Introduction
Bladder cancer is still one of the most common
malignancies in the world (1).
Pathologically, >90% of bladder cancer is transitional cell
carcinoma (TCC) (2). Surgery,
radiation, and chemotherapy are evidence-based treatments depending
on clinical staging (3,4). Chemotherapy with standard regimen
MVAC (methotrexate, vinblastine, adriamycin, and cisplatin) or more
tolerable regimen GC (gemcitabine and cisplatin) for advanced or
metastatic bladder cancer has shown poor response (5). However, once diagnosed as a
muscle-invasive type, the 5-year survival and freedom from relapse
rate under conservative multimodality therapies were 54 and 42%,
respectively (6). Therefore, we
need to investigate new strategies for bladder cancer
treatment.
Thalidomide possesses anti-angiogenic and
immunomodulatory effects (7). It
has been widely used for the standard therapies of multiple myeloma
(MM) (8), based not only on its
anti-angiogenic and immunomodulatory mechanisms but also on
inducing apoptosis (9). Its new
analogues, so called IMiDs (immunomodulatory drugs), lenalidomide
and pomalidomide, have also been approved by US FDA for MM therapy
with satisfactory and tolerable responses (10–12).
Thalidomide has also been used clinically for the treatments of
some solid tumors, such as hepatocellular carcinoma (13,14)
and glioblastoma multiforme (15,16),
but controversial effects were observed in these studies. Beyond
its well-known anti-angiogenic property, only few studies of
cytotoxic effect (17) and
migratory inhibition (18) on
solid tumor cells have been reported. In the present study, we
demonstrated the therapeutic effects of thalidomide via induction
of intracellular reactive oxygen species (ROS) to elicit apoptosis,
inhibition of angiogenesis, and suppression of invasion in
gemcitabine-resistant TCC BFTC905 cells in vitro and in
vivo.
Materials and methods
Cell lines and cell culture
Four TCC cell lines were used: BFTC905 (19), BFTC909 (19), T24, and TSGH8301. Cells were
maintained as described previously (20,21).
BFTC905 cells were gemcitabine-resistant (22). We also used primary human
urothelial cells (HUCs) (ScienCell Research Laboratories, Carlsbad,
CA, USA) (22).
MTT cytotoxicity assay
Procedures as previously described were followed
(21,23). Thalidomide (Sigma-Aldrich, St.
Louis, MO, USA), gemcitabine (Sigma-Aldrich), pomalidomide (kindly
provided by Professor Chinpiao Chen, Department of Chemistry,
National Don Hwa University, Hualien, Taiwan), lenalidomide (LC
Laboratories, Woburn, MA, USA), CRBN siRNA/scrambled siRNA (Santa
Cruz Biotechnology, Santa Cruz, CA, USA), N-acetylcysteine (NAC,
Sigma-Aldrich), and dl-dithiothreitol (DTT, Sigma-Aldrich) were
used in individual experiments. Gemcitabine, NAC, and DTT were
dissolved in distilled water. Dimethyl sulfoxide (DMSO, J.T. Baker,
Phillipsburg, NJ, USA), (2-hydroxypropyl)-β-cyclodextrin
(Sigma-Aldrich), γ-cyclodextrin (Sigma-Aldrich), carboxymethyl
cellulose (CMC, Sigma-Aldrich), and Cremophor-EL (Sigma-Aldrich)
were chosen as the solvent to dissolve thalidomide, and the
viability changes after administration of these solvents were
tested. The protocol for siRNA transfection provided by Santa Cruz
Biotechnology was followed. The cell viability was calculated
according to the following formula: cell viability = (absorbance of
the experimental group)/(absorbance of reference group) × 100%.
Reference group was administered with equal volume of PBS
(phosphate-buffered saline) as control.
Cell proliferation by trypan blue
exclusion test
For cell proliferation assay, thalidomide at 0–200
μM was administered (day 1) after overnight seeding of
1×104 BFTC905 cells in 10-cm dish with complete medium.
Subsequently, the cells were re-cultured with fresh medium and
administration of each concentration of thalidomide on day 5.
Administration of equal volume of vehicle γ-cyclodextrin was used
as control. Viable cells do not take up impermeable trypan blue
(24), so counts of cells with
negative stainings of trypan blue (Sigma-Aldrich) were calculated
by a hemocytometer on days 5 and 9. In another experiment,
thalidomide at 200 μM plus 100 nM recombinant human vascular
endothelial growth factor (VEGF, PreproTech, Rocky Hill, NJ, USA),
100 nM human recombinant basic fibroblast growth factor (bFGF,
Merck Millipore, Darmstadt, Germany), or 100 ng/ml human
recombinant tumor necrosis factor-α (TNF-α, Merck Millipore) were
administered after overnight seeding of 1×104 BFTC905
cells in 10-cm dish (day 1). Administration of equal volume of PBS
was used as control. Trypan blue-negative cells were calculated on
day 3. To determine the appropriate concentration of TNF-α for
experiment of invasion without the proliferative effects, a 48-h
administration of TNF-α at 0–100 ng/ml on BFTC905 cells was
cultured in the medium with 1% FBS. Then trypan blue-negative cells
were counted.
Western blotting
Conventional protocols as described previously were
followed (20). Primary antibodies
were purchased from the following vendors: Cell Signaling
Technology (Danvers, MA, USA), anti-survivin (no. 2808), anti-LC3B
(no. 2775), cell cycle regulation (no. 9932), and cell
cycle/checkpoint antibody sampler kits (no. 9917); Abcam
(Cambridge, MA, USA), anti-securin (ab3305) and anti-cleaved PARP
[poly (ADP-ribose) polymerase, ab4830]; R&D Systems
(Minneapolis, MN, USA), anti-CAIX (carbonic anhydrase 9, AF 2188);
GeneTex (Irvine, CA, USA), anti-HIF-1α (hypoxia-inducible factor 1
alpha, GTX 127309), anti-caspase 3 (GTX110543), anti-Bcl2 (B-cell
lymphoma 2, GTX127958), anti-Bax (Bcl-2-associated X protein,
GTX109683), anti-cIAP1 (cellular inhibitor of apoptosis 1,
GTX110087), anti-cIAP2 (GTX113128), anti-Bcl-xL (B-cell
lymphoma-extra-large, GTX105661), anti-TCTP (translationally
controlled tumor protein, GTX63597), anti-cyclin A1 (GXT103042),
anti-cyclin B1 (GTX100911), anti-cyclin D1 (GTX112874), anti-cyclin
E1 (GTX103045), and anti-cereblon (GTX 45011); and Santa Cruz
Biotechnology (Dallas, TX, USA), anti-Ki-67 (sc-15402), anti-MMP-9
(matrix metalloproteinase 9, sc-6840), anti-ICAM-1 (intercellular
adhesion molecule 1, sc-7891), anti-CD34 (sc-9095), α-tubulin
(sc-8305), and actin (sc-1616). Expression of α-tubulin or actin
was used as the internal standard.
Cell cycle analysis
After treatment, the supernatant and the dead
BFTC905 cells were removed. Only the cell cycle changes in the
viable BFTC905 cells were analyzed. Procedures using flow cytometry
(Bedford, MA, USA), as described, were followed (22).
DNA damage assay
The Cell Death Detection ELISAplus
(Roche, Mannheim, Germany) assay kit was used to differentiate
apoptotic or necrotic condition of BFTC905 cells after thalidomide
treatment. Procedures as previously described were followed
(25). To determine the oxidative
DNA damage, OxiSelect™ Oxidative DNA Damage ELISA kit (Cell
Biolabs, San Diego, CA, USA) was used for the detection and
quantitation of 8-hydroxy-2′-deoxyguanosine (8-OHdG).
Immunofluorescent stainings of DCF (2′,7′
dichlorodihydro-fluorescein)
The procedures using OxiSelect™ Intracellular ROS
(reactive oxygen species) Assay kit (Cell Biolabs) were followed.
The intensity of greenish fluorescence of DCF is proportional to
the levels of ROS production in the cytoplasm of BFTC905 cells. The
cells were photographed (×100) 48 h after exposure to IMiDs or
vehicle.
Total oxidant status (TOS) assay
TOS levels were measured by commercial assay kit
(Rel Assay Diagnostics, Turkey) (26). The oxidants in BFTC905 cells
oxidized the ferrous ion-chelator complex to ferric ion. The
oxidation reaction was prolonged by the enhanced molecule,
glycerol, which was abundant in the reaction medium. The ferric ion
produced a colored complex with xylenol orange in an acidic medium.
The color intensity, measured as OD (optic density) at 530 nm by a
spectrophotometer, was recorded at 6, 12, 24, 48 h after BFTC905
cells were treated with 200 μM thalidomide or vehicle. Due to
significant differences of cell counts between vehicle and
thalidomide treatment, the changes of TOS at each time-point were
calculated according to the following formula: TOS ratio per cell =
[(OD value of thalidomide treatment)/(cell counts of thalidomide
treatment)] ÷ [(OD value of vehicle treatment)/(cell counts of
vehicle treatment)]. Cell counts were calculated conventionally by
a hemocytometer.
Matrigel invasion assay
The BioCoat™ growth factor reduced Matrigel™
Invasion Chamber (BD Biosciences) was used for in vitro
invasion study. TNF-α at 10 ng/ml and/or 50 μM of thalidomide were
added to the chamber with medium containing 1% FBS. Subsequently,
the chambers were put into 24-well plate containing medium plus 10%
FBS for 24 h. The protocol as described previously was followed
(23). BFTC905 cells attached on
the bottom layer of chambers were counted from 10 randomized fields
(×200).
Immunocytochemistry
The protocol as described previously was followed
(20). Primary antibody of
anti-NF-κB (nuclear factor kappa-light-chain-enhancer of activated
B cells) p65 (SC-109, Santa Cruz Biotechnology) was used. After
48-h treatment with thalidomide or vehicle with/without TNF-α
stimulation, BFTC905 cells fixed on the slides with positive
immunostaining for nuclear NF-κB p65 were counted from 5 randomized
fields (×200).
Enzyme-linked immunosorbent assay
(ELISA)
Human BDNF (brain-derived neurotrophic factor)
Quantikine™ ELISA kit purchased from R&D Systems and human VEGF
(vascular endothelial growth factor), as well as bFGF (basic
fibroblast growth factor) and TNF-α ELISA kit purchased from
Millipore were used to detect the above molecules in culture
medium. The procedures were recommended by the manufacturer and
used in our previous study (20).
MMP-9 activity in culture medium was also measured by ELISA method
(Biotrak activity assay system, Amersham Pharmacia Biotech, Little
Chalfont, UK). After 48-h treatment with thalidomide or vehicle
with/without TNF-α stimulation, MMP-9 activity in the medium was
assayed according to the manufacturer's instructions.
