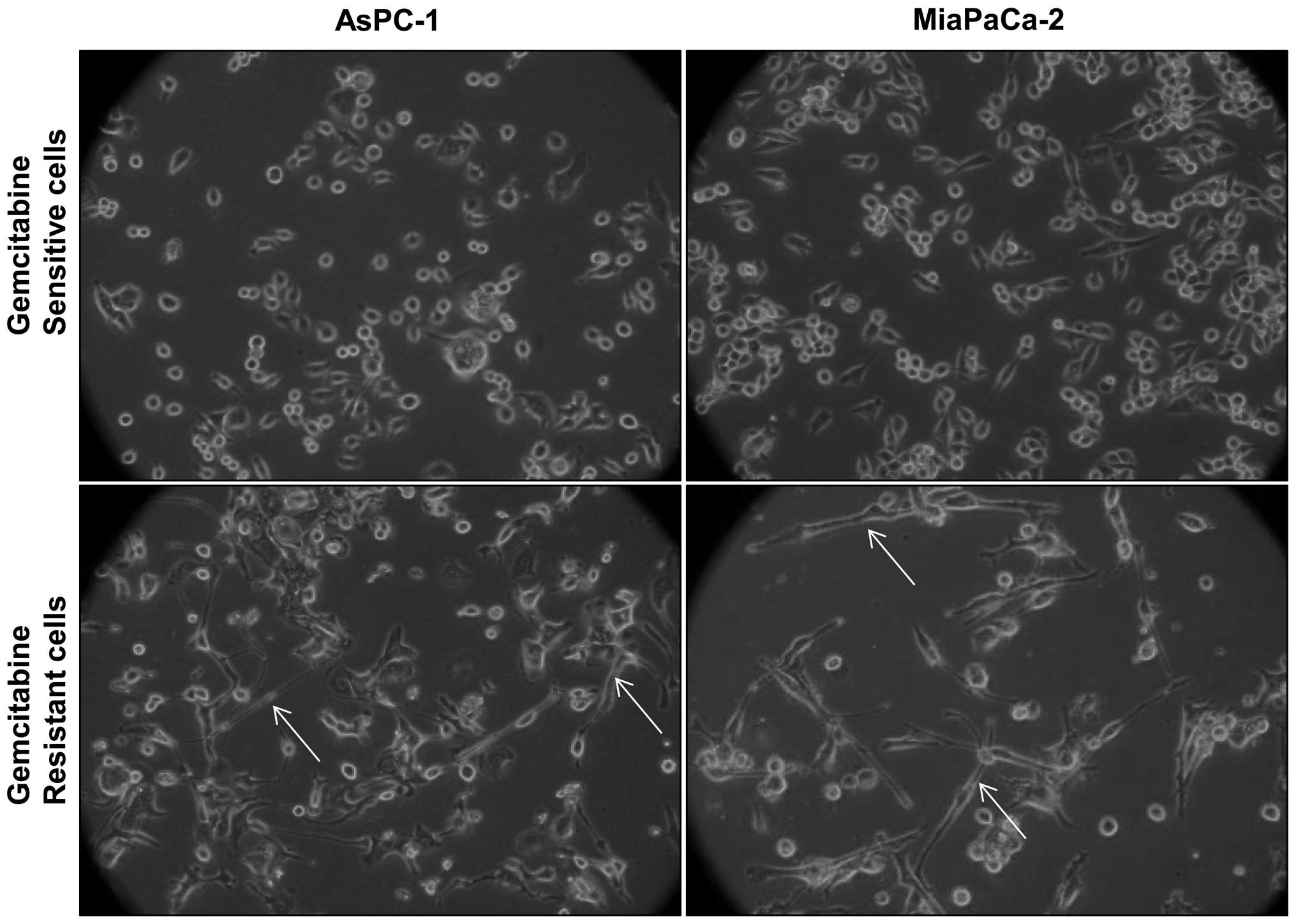
GR cells exhibit distinct morphology
Morphological comparison of gemcitabine-sensitive
(GS) and GR cells revealed that GR AsPC-1 and MiaPaCa-2 cells have
a mixed population of small, round-shaped as well as elongated,
spindle-shaped cells; however, GS counterparts mostly have small,
round-shaped cells, and elongated, spindle-shaped cells were mostly
absent (Fig. 1). Since both AsPC-1
and MiaPaCa-2 cells showed similar morphological features following
gemcitabine exposure, for all future experiments we used AsPC-1 as
a representative GR PanC cell line.
Metabolic and molecular characterization
of GR AsPC-1 cells
To examine the metabolic differences between GS and
GR AsPC-1 cells, Seahorse XF24 Extracellular Flux Analyzer was
employed, and OCR (indicative of OXPHOS) and ECAR (indicative of
glycolysis) were measured. As shown in Fig. 2A (upper left panel), GR AsPC-1
cells showed an increase in baseline OCR (p≤0.05) compared with GS
AsPC-1 cells. To study ATP synthesis in GR cells, OCR was
determined in response to oligomycin addition, and we observed a
distinct increase in ATP synthesis in GR AsPC-1 cells compared to
GS AsPC-1 cells (Fig. 2A, upper
right panel). RRC was also significantly higher in GR AsPC-1 cells
compared to GS cells (Fig. 2A,
lower left panel). Regarding glycolytic rate (indicated by ECAR),
there was an increase, though statistically not significant, in
ECAR in GR AsPC-1 cells compared to GS AsPC-1 cells (Fig. 2A, lower right panel). Overall,
bioenergetic analyses suggested that GR AsPC-1 cells have a higher
metabolic rate to possibly generate more ATP to support the
chemoresistant phenotype.
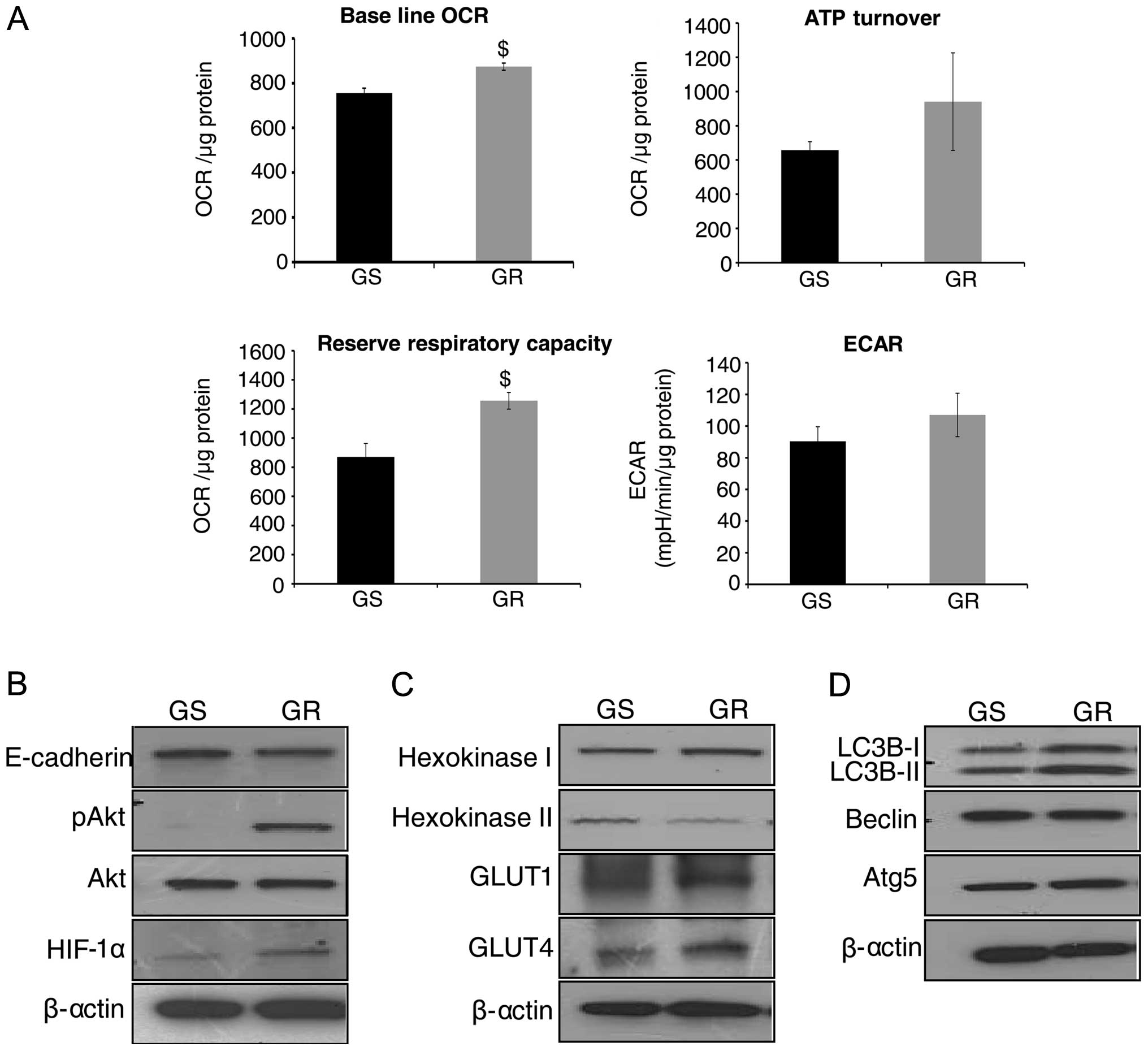 | Figure 2Metabolic and molecular
characterization of gemcitabine-resistant (GR) pancreatic cancer
(PanC) cells. (A) Gemcitabine-sensitive (GS) and GR AsPC-1 cells
were plated in XF24 analyzer microplates for 24 h, and baseline
oxygen consumption rate (OCR), ATP turnover, reserve respiratory
capacity and extracellular acidification rate (ECAR) were measured,
as detailed in Materials and methods. The representative data are
presented as mean ± SEM normalized with respective protein
concentration. Each experiment was performed in triplicate or
quadruplicate at least twice. $P<0.05. (B–D) Whole
cell lysates were prepared from GS and GR AsPC-1 cells, and
analyzed by western blotting for E-cadherin, pAkt, total Akt,
hypoxia inducible factor (HIF)-1α, hexokinase I and II, GLUT1 and
4, LC3B, Beclin and Atg5. Protein loading was confirmed by
re-probing the membranes for β-actin. |
We next characterized GR AsPC-1 cells to understand
their morphological and metabolic differences compared to GS cells.