Mouse xenograft model
BFTC905 xenograft model and protocol in
NOD.CB17-Prkdcscid/Tcu (SCID) male mice were established
in our previous study (20).
BFCT905 cells (1×106) were implanted s.c. into the right
inguinal area of SCID mice, concomitantly 250 mg/kg of thalidomide
or vehicle was administered s.c. into the loading site of cancer
cells when the tumor was impalpable or directly into the tumor 3
times per week (W1, 3, 5) since the day of implantation. In another
experiment to evaluate the therapeutic effects, the same dosage of
thalidomide with the same frequency was administered for 2 weeks
after implantation of BFTC905 cells for 3 weeks. Body weight was
measured after sacrifice, and necropsy of xenografts was performed
immediately. Tumor volumes were calculated using the formula: [1/2]
× a × b2, where a and b represent the largest and
smallest tumor diameters, respectively. Expression of cleaved PARP
and CD34 was determined by western blotting, and expression of
human VEGF was measured by ELISA.
Locomotor activity
Locomotor activity as described were followed
(27). Briefly, a 30-min
habituation period was used prior to the first episode of
administration of thalidomide or vehicle. Images of traveled
distance, the so-called locomotor activity, were captured by a
video camera and the recorded images were analyzed by TrackMot
software (Diagnostic & Research Instruments Co., Taoyuan,
Taiwan). The activity was summated consecutively for two 10-min
intervals following the drug injection. All animals without
xenograft implantation were used only once with 3 drug injections
(W1, 3, 5).
Statistical analysis
Data were analyzed by Student's t-test, Mann-Whitney
U test, or one-way/two-way ANOVA based on individual data, and
presented as mean ± SEM (standard error of mean). In all cases,
p<0.05 was considered statistically significant, and indicated
in the figures as: *0.01≤p<0.05,
**0.005≤p<0.01 and ***p<0.005,
respectively.
Results
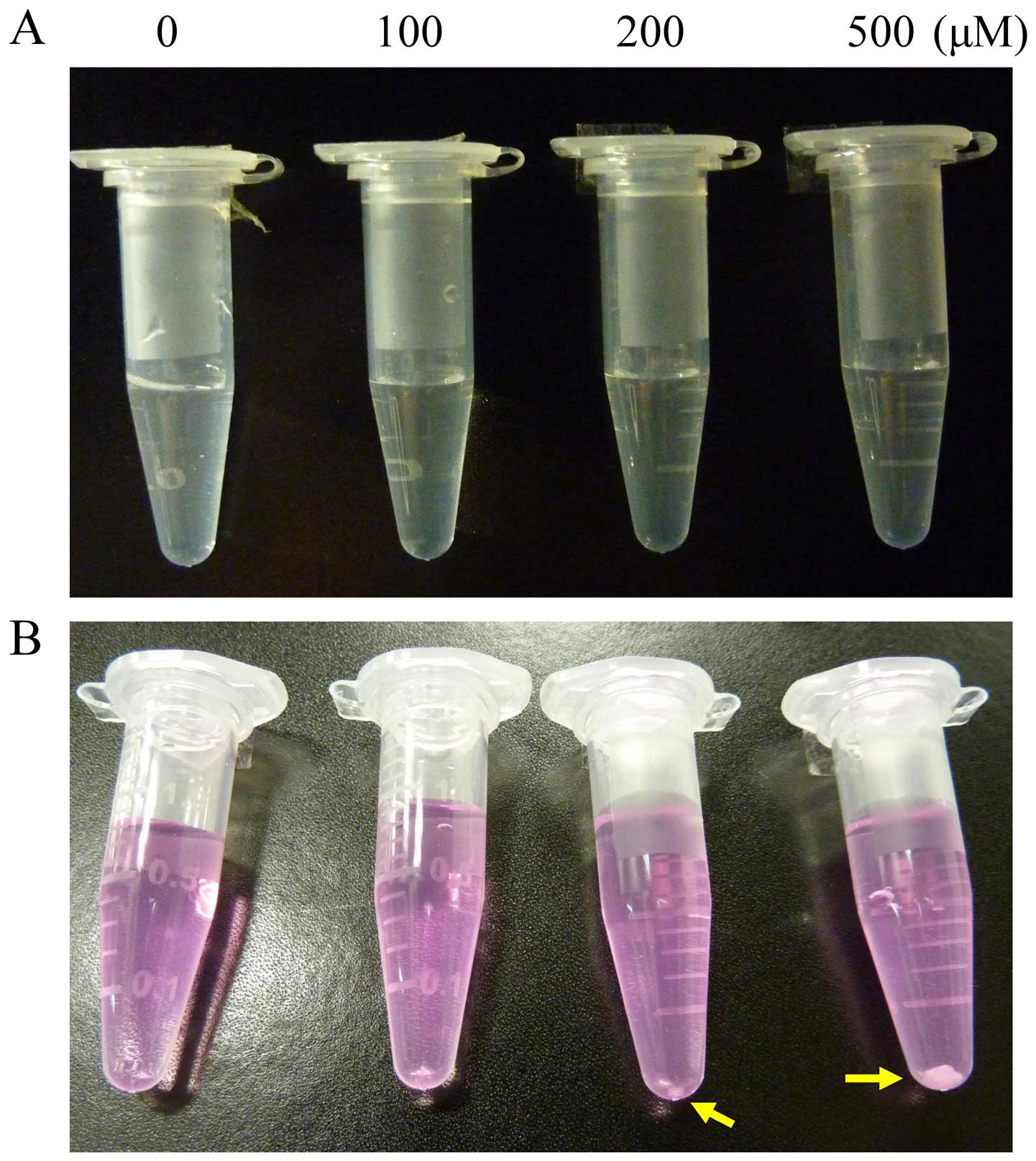
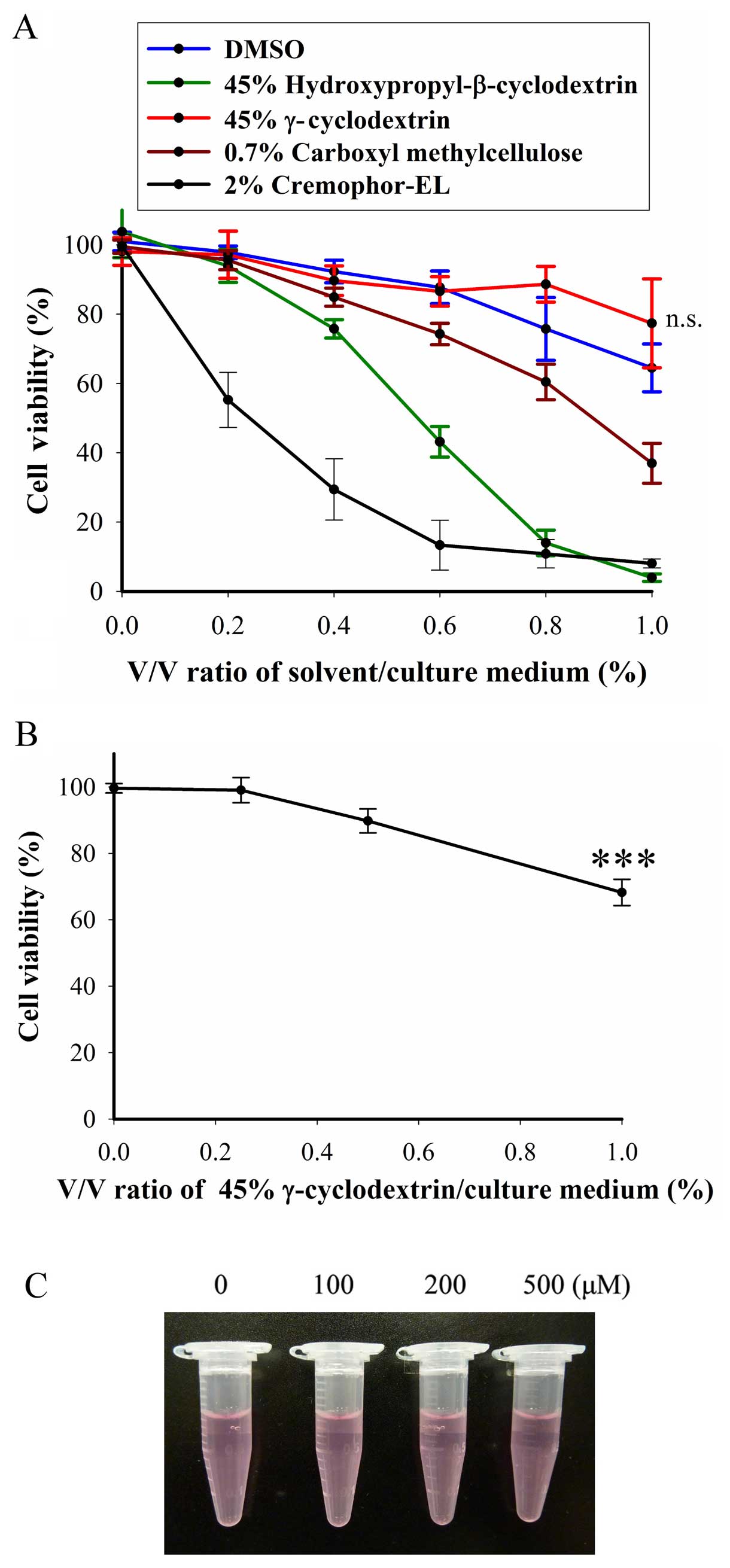
Only the γ-cyclodextrin did not elicit
cytotoxicity in BFTC905 cells
Based on the literature, initially we used DMSO as
the solvent for thalidomide (Fig.
1A). However, precipitation of thalidomide was observed
immediately following addition of thalidomide-DMSO solution into
culture medium (Fig. 1B).
Therefore, several solvents or substances were tested for their
ability to carry thalidomide into culture medium without
precipitation. We observed that cremophor-EL,
(2-hydroxypropyl)-β-cyclodextrin and γ-cyclodextrin can dissolve
thalidomide, while carboxymethyl cellulose (CMC) can keep
thalidomide in a suspension state. To test the cytotoxicity of
these solvents, BFTC905 cells were exposed to different v/v ratios
of solvent/culture medium (0–1%) for 48 h. At 1% of v/v, only 45%
γ-cyclodextrin did not exert cytotoxicity in BFTC905 cells
(p=0.198, n=3; Fig. 2A). Whereas,
γ-cyclodextrin did not elicit significant cytotoxicity in primary
HUC cells, except at the concentration of 1% v/v (p=0.002, n=3;
Fig. 2B). No precipitation was
observed when thalidomide/45% γ-cyclodextrin solution was added
into the culture medium (Fig. 2C).