The elongated, spindle-shaped structures in GR AsPC-1 cells
suggested an epithelial-mesenchymal transition (EMT) phenotype;
therefore, first we compared E-cadherin expression and found it to
be slightly lower in GR compared to GS AsPC-1 cells (Fig. 2B). Akt is an important regulator of
both EMT and cellular metabolism, and therefore, next we analyzed
Akt phosphorylation. As shown in Fig.
2B, Akt phosphorylation (at Ser-473 site) was strongly
activated in GR AsPC-1 cells with no detectable level in sensitive
cells; no difference in total Akt was observed between GS and GR
AsPC-1 cells. Furthermore, the expression of HIF-1α, which is
downstream of Akt and is known to reduce sensitivity of PanC cells
towards gemcitabine (31), was
also higher in GR AsPC-1 cells (Fig.
2B).
As mentioned above, higher ATP synthesis is required
to afford drug efflux from cells (7). Hexokinase is a key regulator of
glycolytic flux (32), hence, we
also evaluated hexokinase I and II expression. As shown in Fig. 2C, we observed a higher hexokinase I
and lower hexokinase II expression in GR AsPC-1 cells compared to
GS AsPC-1 cells, suggesting a preference for hexokinase I enzyme in
GR cells. We also observed an increase in the protein levels of
GLUT1 and 4 in GR AsPC-1 cells (Fig.
2C). To further characterize and evaluate the mechanism of
gemcitabine resistance, we studied the autophagy markers in both GS
and GR AsPC-1 cells. Our results indicated that LC3B-I and II
protein levels were increased in GR compared to GS AsPC-1 cells
without any change in the protein levels of Beclin and Atg5
(Fig. 2D).
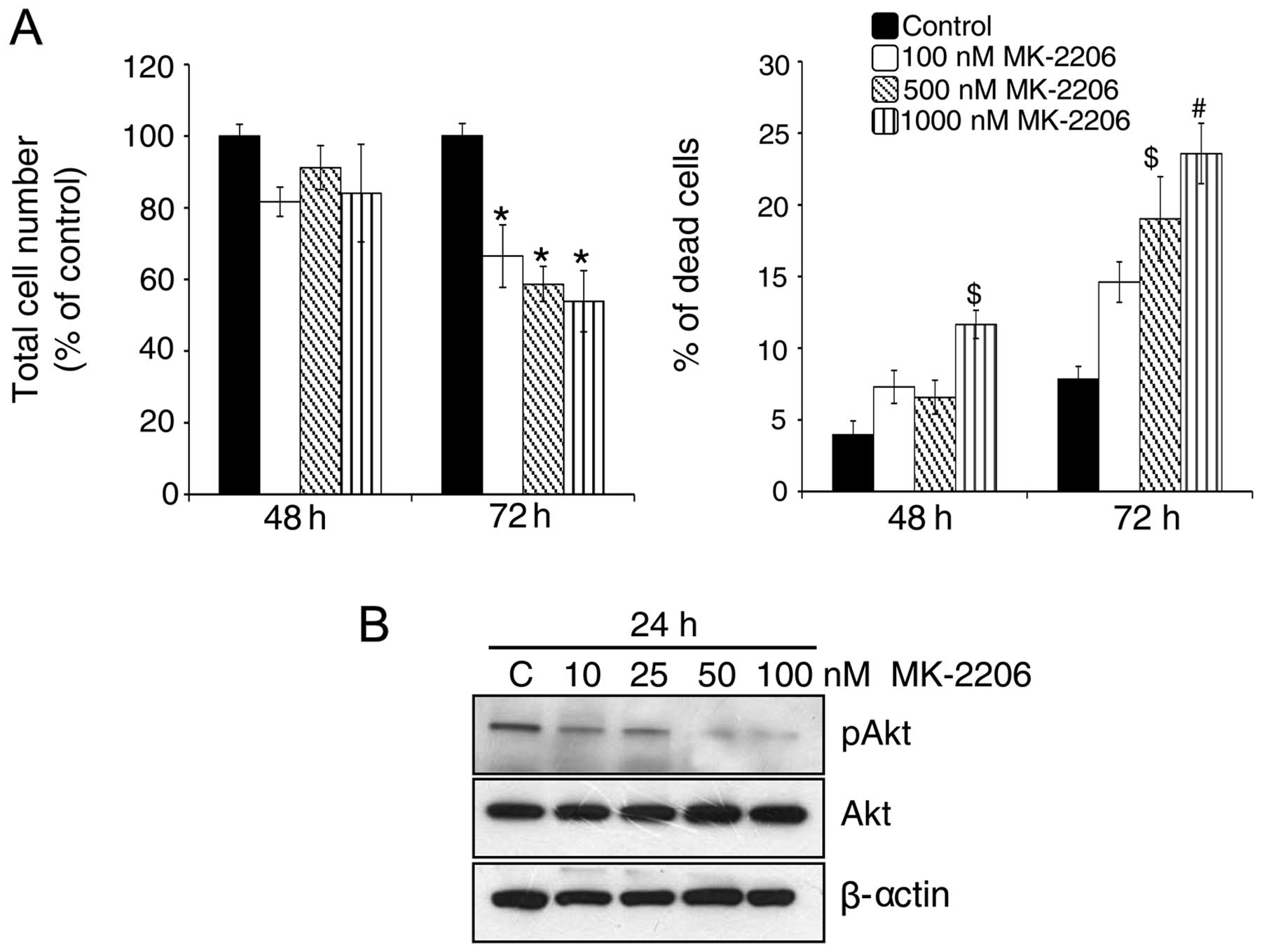
Akt inhibitor decreases growth and
induces death in GR AsPC-1 cells
Akt is a key regulator of the balance between cell
survival and apoptosis (33). As
mentioned above, we observed a strong increase in Akt
phosphorylation in GR AsPC-1 cells which might play an important
role in drug resistance in these cells. Therefore, next, we treated
GR AsPC-1 cells with an Akt inhibitor, i.e., MK-2206 (100–1,000
nM). As shown in Fig. 3A, Akt
inhibition led to a significant decrease in total cell number
together with an increase in dead cells in GR AsPC-1 cells
especially after 72 h of its treatment. These results were
consistent with a dose-dependent decrease in Akt phosphorylation
levels in GR AsPC-1 cells by MK-2206 treatment (Fig. 3B), suggesting a relationship
between elevated Akt phosphorylation and the resistance of AsPC-1
cells to gemcitabine.
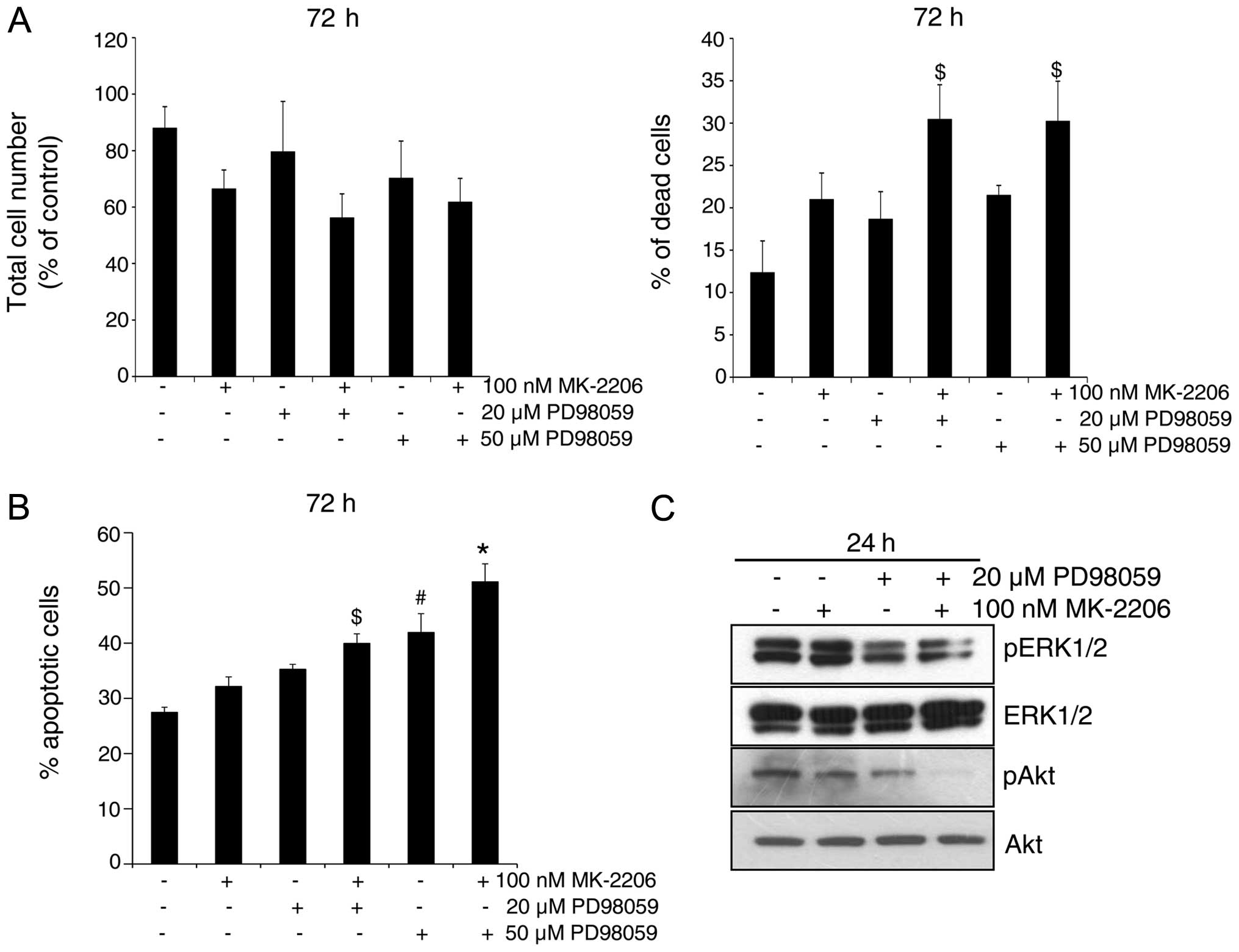
Akt and MEK inhibitors in combination
induce cell death in GR AsPC-1 cells
Since we did not observe a strong growth inhibitory
and cell death effect of Akt inhibitor even though the Akt
phosphorylation was completely inhibited by MK-2206 at 100 nM
concentration (Fig. 3), we next
assessed the involvement of both Akt and MEK-ERK1/2 pathways in
regulating apoptosis in GR AsPC-1 cells, by employing both Akt and
MEK inhibitors MK-2206 and PD98059, respectively, alone and in
combination. As shown in Fig. 4A,
in general, compared to each inhibitor alone, their combination
resulted in a stronger cell growth inhibition and cell death in GR
AsPC-1 cells. Similar observation was also evident in apoptotic
cell death following MK-2206 and PD98059 treatment of GR AsPC-1
cells and a combination was better than either agent alone
(Fig. 4B). Western blotting showed
that indeed both ERK1/2 and Akt are strongly phosphorylated in GR
AsPC-1 cells, and that treatment with MK-2206 and PD98059 reduces
the phosphorylation of Akt and ERK1/2, respectively (Fig. 4C). Importantly, the combination of
MK-2206 and PD98059 caused a maximum inhibition of Akt
phosphorylation; however, no additional decrease in ERK1/2
phosphorylation was observed in combination compared with PD98059
alone (Fig. 4C).