Thalidomide dissolved in 45% γ-cyclodextrin was used for the
subsequent experiments.
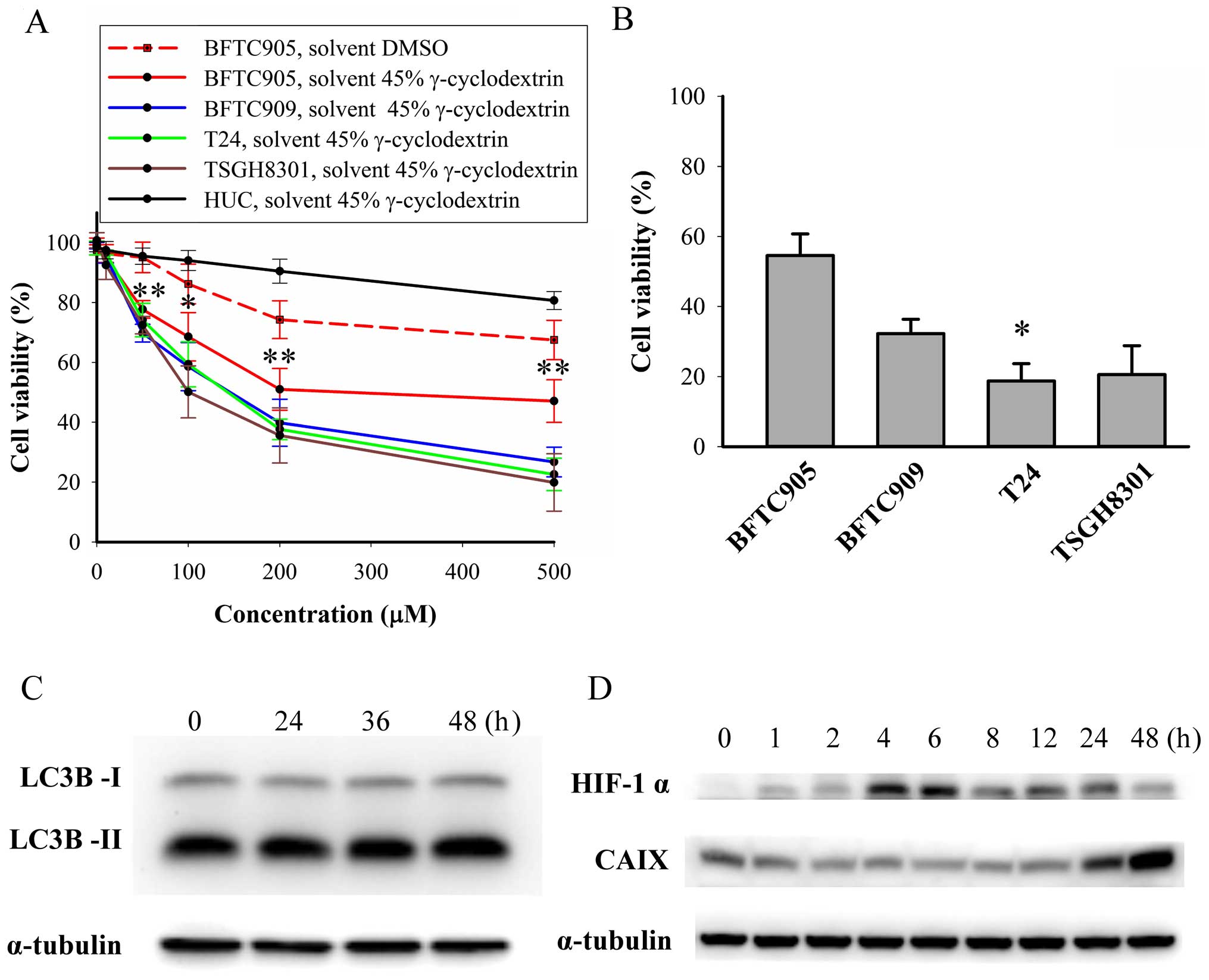
Cytotoxicity of thalidomide on TCC and
primary human urothelial cells
After 48-h treatment, thalidomide (dissolved in 45%
γ-cyclodextrin) caused a concentration-dependent cytotoxicity on
TCC cells (p<0.05 in all TCC cell lines by one-way ANOVA, n=3;
Fig. 3A). Thalidomide exerted
significant less cytotoxic effect in BFTC905 cells compared to
other TCC cell lines (all p<0.05 by two-way ANOVA). The
viability of BFTC905 cells after administration of thalidomide
dissolved in γ-cyclodextrin appeared to plateau at 200 μM
(51.0±7.0% vs. 47.1±7.1% for 200 and 500 μM, respectively;
p=0.714). The v/v of thalidomide and 45% γ-cyclodextrin mixture at
200 and 500 μM was 0.4 and 0.8%, respectively. The viability of
HUCs was not altered after administration of 200 μM thalidomide
dissolved in 45% γ-cyclodextrin (p=0.2, n=3), but 500 μM
thalidomide elicited significant cytotoxicity in HUC cells
(p=0.006). Therefore, 200 μM thalidomide dissolved in 45%
γ-cyclodextrin was used for the subsequent experiments.
Furthermore, we examined the cytotoxicity of
gemcitabine, the main current therapy for bladder cancer plus
thalidomide on TCC cells (Fig.
3B). After 48-h treatment, the viability of BFTC905, BFTC909,
and TSGH8301 cells was similar between thalidomide alone (200 μM)
and thalidomide plus gemcitabine (100 nM) group. Significant
additive cytotoxicity for the combination of gemcitabine and
thalidomide was only observed in T24 cells (37.6±3.4 vs. 18.7±5.0%
for thalidomide alone vs. thalidomide plus gemcitabine treatment,
respectively; p=0.03, n=3). Therefore, gemcitabine-resistant TCC
cell line BFTC905 (22) was chosen
to investigate the therapeutic effects of thalidomide in 45%
γ-cyclodextrin in the following experiments.
Autophagy mediated survival of cancer cells by
overcoming hypoxia and a shortage of nutrients (28). To delineate why thalidomide
elicited less efficacy on BFTC905 cells, the expression of LC3B-II,
HIF-1α, and CAIX was determined after thalidomide treatment.
Expression of LC3B-II, a marker of autophagy and converted from
LC3B-I, was not changed after thalidomide treatment (Fig. 3C). CAIX, a marker of hypoxia
induced by HIF-1α, may be a factor contributing to drug resistance
in cancer cells (29).
Upregulation of HIF-1α started 4 h after thalidomide treatment and
returned to basal level at 48 h (Fig.
3D). Increase in CAIX expression was observed 48 h after
thalidomide treatment (Fig.
3D).
Thalidomide inhibition of BFTC905 cell
growth is not via VEGF, bFGF, or TNF-α
Treatment with 100 and 200 μM thalidomide for 8 days
significantly inhibited the number of viable BFTC905 cells (p=0.02
and <0.001, respectively, in two-way ANOVA; n=3; Fig. 4A). The difference between 100 and
200 μM thalidomide treatment was statistically significant
(p=0.003). Thalidomide at 200 μM maintained the static growth of
BFTC905 cells. VEGF (30) and bFGF
(31) were secreted from TCC cells
as growth factors, and thalidomide has been reported to decrease
the expression of both (32).
Exogenous administration of TNF-α promoted proliferation of TCC
cells (33), and thalidomide
enhanced the degradation of TNF-α mRNA (34). Besides, BDNF was also a survival
factor for TCC in our previous study (20). Therefore, the secretion of these
molecules after thalidomide treatment for 48 h was determined.
Thalidomide at 200 and 500 μM significantly inhibited the secretion
of TNF-α and VEGF (n=3, Fig. 4B).
Furthermore, exogenous VEGF (100 nM), bFGF (100 nM), or TNF-α (100
ng/ml) did not reverse thalidomide induced growth inhibition
(Fig. 4C).
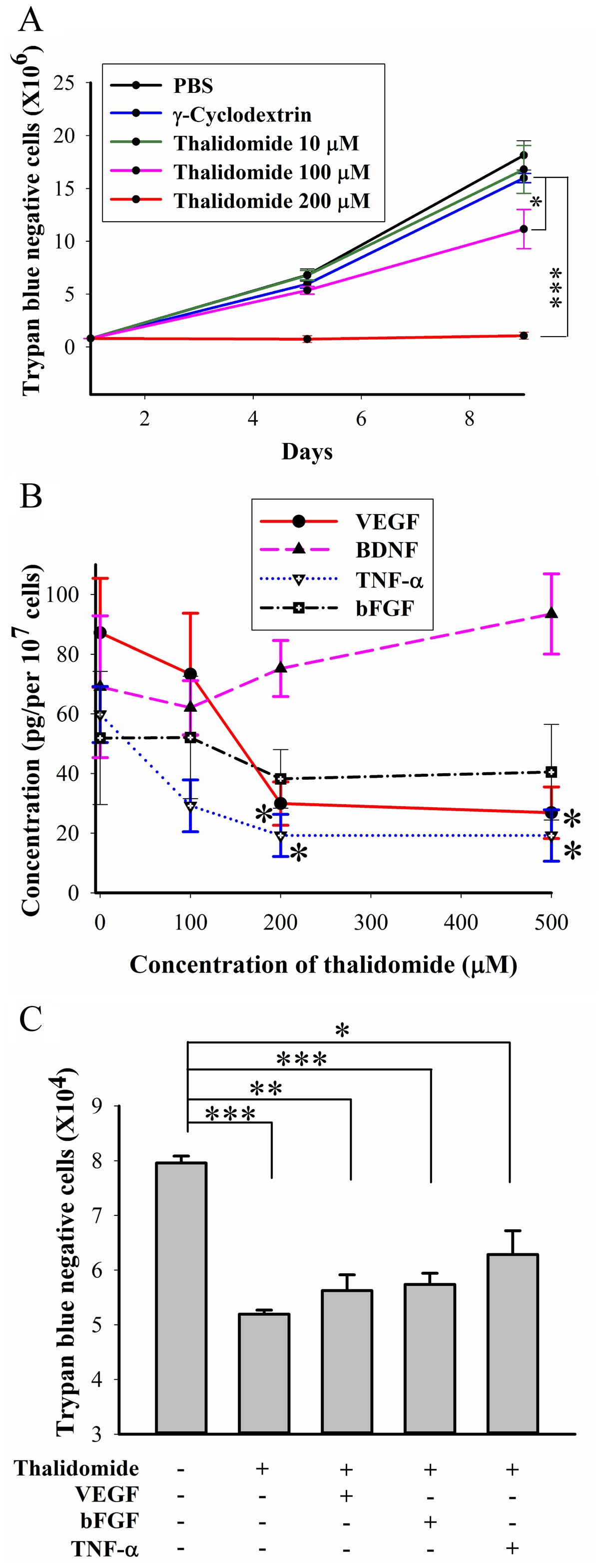 | Figure 4Roles of growth factors in BFTC905
cells treated with thalidomide (200 μM, dissolved in 45%
γ-cyclodextrin). (A) After administration of thalidomide on days 1
and 5, trypan blue-negative (viable) cell counts of BFTC905 cells
were calculated on days 5 and 9. (B) Secretion of BDNF, VEGF, bFGF,
and TNF-α from BFTC905 cells treated with thalidomide for 48 h was
measured by ELISA. (C) After co-adminstration of thalidomide and
100 nM VEGF, 100 nM BDNF, 100 nM bFGF, or 100 ng/ml TNF-α in
culture medium with 1% FBS for 48 h, trypan blue-negative cell
counts of BFTC905 cells were calculated.
*0.01≤p<0.05, **0.005≤p<0.01,
***p<0.005. |
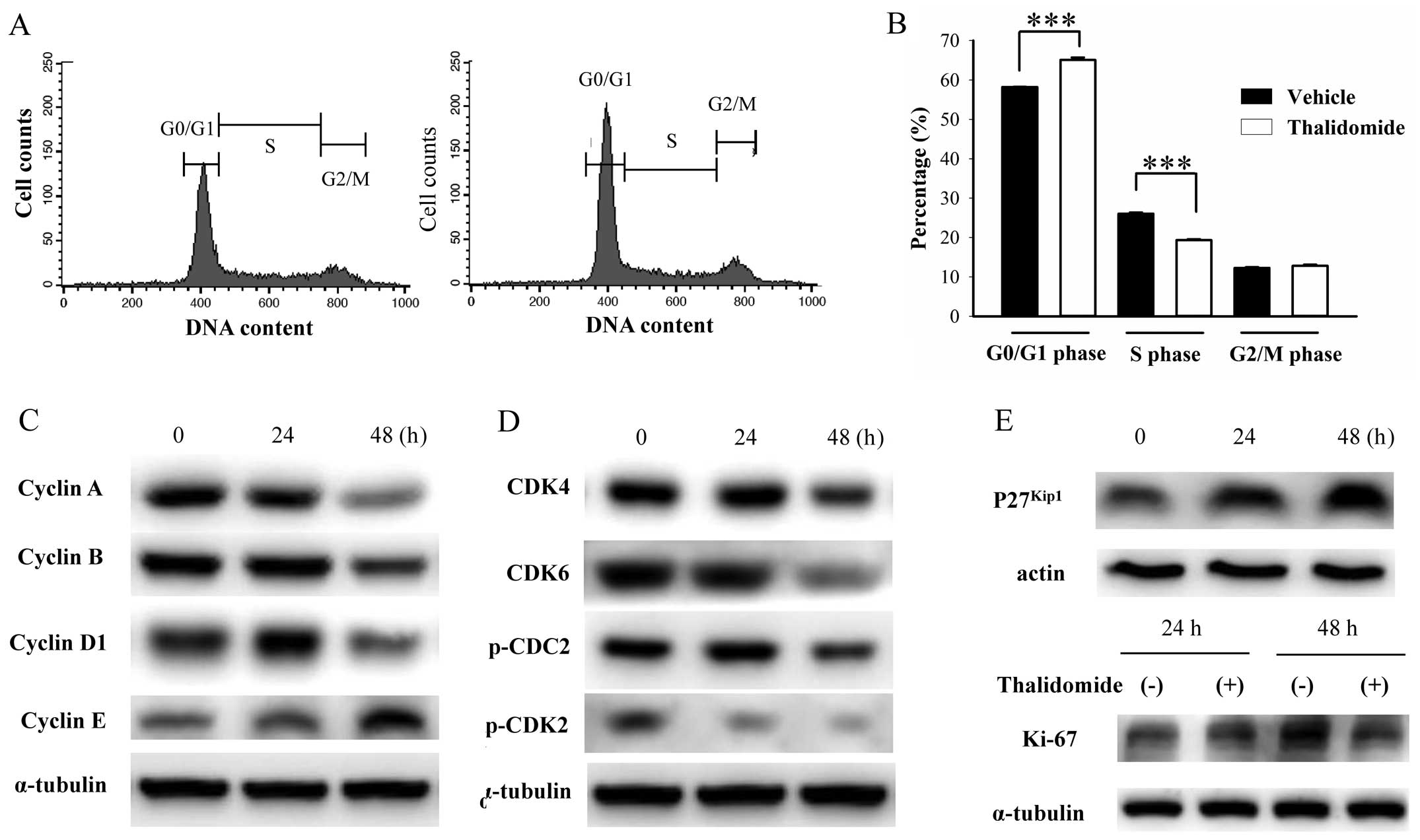
Thalidomide elicits quiescence in BFTC905
cells
After 48-h treatment, a significant accumulation of
G0/G1 phase [58.2±0.1 vs. 65.1±0.6% after vehicle (γ-cyclodextrin)
vs. 200 μM thalidomide treatment, n=3, p=0.002, Fig. 5A and B] was observed. Cell cycle
analysis by flow cytometry only reveals the proportional changes in
each phase, so we further examined the specific cell cycle related
proteins by western blotting (Fig. 5C
and D). The dynamic expression of cyclins, cyclin dependent
kinases, and related molecules in cell cycles is well established
(35,36). Downregulation of cyclin B and
phospho-CDC2, the markers of G2/M phase marker, indicated no G/M
arrest. Downregulation of cyclin A, which is expressed in S phase,
indicated no S phase arrest. Decrease in phospho-CDK2 expression,
which is expressed in late G1 phase or S phase, indicated no cell
cycle stasis in late G1 and S phase. Decrease in expression of
cyclin D1 and CDK4/6 (expressed in G1 phase before R point) and
cyclin E (expressed in late G1 phase after R point) indicated that
thalidomide did not induce G1 phase arrest. Due to the increased
ratio of G0/G1 phase without G1 arrest, we substantiated that
thalidomide induced G0 phase stasis, the so-called quiescence. The
increased expression of p27kip1, a potent inhibitor of
cyclin-CDK complex formation, supported this idea (Fig. 5E). Ki-67 protein is present in the
nucleus during all active phases of the cell cycle (G1, S, G2, and
mitotic phase) but absent from quiescent cells (G0), so Ki-67 is
the most referenced marker for cellular proliferation (37,38).
As shown in Fig. 5E, expression of
Ki-67 after 48-h thalidomide treatment was lower than control.
Thus, we found that thalidomide promoted quiescence in BFTC905
cells.
Thalidomide induces apoptosis in BFTC905
cells
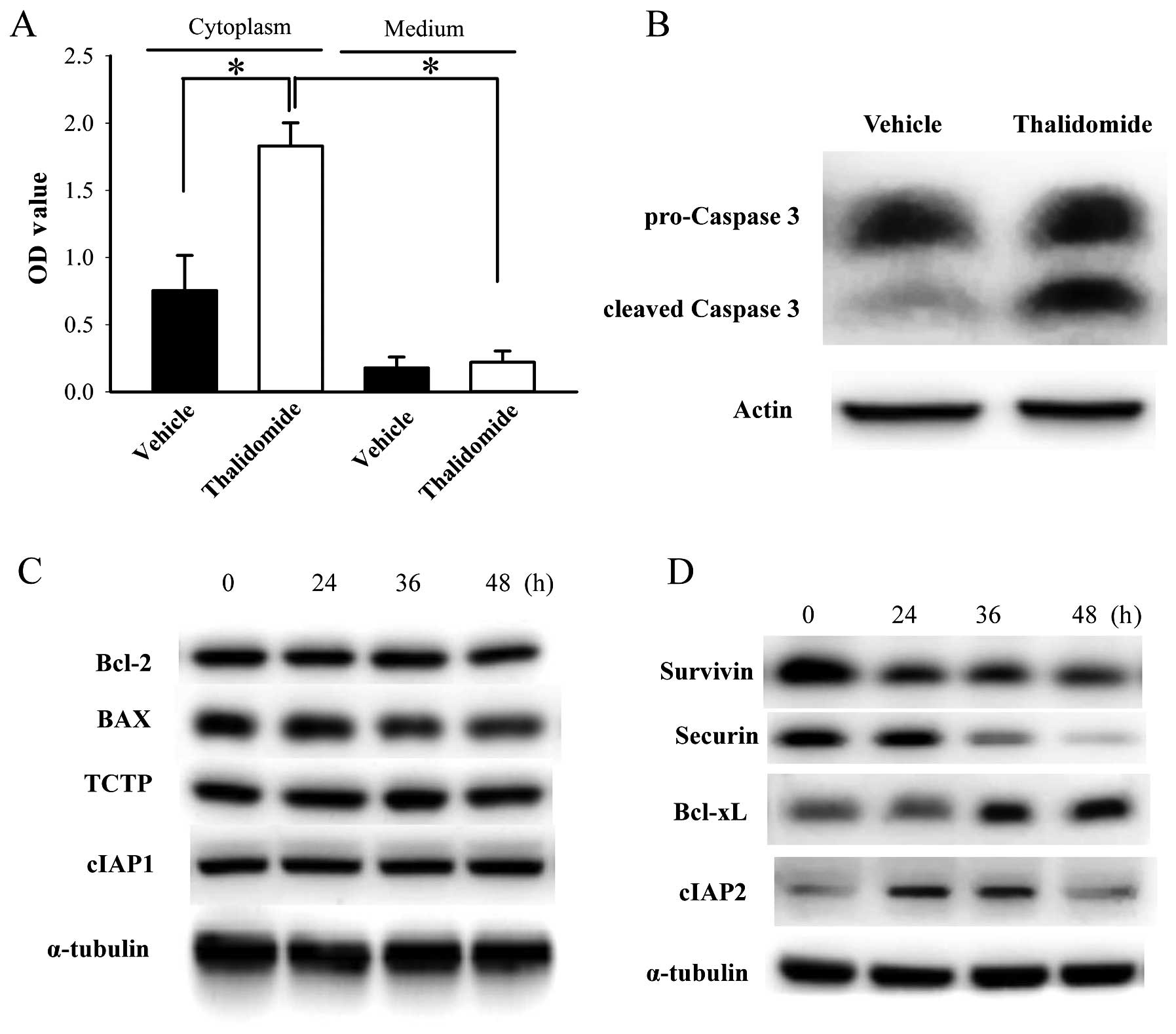
Cytoplasmic DNA fragments in BFTC905 cells, detected
by binding of anti-histone plus anti-DNA antibodies, was
significantly increased 48-h after 200 μM thalidomide treatment
compared to vehicle (n=3, p=0.03; Fig.