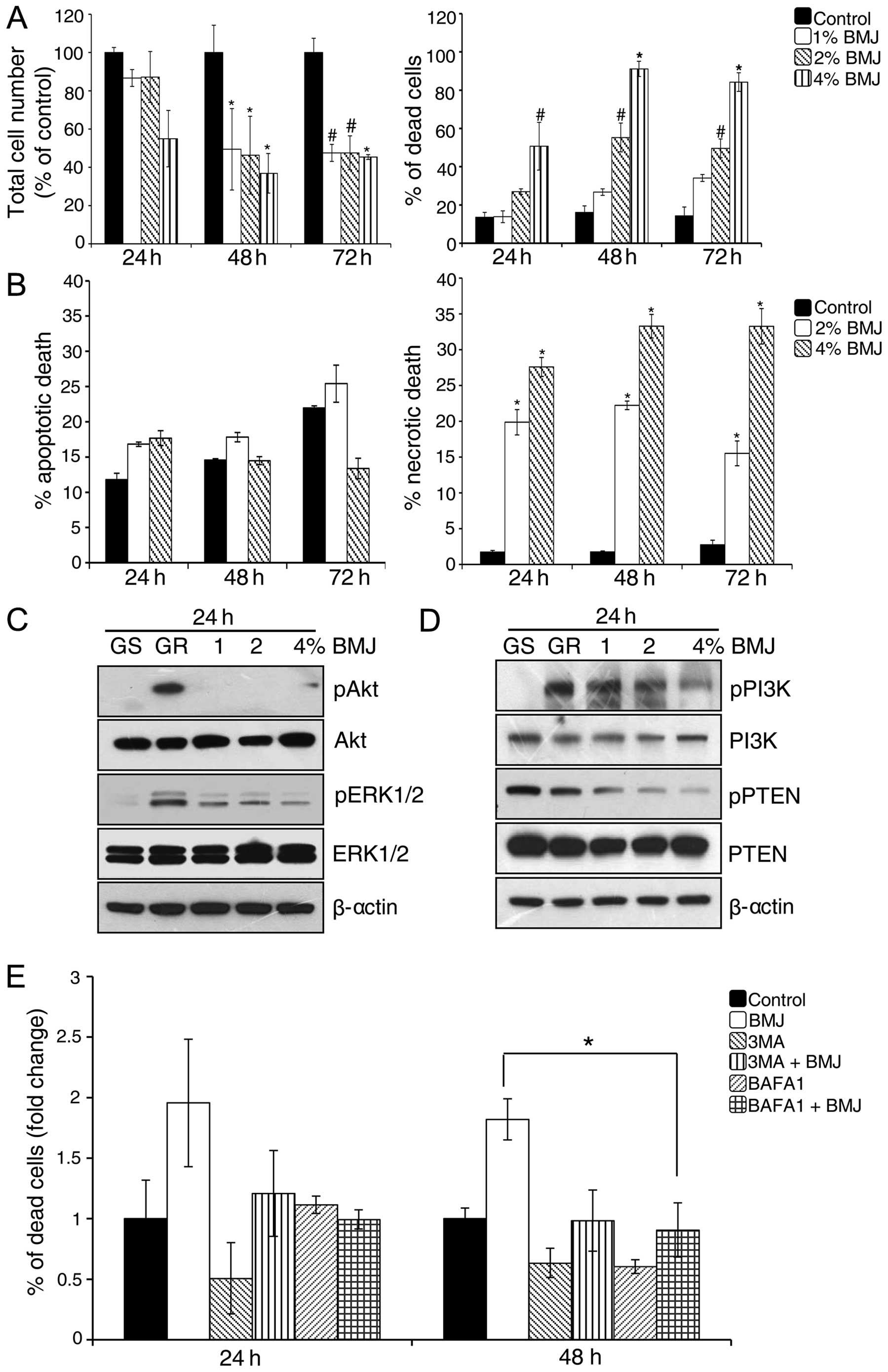
BMJ inhibits the viability of GR AsPC-1
cells via targeting PI3K/Akt pathway
Next, we examined the effect of BMJ (1–4%) treatment
on the viability of GR AsPC-1 cells. As shown in Fig. 5A, BMJ treatment significantly
reduced the total cell number and increased cell death in GR AsPC-1
cells. To further characterize the BMJ-induced cell death in GR
AsPC-1 cells, we stained the cells with Annexin V/PI and analyzed
by FACS (Fig. 5B). Results showed
that BMJ treatment did not significantly affect the apoptotic cell
death but significantly increased the necrotic cell death in GR
AsPC-1 cells (Fig. 5B).