6A). Low basal levels and no significant changes of DNA
fragments were detected in the culture medium of BFTC905 cells 48 h
after thalidomide treatment (p=0.73; Fig. 6A). Expression of cleaved caspase 3,
activated both by extrinsic and intrinsic pathways in apoptotic
cells, was also increased after thalidomide treatment (Fig. 6B). These results indicated that
thalidomide induced apoptosis of BFTC905 cells. Furthermore, we
examined the changes of several anti-apoptosis or apoptosis related
proteins. Expression of Bcl-2, BAX, TCTP, and cIAP1 were not
changed (Fig. 6C). In contrast,
expression of survivin and securin were downregulated. Expression
of cIAP2 increased 24 and 36 h after thalidomide treatment, and
returned to baseline at 48 h (Fig.
6D). Upregulation of Bcl-xL was observed after thalidomide
treatment (Fig. 6D).
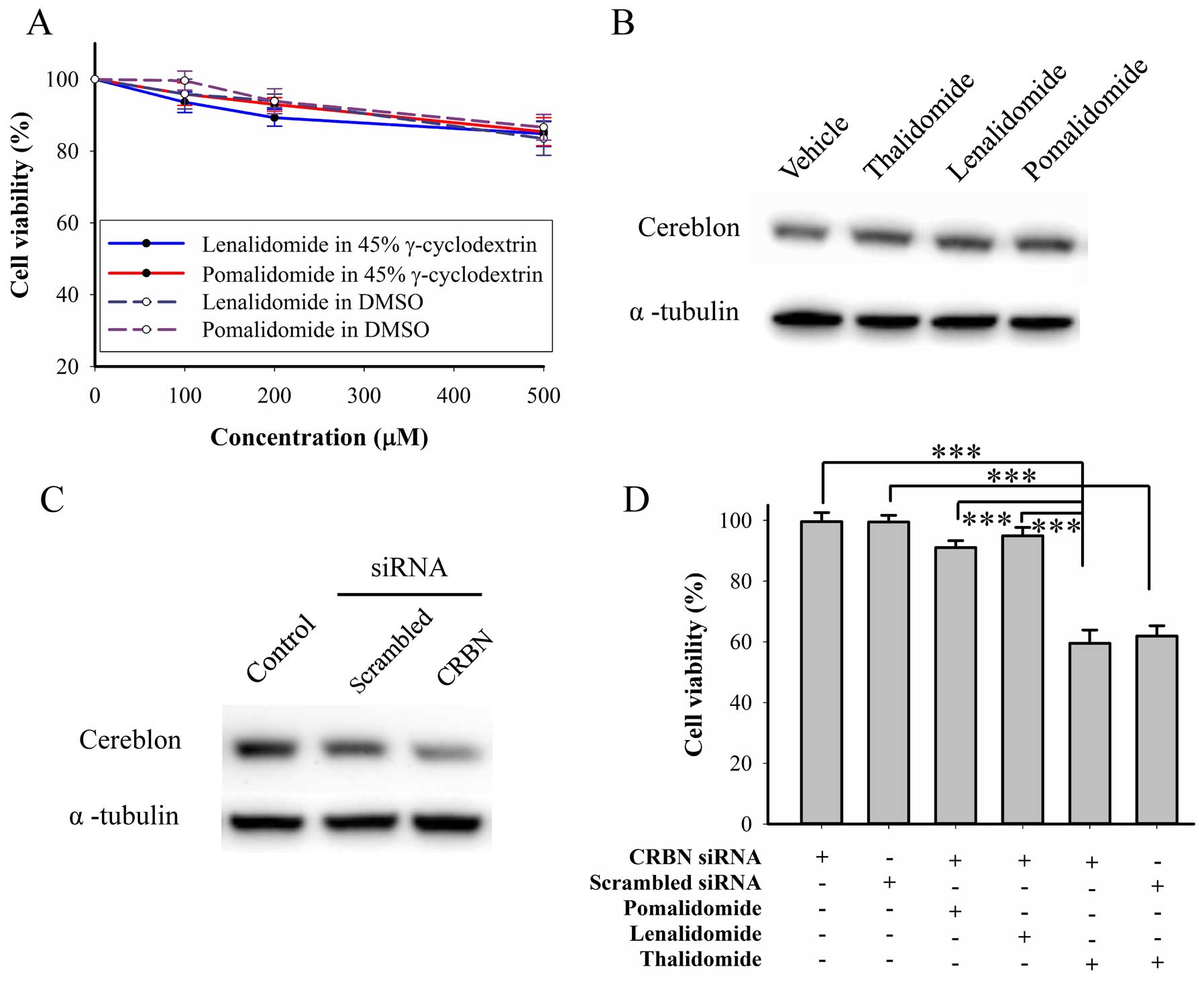
Lenalidomide and pomalidomide do not
elicit cytotoxicity in BFTC905 cells
We further evaluated cytotoxicity of other IMiDs,
lenalidomide and pomalidomide, on BFTC905 cells. Both drugs can be
dissolved in DMSO and γ-cyclodextrin. After 48-h treatment, 200 μM
of both drugs dissolved in both solvents did not elicit significant
cytotoxicity, but both at 500 μM elicited significant cytotoxicity
(~15%, Fig. 7A). Thalidomide and
other IMiDs were shown to bind to cereblon and inhibited the
associated E3 ubiquitin ligase activity (39), so we determined the roles of
cereblon in the mechanism of the thalidomide effects. After 48-h
treatment with IMiDs, no change of cereblon expression was observed
(Fig. 7B). Furthermore, knockdown
of cereblon by RNAi (Fig. 7C) did
not change the cytotoxicity elicited by thalidomide, lenalidomide,
or pomalidomide alone at 200 μM (Fig.
7C). Therefore, cereblon plays no role in thalidomide for
bladder cancer therapy.
Reactive oxidative species is produced
after thalidomide treatment
Free radical-mediated oxidative damage was involved
in the teratogenicity of thalidomide (40), so we evaluated the role of reactive
oxidative species (ROS) in thalidomide-induced cytotoxicity. After
48-h treatment, only thalidomide, neither lenalidomide nor
pomalidomide, triggered the production of ROS in BFTC905 cells
(Fig. 8A). A progressive
significant increase in total oxidant status (TOS) was found
following 48-h treatment with thalidomide (p=0.02 by one-way ANOVA,
n=3; Fig. 8B). Administration of
anti-oxidant catalase (10,000 U/ml), NAC (500 μM), and DTT (500 nM)
for 48 h all ameliorated thalidomide-induced cytotoxicity (p=0.04,
0.03, and 0.02 in catalase, NAC, DTT treatment group, respectively;
n=3; Fig. 8C). Thalidomide
significantly elicited oxidative DNA damage detected by the
formation of 8-OHdG (p=0.00004, n=3; Fig. 8D), and NAC significantly reversed
the effect (p=0.0005; Fig.
8D).
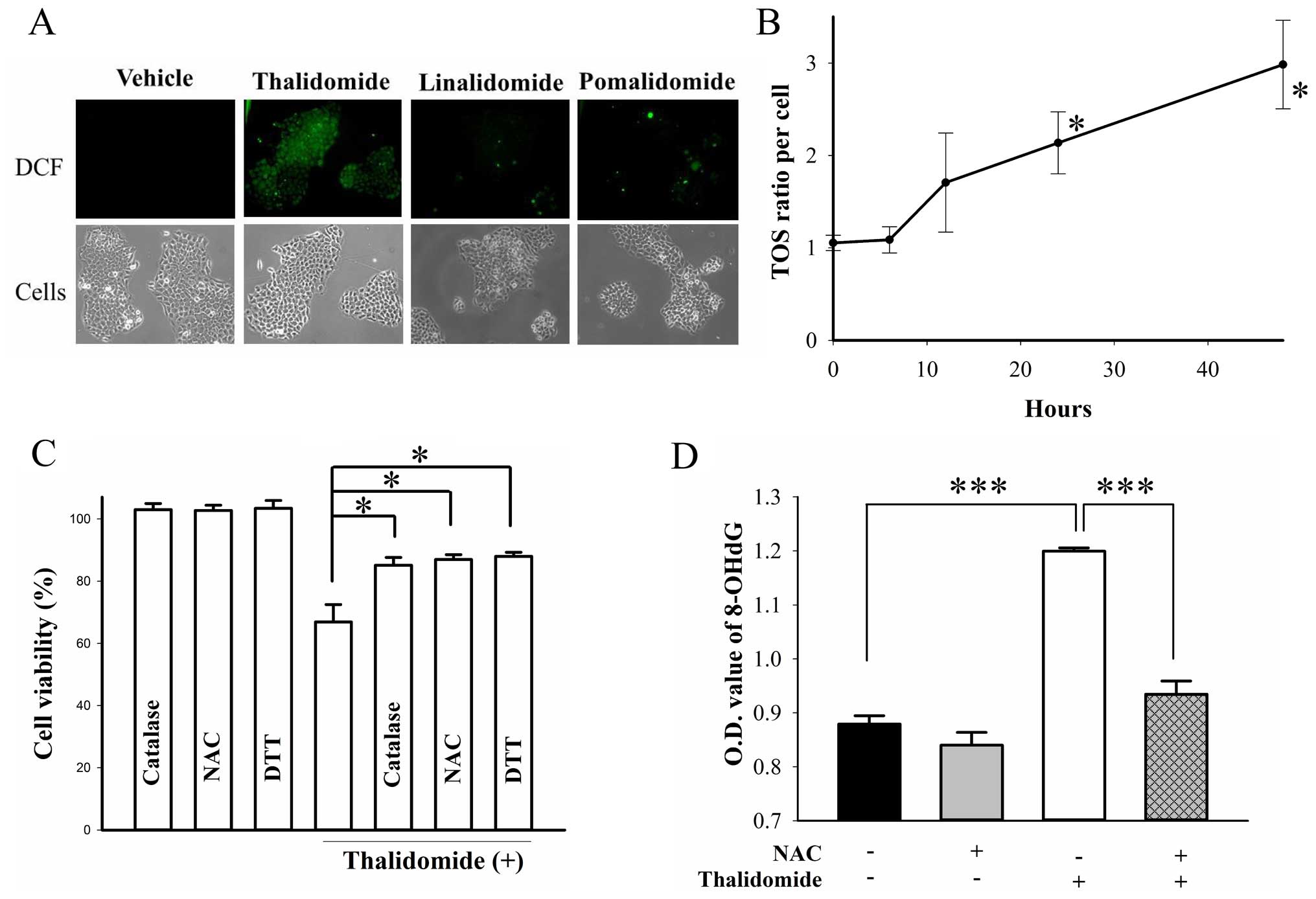 | Figure 8Role of ROS in BFCT905 cells treated
with IMiDs (all at 200 μM, dissolved in 45% γ-cyclodextrin). (A)
Expression of DCF, proportional to the ROS level within the cell
cytosol, was photographed 48 h after thalidomide, lenalidomide, or
pomalidomide treatment (×100). (B) Ratios of total oxidant status
(TOS) compared to vehicle exposure were measured after thalidomide
treatment within 48 h. (C) The viability of BFTC905 cells was
measured by MTT assay 48 h after administration with thalidomide
and/or anti-oxidants, including catalase (10,000 U/ml), NAC (500
μM), and DTT (500 nM). (D) Formation of 8-OHdG, the marker of
oxidative DNA damage after administration of thalidomide and/or
NAC. Vehicle, 45% γ-cyclodextrin. *0.01≤p<0.05,
***p<0.005. |
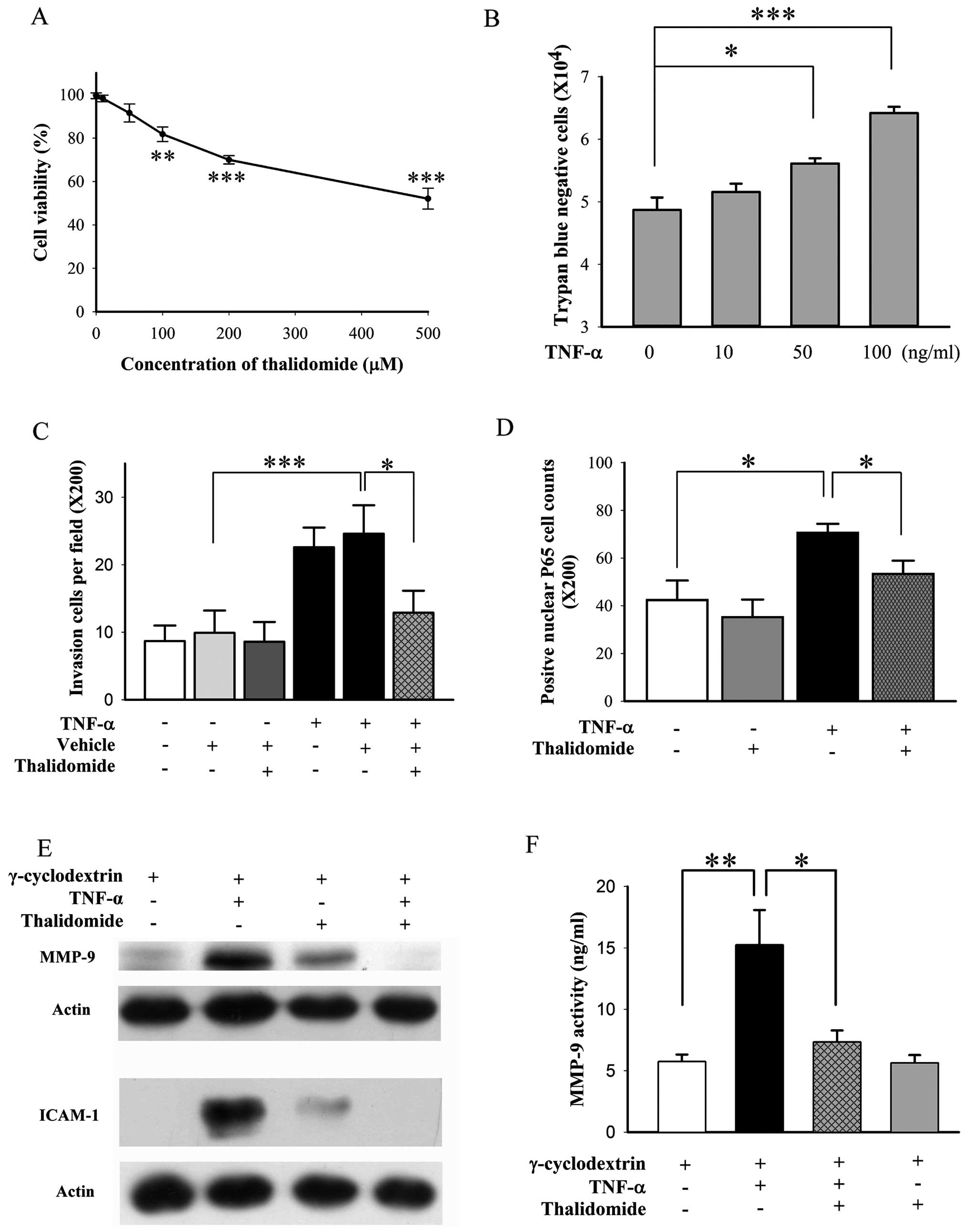
Thalidomide suppresses the invasion of
BFTC905 cells induced by TNF-α
For Matrigel invasion assay, chamber containing
BFTC905 cells with medium deficient in nutrient is placed above the
well containing full nutrient medium to create a nutrient gradient
for chemotaxis of invasion by BFTC905 cells. Thalidomide exerted
less cytotoxicity on BFTC905 cells in the medium containing 1% FBS
compared to normal medium containing 10% FBS (Fig. 9A vs. Fig. 3A). We found that 50 μM of
thalidomide did not alter the viability of BFTC905 cells (Fig. 9A; p=0.142, n=3), and this
concentration was chosen for the invasion experiment. However, no
changes of invasion ability and related signalings were noted.
Therefore, TNF-α induced invasion of TCC cells was followed
(33). In agreement with a
previous report (33), we found
that exogenous administration of 100 ng/ml TNF-α for 48 h, promoted
proliferation of BFTC905 cells with medium containing 1% FBS
(p=0.002, n=3; Fig. 9B). However,
in order to avoid the effects of proliferation in invasion
experiment, TNF-α at 10 ng/ml, which did not increase the number of
BFTC905 cells in 1% FBS, was used (p=0.4, n=3; Fig. 9B). TNF-α promoted the invasive
ability of BFTC905 cells in Matrigel assay (p=0.004, n=10), and
thalidomide significantly suppressed it (p=0.03, Fig. 9C). Transcription of NF-κB is
involved in TNF-α triggered signaling pathways, the nuclear
translocation of P65 after treatment with TNF-α and/or thalidomide
was examined. Nuclear P65-positive cells were increased after TNF-α
stimulation (p=0.01, n=5), which were reversed to baseline by
thalidomide treatment (p=0.03, Fig.
9D). The levels of MMP-9 and ICAM-1 have been found to be
higher in high grade and more invasive type of bladder cancer
(41,42). Thus, we examined the total
expression of MMP-9 and ICAM-1 and MMP-9 activity in the culture
medium. TNF-α upregulated the expression of MMP-9 and ICAM-1, and
thalidomide ameliorated their expression after TNF-α stimulation
(Fig. 9E). MMP-9 activity was also
significantly increased by TNF-α stimulation (p=0.008, n=5;
Fig. 9F). Thalidomide suppressed
MMP-9 activity after TNF-α stimulation (p=0.03, Fig. 9F).
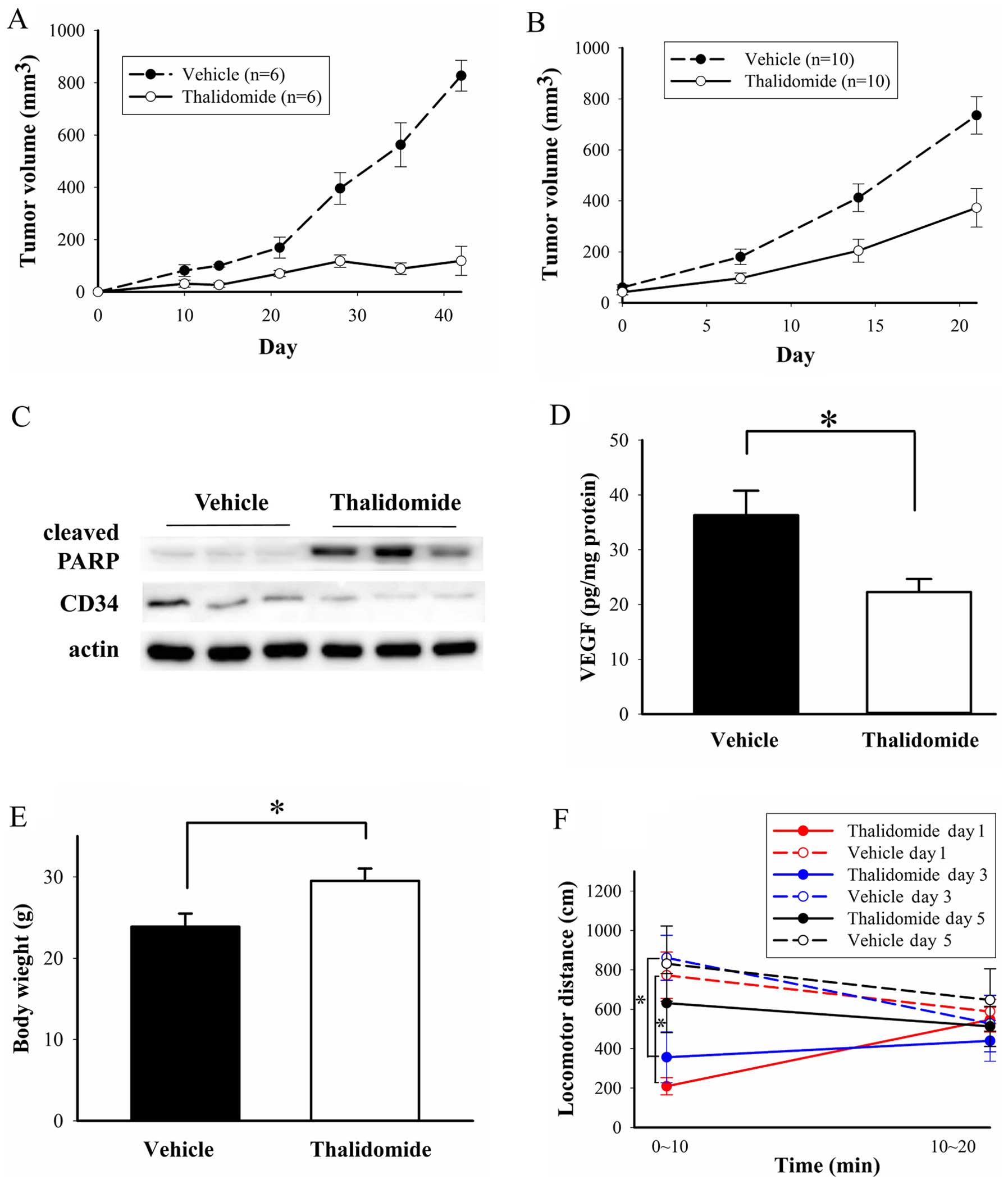
Effects of thalidomide on BFTC905
xenografts in vivo
When thalidomide treatment was initiated on the day
of BFTC905 cell implantation, almost no xenograft tumor formation
was observed (Fig. 10A). The
difference of tumor growth curve between thalidomide and vehicle
treatment revealed statistical significance (p<0.0001 by two-way
ANOVA, n=6). To evaluate the possible therapeutic effect,
thalidomide treatment was administered after tumor formation (2
weeks after BFTC905 cell implantation) in another experiment
(post-treatment strategy). After thalidomide treatment for 3 weeks,
the difference of tumor volume between thalidomide and vehicle
treatment also revealed statistical significance (p<0.0001 by
two-way ANOVA, n=10; Fig. 10B).