PI3K/Akt signaling plays an important role in
developing chemoresistance in a variety of cancer cell lines
(34). We also observed an
increase in Akt phosphorylation in GR AsPC-1 cells (Fig. 2B). Since we observed a significant
growth inhibition by BMJ in AsPC-1 cells, we next examined the BMJ
effect on Akt and related signaling molecules. Western blotting
results illustrated that GR AsPC-1 cells have higher Akt, ERK1/2
and PI3K phosphorylation and a lower phosphorylated PTEN compared
with GS AsPC-1 cells, and that BMJ treatment strongly reduces the
Akt, ERK1/2, PI3K and PTEN phosphorylation in a dose-dependent
manner without significantly affecting the total level of these
molecules (Fig. 5C and D).
BMJ induces cell death by autophagy
mechanism in GR AsPC-1 cells
Since we did not observe apoptosis following BMJ
treatment, we next sought to determine the role of autophagy in
BMJ-induced cell death in GR AsPC-1 cells. For this purpose, we
used two autophagy inhibitors, an early autophagy inhibitor 3-MA
and a late autophagy inhibitor BAFA1. Results showed that the
BMJ-induced cell death was compromised in the presence of both
autophagy inhibitors, with stronger effect at 48 h (Fig. 5E).
Discussion
PanC is an aggressive disease and is usually
advanced at the time of diagnosis. Median survival of PanC patients
post-diagnosis is <6 months and an overall 5-year survival rate
is 3–5%. In 2013, ~43,920 new cases of PanC were reported in US
with ~37,390 deaths (35). These
statistics show that PanC is untreatable malignancy; therefore more
emphasis should be placed on PanC management and control. In most
PanC cases, disease relapse occurs due to chemoresistance towards
drugs like gemcitabine that is the front-line therapy for PanC.
Therefore, there is a critical need to understand and target
mechanisms responsible for gemcitabine resistance in PanC cells. In
the present study, we investigated the possible bioenergetic and
molecular mechanisms underlying the gemcitabine resistance in PanC
cells. Since, it is also important to identify agents that could
target the molecular pathways responsible for gemcitabine
resistance in PanC cells, we, for the first time, also tested the
efficacy of a natural agent BMJ to target the survival of GR PanC
cells. Our results are quite encouraging as BMJ effectively
inhibited the proliferation and induced death in GR AsPC-1 cells.
It is important to mention here that our recent studies have shown
that BMJ also strongly inhibits the growth and induces apoptotic
death in several PanC cell lines (GS) in culture and nude mouse
xenografts (26). Therefore, BMJ
could be useful against both GS and GR PanC cells.
Drug resistant cells are known to produce more ATP
in comparison to the drug-sensitive cells (7); therefore, cellular bioenergetic
pathways seem logical targets to overcome drug resistance in cancer
cells. Role of bioenergetic pathways in gemcitabine resistance in
PanC cells is not well defined; therefore, we compared the
glycolytic and OXPHOS rate in GR AsPC-1 cells. We observed higher
glycolysis and OXPHOS in GR AsPC-1 cells, suggesting significantly
higher metabolic rate in these cells. The observed higher level of
GLUT1 and GLUT4, facilitating higher glucose uptake, as well as
higher hexokinase I (rate limiting enzyme during glycolysis)
expression also support increased glucose metabolism in GR cells to
meet higher ATP demand. In an earlier study, we reported that BMJ
acts against PanC cells via activating cellular energy sensor AMPK
(26); therefore, it is possible
that BMJ inhibits the proliferation of GR PanC cells via enforcing
energy restriction in these cells; however, further studies are
needed in future to support this assumption.
Akt is a serine threonine kinase known to exert
anti-apoptotic and pro-survival effects through several downstream
pathways in cancer cells (36). It
has been reported earlier that the inhibition of Akt activation
enhances the gemcitabine sensitivity in PanC cells (4). We observed significantly higher Akt
phosphorylation in GR AsPC-1 cells, and its inhibition by MK-2206
resulted in growth inhibition and induction of cell death. Besides
Akt, increased level of ERK1/2 phosphorylation is also considered
responsible for chemoresistance in cancer cells (37). For example, Mirmohammadsadegh et
al reported on ERK1/2 in inducing chemoresistance in melanoma
cells (38). Our data demonstrated
that ERK1/2 phosphorylation is also enhanced in GR PanC cells.
Importantly, inhibition of ERK1/2 phosphorylation by MEK inhibitor
PD98059 also decreased the Akt phosphorylation, and we observed
additional Akt inhibition when we used both MK-2206 and PD98059,
suggesting crosstalk between these two pathways and that possibly
Akt is downstream of MEK/ERK pathway in GR AsPC-1 cells.
Importantly, the combined inhibition of both Akt and MEK/ERK
pathways induced maximal apoptosis in GR AsPC-1 cells. Also
notably, BMJ treatment targeted both PI3K/Akt and ERK1/2 pathways;
therefore, BMJ seems to be a broad-spectrum inhibitor
simultaneously targeting several signaling pathways responsible for
gemcitabine resistance in PanC cells.