In addition, the significant difference was observed at 7 days
after treatment (96.4±20.7 and 180.3±30.1 mm3 for
thalidomide and vehicle treatment, respectively; p=0.03). Although
the tumor sizes were significantly increased after thalidomide
treatment (p<0.001 in one-way ANOVA), the average tumor sizes in
the thalidomide group were only half of those in the control group
(p=0.003). After 3-week treatment, the mean tumor volume in
thalidomide and vehicle group was 372.7±75.7 and 735.7±73.6
mm3, respectively. In the following results, we examined
the changes of cleaved PARP, CD34 and VEGF in the xenograft tumors
for the post-treatment strategy. Cleaved PARP facilitates cellular
disassembly and promotes apoptosis (43), and thalidomide treatment increased
the expression of cleaved PARP in three repeated samples (Fig. 10C). CD34 is a marker for
angiogenesis in bladder cancer (44). Expression of CD34 was decreased in
three repeated samples of thalidomide therapy (Fig. 10C). VEGF levels detected by ELISA
were also significantly decreased after thalidomide treatment
(p=0.02, n=5; Fig. 10D). Grossly,
no overt activity changes and no distal organ metastases were
observed.
Cachexia, regardless of disease category or
treatment modality, is a problem for cancer patients (45). However, in our study, average body
weight in thalidomide group was significantly higher than vehicle
group (29.5±1.5 vs. 23.9±1.6 g for thalidomide and vehicle group,
respectively; n=10, p=0.01, Fig.
10E). Sedative effects of thalidomide have been reported in
human (46), so we also examined
this adverse effect in mice. A significantly sedative effect with
lower locomotor distance (209.3±43.7 and 772.9±117.7 cm for
thalidomide and vehicle treatment, respectively; n=3, p=0.01) was
observed within the first 10 min after injecting the first dose of
thalidomide to mice (Fig. 10F).
The locomotor distance in the first 10 min after the second
administration of thalidomide also significantly less (356.6±129.6
and 861.7±114.0 cm for thalidomide and vehicle treatment,
respectively; n=3, p=0.04). The locomotor distance in the first 10
min after the third administration showed no difference in either
group (p=0.21). The locomotor distance in the 10–20 min after the
first, second, and third administration was similar for both groups
(p=0.66, 0.65 and 0.52, respectively).
Discussion
In previous studies, thalidomide was usually
dissolved in DMSO (18,47–49)
or CMC (50–52). However, there are only a few in
vitro thalidomide studies, and few researchers pay attention to
the solubility of thalidomide. Some in vitro studies
reported that the inhibitory effect of thalidomide may be partially
attributed to the solvent DMSO alone (53,54),
and our results were compatible with these studies. Cyclodextrins,
cyclic oligosaccharides with a hydrophilic outer surface and a
lipophilic central cavity to form inclusion complexes, can improve
solubility and permeability (55).
Some studies reported that thalidomide dissolved in ether-7
β-cyclodextrin or hydroxypropyl-β-cyclodextrin improved the aqueous
solubility (56–58). Kratz et al tested the
solubility of thalidomide with α-cyclodextrin, β-cyclodextrin, and
γ-cyclodextrin (56). Thalidomide
dissolved in β-cyclodextrin showed the best solubility, however,
cytotoxicity was not determined.
There are few in vitro studies of thalidomide
on solid malignancy cells. The concentration of thalidomide used in
this study might be relative high for BFTC905 cells, so we tried to
find the reasons. Release rate of drugs using cyclodextrin as a
carrier might be rapid, so insufficient intracellular concentration
due to thalidomide γ-cyclodextrin complex could be excluded
(59). Compensatory increase in
the expression of anti-apoptotic protein Bcl-xL was found in our
experiment, but overexpression of Bcl-xL was not correlated with
the recurrence and survival of bladder cancer patients (60). In our previous study, upregulation
of securin and Bcl-2 in BFTC905 cells may be a compensatory
mechanism for gemcitabine resistance (22). Downregulated expression of securin,
but no changes of Bcl-2 expression were observed after thalidomide
treatment. This may explain the lack of additively therapeutic
effects of gemcitabine plus thalidomide combination in this study,
and the much less potency of thalidomide than YM155, the survivin
inhibitor which decreased both securin and Bcl-2 expression in our
previous study (22). However,
thalidomide has some advantages for the potential therapy of
bladder cancer clinically, for examples, available in the market
and possible oral administration. Second, hypoxia-inducible CAIX
generates protons that contribute to acidification of external pH
and provides bicarbonate to neutralize intracellular pH through the
inward transporter. Such pH maintenance plays a role in the
regulation of cell proliferation, cell adhesion, and tumor
progression to mediate drug resistance (61). Any treatment aimed to inhibit
HIF-1α-CAIX axis signaling pathways as a therapy potential for
bladder cancer has been investigated (62). HIF-1α induced CAIX upregulation was
observed after thalidomide treatment in this study. This may also
contribute to the decreased potency of thalidomide on BFTC905
cells. Third, non-cycling quiescent cells can take the time to
repair the damage after cancer therapy (63,64).
The promoting effects of thalidomide to increase quiescent TCC
cells, although only 7% in magnitude, may also partially explain
the relatively high concentration of thalidomide used in this
study.
In the present study, the growth inhibition of
thalidomide on TCC cells resulted mainly from apoptotic effects.
The cytotoxic effects of 500 μM thalidomide on primary HUC cells
may result from γ-cyclodextrin per se. Our result
demonstrated the safety to normal urothelial cells with
administration of 200 μM thalidomide dissolved in 45%
γ-cyclodextrin. Some studies also reported the apoptotic activity
of thalidomide (17,65). In agreement with a previous study
(66), we found no changes of cell
cycle after thalidomide treatment. Downregulation of survivin and
securin by thalidomide is the novel findings in our study.
Survivin, a member of the inhibitor of apoptosis protein family,
has been shown to be a prognostic parameter of bladder cancer
(67). Previously we reported that
securin was overexpressed in human TCC specimens (68). Therefore, decreased expression of
survivin and securin is a good indicator of thalidomide therapy for
bladder cancer. Jian et al demonstrated that inhibition of
low-grade TCC cells by lenalidomide was attributable to not only
its direct tumor apoptotic but also anti-angiogenic activity
(69), so we compared cytotoxicity
of three IMiDs on gemcitabine-resistant TCC cells. Previous
research demonstrated cereblon-dependent anti-neoplastic activity
of IMiDs in cancer cells (70,71).
Depletion of cereblon endowed cancer cells with therapeutic
resistance to IMiDs. However, cereblon plays no role in
thalidomide-induced cytotoxicity in BFTC905 cells. Lack of
correlation with cereblon and IMiDs in human myeloma cell lines has
also been reported (72). ROS is a
double-edged sword in cancer biology. Puskás et al
demonstrated that lipid droplet binding thalidomide analogs induced
oxidative stress in cancer cells (73). Teratogenetic mechanism of
thalidomide may be caused by enzymes catalyzing thalidomide to a
free radical intermediate, and further generating ROS to modify DNA
in many ways, including the formation of 8-OHdG (40). Our results were consistent with the
pathophysiology of thalidomide-induced damage in embryos.
BFTC905 cells possess greater invasive ability
(23), so any treatment which
suppresses their invasion may be a good strategy for bladder cancer
therapy. The limitation of cell culture experiments is that
sometimes the in vitro environment may not mimic the in
vivo one. Due to lack of stimulated or stress condition in cell
culture system, we followed the model of TNF-α induced invasion in
TCC cells (33). We used only 10%
of TNF-α stimulation compared to Lee et al (33) to avoid the proliferative effects on
invasion. Thalidomide exerted its effect on downregulation of
NF-κB-induced ICAM-1 through inhibition of degradation of IκB
(18,74). Thalidomide decreased the mRNA,
protein synthesis, and secretion of MMP-9 (75). This evidence was also found in our
thalidomide-treated BFTC905 cells.
Although thalidomide significantly induced apoptosis
and inhibited angiogenesis in our animal experiments, thalidomide
only suppressed the growth rates of BFTC905 xenografts.
Nonetheless, it is consistent with the in vitro findings
that some compensatory or resistant mechanisms were found in
thalidomide-treated BFTC905 cells. Comparing to other studies (200
mg/kg/d = 1,400 mg/kg/w), less weekly administration of thalidomide
(250 mg/kg three times per week = 750 mg/kg/w) in our study might
be another explanation (76,77).
Therefore, combination of thalidomide with other drug(s) might be
the next strategy. Unfortunately, gemcitabine plus thalidomide did
not show additive cytotoxicity on BFTC905 cells in vitro.
Better drug combinations should be tested in the future. Beyond the
detection of VEGF in tumors, immunostaining of CD34 to analyze
microvascular density, a specific marker of endothelial cells, is a
common method for the evaluation of angiogenesis (78). We have also examined the
localization of CD34 in BFTC905 xenograft tumors by
immunohistochemistry, but few and scattered distribution of
positive CD34 endothelial cells were found (data not shown). So
western blotting was used instead of immunohistochemistry for CD34
in xenograft tumors. Low expression of CD34 measured by western
blotting was correlated with the findings by immunohistochemistry.
Thalidomide was effective in attenuating weight loss in cancer
patients with cachexia (79), and
our study also showed similar results. No animal studies report the
sedative effect after thalidomide treatment. Our observation that
thalidomide exerted significant somnolence in mice may be due to
the higher dose of injections. Patients developed tolerance to the
side effect of sedation (80), and
the present study is compatible with clinical usage of
thalidomide.