Previous reports have demonstrated that autophagy
also contributes resistance of cancer cells towards
chemotherapeutic agents by enhancing their survival and decreasing
their apoptotic potential (39–41).
Therefore, autophagy inhibitors have been tested in combination
with chemotherapy to suppress tumor growth both in vitro and
in vivo (42). We observed
an increase in LC3B-I and II in GR AsPC-1 cells suggesting that
autophagy could be involved in drug resistance in these cells.
However, BMJ seems to induce cell death in GR AsPC-1 cells via
enhancing the autophagy, as autophagy inhibitors compromised
BMJ-induced cell death. Collectively, these observations suggest
that further studies are needed to clearly understand the role of
autophagy in conferring resistance towards gemcitabine in PanC
cells as well as to understand the molecular mechanisms through
which BMJ induces autophagy in these cells, as the observed effect
of BMJ could also be linked to AMPK activation (26) and the resultant mTOR inhibition in
PanC cells.
In summary, GR AsPC-1 cells showed an increased
level of OCR and ECAR corresponding to higher ATP production in
these cells. The higher expression of glycolytic proteins further
confirms the increase in glucose metabolism in GR cells. Our
results also revealed the important role of Akt and ERK1/2 in
regulating the survival and proliferation of GR PanC cells. Present
study also demonstrated the efficacy of a natural agent BMJ against
GR PanC cells by targeting multiple signaling pathways including
PI3K/Akt and ERK1/2. Hence, BMJ that is widely consumed as a
vegetable and for health benefits could have significant efficacy
against GR PanC cells. Overall, the present study reveals novel
mechanisms of gemcitabine resistance in PanC cells which are
targeted by BMJ; considering the poor survival rate in PanC
patient, our findings could have high translational potential in
controlling this deadly malignancy.
Acknowledgements
This study was supported by NCI RO1 grant CA195708
(to R.A.). Authors also acknowledge the CCSG P30CA046934 grant for
supporting the shared resources used in this study. Funding sources
had no involvement in the design of experiments and
interpretation/presentation of the data.
References
|
1
|
Guillermet-Guibert J, Davenne L,
Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C,
Delisle MB, Cuvillier O, Susini C and Bousquet C: Targeting the
sphingolipid metabolism to defeat pancreatic cancer cell resistance
to the chemotherapeutic gemcitabine drug. Mol Cancer Ther.
8:809–820. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huanwen W, Zhiyong L, Xiaohua S, Xinyu R,
Kai W and Tonghua L: Intrinsic chemoresistance to gemcitabine is
associated with constitutive and laminin-induced phosphorylation of
FAK in pancreatic cancer cell lines. Mol Cancer. 8:1252009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Marco M, Di Cicilia R, Macchini M,
Nobili E, Vecchiarelli S, Brandi G and Biasco G: Metastatic
pancreatic cancer: Is gemcitabine still the best standard
treatment? (Review). Oncol Rep. 23:1183–1192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kagawa S, Takano S, Yoshitomi H, et al:
Akt/mTOR signaling pathway is crucial for gemcitabine resistance
induced by Annexin II in pancreatic cancer cells. J Surg Res.
178:758–767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fanciulli M, Bruno T, Giovannelli A,
Gentile FP, Di Padova M, Rubiu O and Floridi A: Energy metabolism
of human LoVo colon carcinoma cells: correlation to drug resistance
and influence of lonidamine. Clin Cancer Res. 6:1590–1597.
2000.PubMed/NCBI
|
|
7
|
Zhou M, Zhao Y, Ding Y, et al: Warburg
effect in chemosensitivity: Targeting lactate dehydrogenase-A
re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer.
9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tokunaga E, Oki E, Egashira A, Sadanaga N,
Morita M, Kakeji Y and Maehara Y: Deregulation of the Akt pathway
in human cancer. Curr Cancer Drug Targets. 8:27–36. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
11
|
Elstrom RL, Bauer DE, Buzzai M, et al: Akt
stimulates aerobic glycolysis in cancer cells. Cancer Res.
64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gottlob K, Majewski N, Kennedy S, Kandel
E, Robey RB and Hay N: Inhibition of early apoptotic events by
Akt/PKB is dependent on the first committed step of glycolysis and
mitochondrial hexokinase. Genes Dev. 15:1406–1418. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plas DR, Talapatra S, Edinger AL, Rathmell
JC and Thompson CB: Akt and Bcl-xL promote growth
factor-independent survival through distinct effects on
mitochondrial physiology. J Biol Chem. 276:12041–12048. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Majewski N, Nogueira V, Bhaskar P, Coy PE,
Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB and Hay N:
Hexokinase-mitochondria interaction mediated by Akt is required to
inhibit apoptosis in the presence or absence of Bax and Bak. Mol
Cell. 16:819–830. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang WC and Hung MC: Induction of Akt
activity by chemotherapy confers acquired resistance. J Formos Med
Assoc. 108:180–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duxbury MS, Ito H, Zinner MJ, Ashley SW
and Whang EE: siRNA directed against c-Src enhances pancreatic
adenocarcinoma cell gemcitabine chemosensitivity. J Am Coll Surg.
198:953–959. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yibchok-anun S, Adisakwattana S, Yao CY,
Sangvanich P, Roengsumran S and Hsu WH: Slow acting protein extract
from fruit pulp of Momordica charantia with insulin secretagogue
and insulinomimetic activities. Biol Pharm Bull. 29:1126–1131.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ganguly C, De S and Das S: Prevention of
carcinogen-induced mouse skin papilloma by whole fruit aqueous
extract of Momordica charantia. Eur J Cancer Prev. 9:283–288. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Claflin AJ, Vesely DL, Hudson JL, Bagwell
CB, Lehotay DC, Lo TM, Fletcher MA, Block NL and Levey GS:
Inhibition of growth and guanylate cyclase activity of an
undifferentiated prostate adenocarcinoma by an extract of the
balsam pear (Momordica charantia abbreviata). Proc Natl Acad Sci
USA. 75:989–993. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagasawa H, Watanabe K and Inatomi H:
Effects of bitter melon (Momordica charantia L.) or ginger rhizome
(Zingiber offifinale rosc) on spontaneous mammary tumorigenesis in
SHN mice. Am J Chin Med. 30:195–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh A, Singh SP and Bamezai R: Momordica
charantia (Bitter Gourd) peel, pulp, seed and whole fruit extract
inhibits mouse skin papillomagenesis. Toxicol Lett. 94:37–46. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weng JR, Bai LY, Chiu CF, Hu JL, Chiu SJ
and Wu CY: Cucurbitane triterpenoid from Momordica charantia
induces apoptosis and autophagy in breast cancer cells, in part,
through peroxisome proliferator-activated receptor γ activation.
Evid Based Complement Alternat Med. 2013:9356752013. View Article : Google Scholar
|
|
23
|
Kwatra D, Subramaniam D, Ramamoorthy P,
Standing D, Moran E, Velayutham R, Mitra A, Umar S and Anant S:
Methanolic extracts of bitter melon inhibit colon cancer stem cells
by affecting energy homeostasis and autophagy. Evid Based
Complement Alternat Med. 2013:7028692013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hsiao PC, Liaw CC, Hwang SY, Cheng HL,
Zhang LJ, Shen CC, Hsu FL and Kuo YH: Antiproliferative and
hypoglycemic cucurbitane-type glycosides from the fruits of
Momordica charantia. J Agric Food Chem. 61:2979–2986. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Limtrakul P, Khantamat O and Pintha K:
Inhibition of P-glycoprotein activity and reversal of cancer
multidrug resistance by Momordica charantia extract. Cancer
Chemother Pharmacol. 54:525–530. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaur M, Deep G, Jain AK, Raina K, Agarwal
C, Wempe MF and Agarwal R: Bitter melon juice activates cellular
energy sensor AMP-activated protein kinase causing apoptotic death
of human pancreatic carcinoma cells. Carcinogenesis. 34:1585–1592.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Agarwal C, Tyagi A, Kaur M and Agarwal R:
Silibinin inhibits constitutive activation of Stat3, and causes
caspase activation and apoptotic death of human prostate carcinoma
DU145 cells. Carcinogenesis. 28:1463–1470. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu M, Neilson A, Swift AL, et al:
Multiparameter metabolic analysis reveals a close link between
attenuated mitochondrial bioenergetic function and enhanced
glycolysis dependency in human tumor cells. Am J Physiol Cell
Physiol. 292:C125–C136. 2007. View Article : Google Scholar
|
|
29
|
Brand MD and Nicholls DG: Assessing
mitochondrial dysfunction in cells. Biochem J. 435:297–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Potter VR and Reif AE: Inhibition of an
electron transport component by antimycin A. J Biol Chem.
194:287–297. 1952.PubMed/NCBI
|
|
31
|
Kasuya K, Tsuchida A, Nagakawa Y, Suzuki
M, Abe Y, Itoi T, Serizawa H, Nagao T, Shimazu M and Aoki T:
Hypoxia-inducible factor-1α expression and gemcitabine chemotherapy
for pancreatic cancer. Oncol Rep. 26:1399–1406. 2011.PubMed/NCBI
|
|
32
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao T, Furnari F and Newton AC: PHLPP: a
phosphatase that directly dephosphorylates Akt, promotes apoptosis,
and suppresses tumor growth. Mol Cell. 18:13–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao AM, Ke ZP, Shi F, Sun GC and Chen H:
Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin
by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol
Interact. 206:100–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang D, Okada H, Ruland J, Liu L,
Stambolic V, Mak TW and Ingram AJ: Akt is activated in response to
an apoptotic signal. J Biol Chem. 276:30461–30466. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeong EK, Lee SY, Jeon HM, Ju MK, Kim CH
and Kang HS: Role of extracellular signal-regulated kinase (ERK)1/2
in multicellular resistance to docetaxel in MCF-7 cells. Int J
Oncol. 37:655–661. 2010.PubMed/NCBI
|
|
38
|
Mirmohammadsadegh A, Mota R, Gustrau A,
Hassan M, Nambiar S, Marini A, Bojar H, Tannapfel A and Hengge UR:
ERK1/2 is highly phosphorylated in melanoma metastases and protects
melanoma cells from cisplatin-mediated apoptosis. J Invest
Dermatol. 127:2207–2215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao
L, Sun K, Shen F, Wu M and Wei L: Hypoxia-induced autophagy
contributes to the chemoresistance of hepatocellular carcinoma
cells. Autophagy. 5:1131–1144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sui X, Chen R, Wang Z, et al: Autophagy
and chemotherapy resistance: a promising therapeutic target for
cancer treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|