There are limitations in this study. We cannot
characterize the thalidomide γ-cyclodextrin complex, for example,
using X-ray powder diffractometry. In addition, we cannot detect
the concentration of thalidomide in cells. Lack of co-culture of
natural killer cells and TCC cells to examine the effects of IMiDs
on immune cells (81) is another
limitation. However, in the present study, we demonstrated the
potential effects of thalidomide therapy, including induction of
apoptosis through oxidative stress, inhibition of angiogenesis
through decreases in VEGF production and secretion, and suppression
of invasive ability via downregulation of MMP-9 and ICAM-1, for
gemcitabine-resistant bladder cancer cells in vitro and
in vivo.
Acknowledgements
This study was supported by a grant-in-aid from Tzu
Chi General Hospital, Hualien 970, Taiwan.
References
|
1
|
Murta-Nascimento C, Schmitz-Dräger BJ,
Zeegers MP, Steineck G, Kogevinas M, Real FX and Malats N:
Epidemiology of urinary bladder cancer: From tumor development to
patient's death. World J Urol. 25:285–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tavora F and Epstein JI: Bladder cancer,
pathological classification and staging. BJU Int. 102B:1216–1220.
2008. View Article : Google Scholar
|
|
3
|
Hall MC, Chang SS, Dalbagni G, Pruthi RS,
Seigne JD, Skinner EC, Wolf JS Jr and Schellhammer PF: Guideline
for the management of nonmuscle invasive bladder cancer (stages Ta,
T1, and Tis): 2007 update. J Urol. 178:2314–2330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Witjes JA, Compérat E, Cowan NC, De Santis
M, Gakis G, Lebret T, Ribal MJ, Van der Heijden AG and Sherif A:
European Association of Urology: EAU guidelines on muscle-invasive
and metastatic bladder cancer: Summary of the 2013 guidelines. Eur
Urol. 65:778–792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
von der Maase H, Hansen SW, Roberts JT,
Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A,
Lippert CM, et al: Gemcitabine and cisplatin versus methotrexate,
vinblastine, doxorubicin, and cisplatin in advanced or metastatic
bladder cancer: Results of a large, randomized, multinational,
multi-center, phase III study. J Clin Oncol. 18:3068–3077.
2000.PubMed/NCBI
|
|
6
|
George L, Bladou F, Bardou VJ, Gravis G,
Tallet A, Alzieu C, Serment G and Salem N: Clinical outcome in
patients with locally advanced bladder carcinoma treated with
conservative multimodality therapy. Urology. 64:488–493. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartlett JB, Dredge K and Dalgleish AG:
The evolution of thalidomide and its IMiD derivatives as anticancer
agents. Nat Rev Cancer. 4:314–322. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kumar S and Rajkumar SV: Thalidomide and
dexamethasone: Therapy for multiple myeloma. Expert Rev Anticancer
Ther. 5:759–766. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hideshima T, Chauhan D, Shima Y, Raje N,
Davies FE, Tai YT, Treon SP, Lin B, Schlossman RL, Richardson P, et
al: Thalidomide and its analogs overcome drug resistance of human
multiple myeloma cells to conventional therapy. Blood.
96:2943–2950. 2000.PubMed/NCBI
|
|
10
|
Gras J: Pomalidomide for patients with
multiple myeloma. Drugs Today (Barc). 49:555–562. 2013. View Article : Google Scholar
|
|
11
|
Andhavarapu S and Roy V: Immunomodulatory
drugs in multiple myeloma. Expert Rev Hematol. 6:69–82. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chanan-Khan AA, Swaika A, Paulus A, Kumar
SK, Mikhael JR, Rajkumar SV, Dispenzieri A and Lacy MQ:
Pomalidomide: The new immunomodulatory agent for the treatment of
multiple myeloma. Blood Cancer J. 3:e1432013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pinter M, Wichlas M, Schmid K, Plank C,
Müller C, Wrba F and Peck-Radosavljevic M: Thalidomide in advanced
hepatocellular carcinoma as antiangiogenic treatment approach: A
phase I/II trial. Eur J Gastroenterol Hepatol. 20:1012–1019. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yau T, Chan P, Wong H, Ng KK, Chok SH,
Cheung TT, Lam V, Epstein RJ, Fan ST and Poon RT: Efficacy and
tolerability of low-dose thalidomide as first-line systemic
treatment of patients with advanced hepatocellular carcinoma.
Oncology. 72(Suppl 1): S67–S71. 2007. View Article : Google Scholar
|
|
15
|
Fadul CE, Kingman LS, Meyer LP, Cole BF,
Eskey CJ, Rhodes CH, Roberts DW, Newton HB and Pipas JM: A phase II
study of thalidomide and irinotecan for treatment of glioblastoma
multiforme. J Neurooncol. 90:229–235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Puduvalli VK, Giglio P, Groves MD, Hess
KR, Gilbert MR, Mahankali S, Jackson EF, Levin VA, Conrad CA, Hsu
SH, et al: Phase II trial of irinotecan and thalidomide in adults
with recurrent glioblastoma multiforme. Neurooncol. 10:216–222.
2008.
|
|
17
|
Sun P, Zhang LM, Sun DJ and Dong LL:
Inhibitory effect of thalidomide on growth of human hepatoma cell
line SMMC-7721 cells. Zhonghua Zhong Liu Za Zhi. 31:582–586.
2009.(In Chinese). PubMed/NCBI
|
|
18
|
Lin YC, Shun CT, Wu MS and Chen CC: A
novel anticancer effect of thalidomide: Inhibition of intercellular
adhesion molecule-1-mediated cell invasion and metastasis through
suppression of nuclear factor-kappaB. Clin Cancer Res.
12:7165–7173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tzeng CC, Liu HS, Li C, Jin YT, Chen RM,
Yang WH and Lin JS: Characterization of two urothelium cancer cell
lines derived from a blackfoot disease endemic area in Taiwan.
Anticancer Res. 16A:1797–1804. 1996.
|
|
20
|
Huang YT, Lai PC, Wu CC, Hsu SH, Cheng CC,
Lan YF and Chiu TH: BDNF mediated TrkB activation is a survival
signal for transitional cell carcinoma cells. Int J Oncol.
36:1469–1476. 2010.PubMed/NCBI
|
|
21
|
Lai PC, Yang YC, Cheng CC, Chiu TH and
Huang YT: Brain-derived neurotrophic factor plus vascular
endothelial growth factor additively promotes early growth of the
transitional cell carcinoma cell line BFTC905 in vitro and in vivo.
Tzu Chi Med J. 25:155–160. 2013. View Article : Google Scholar
|
|
22
|
Huang YT, Cheng CC, Lin TC, Chiu TH and
Lai PC: Therapeutic potential of sepantronium bromide YM155 in
gemcitabine-resistant human urothelial carcinoma cells. Oncol Rep.
31:771–780. 2014.
|
|
23
|
Huang YT, Lai PC, Wu CC, Cheng CC and Chiu
TH: TrkB antibody elicits cytotoxicity and suppresses
migration/invasion of transitional cell carcinoma cells. Int J
Oncol. 37:943–949. 2010.PubMed/NCBI
|
|
24
|
Strober W: Trypan blue exclusion test of
cell viability. Curr Protoc Immunol. 3(Appendix): 3B2001.
|
|
25
|
Lai PC, Fang TC, Cheng CC, Chiu TH and
Huang YT: Lestaurtinib is cytotoxic to oxaliplatin-resistant
transitional cell carcinoma cell line T24 in vitro. Tzu Chi Med J.
22:125–130. 2010. View Article : Google Scholar
|
|
26
|
Erel O: A new automated colorimetric
method for measuring total oxidant status. Clin Biochem.
38:1103–1111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsu CW, Chen CY, Wang CS and Chiu TH:
Caffeine and a selective adenosine A2A receptor antagonist induce
reward and sensitization behavior associated with increased
phospho-Thr75-DARPP-32 in mice. Psychopharmacology (Berl).
204:313–325. 2009. View Article : Google Scholar
|
|
28
|
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM
and Lotze MT: Cell-mediated autophagy promotes cancer cell
survival. Cancer Res. 72:2970–2979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng G, Zhou M, Ou X, Peng B, Yu Y, Kong
F, Ouyang Y and He Z: Identification of carbonic anhydrase 9 as a
contributor to pingyangmycin-induced drug resistance in human
tongue cancer cells. FEBS J. 277:4506–4518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu W, Shu X, Hovsepyan H, Mosteller RD and
Broek D: VEGF receptor expression and signaling in human bladder
tumors. Oncogene. 22:3361–3370. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Allen LE and Maher PA: Expression of basic
fibroblast growth factor and its receptor in an invasive bladder
carcinoma cell line. J Cell Physiol. 155:368–375. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li X, Liu X, Wang J, Wang Z, Jiang W, Reed
E, Zhang Y, Liu Y and Li QQ: Thalidomide down-regulates the
expression of VEGF and bFGF in cisplatin-resistant human lung
carcinoma cells. Anticancer Res. 23B:2481–2487. 2003.
|
|
33
|
Lee EJ, Kim WJ and Moon SK: Cordycepin
suppresses TNF-alpha-induced invasion, migration and matrix
metalloproteinase-9 expression in human bladder cancer cells.
Phytother Res. 24:1755–1761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moreira AL, Sampaio EP, Zmuidzinas A,
Frindt P, Smith KA and Kaplan G: Thalidomide exerts its inhibitory
action on tumor necrosis factor alpha by enhancing mRNA
degradation. J Exp Med. 177:1675–1680. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hunter T and Pines J: Cyclins and cancer.
II: Cyclin D and CDK inhibitors come of age. Cell. 79:573–582.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scholzen T and Gerdes J: The Ki-67
protein: From the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gerdes J, Li L, Schlueter C, Duchrow M,
Wohlenberg C, Gerlach C, Stahmer I, Kloth S, Brandt E and Flad HD:
Immunobiochemical and molecular biologic characterization of the
cell proliferation-associated nuclear antigen that is defined by
monoclonal antibody Ki-67. Am J Pathol. 138:867–873.
1991.PubMed/NCBI
|
|
39
|
Ito T, Ando H, Suzuki T, Ogura T, Hotta K,
Imamura Y, Yamaguchi Y and Handa H: Identification of a primary
target of thalidomide teratogenicity. Science. 327:1345–1350. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Parman T, Wiley MJ and Wells PG: Free
radical-mediated oxidative DNA damage in the mechanism of
thalidomide teratogenicity. Nat Med. 5:582–585. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Donmez G, Sullu Y, Baris S, Yildiz L,
Aydin O, Karagoz F and Kandemir B: Vascular endothelial growth
factor (VEGF), matrix metalloproteinase-9 (MMP-9), and
thrombospondin-1 (TSP-1) expression in urothelial carcinomas.
Pathol Res Pract. 205:854–857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ozer G, Altinel M, Kocak B, Balci M, Altan
A and Gonenc F: Potential value of soluble intercellular adhesion
molecule-1 in the serum of patients with bladder cancer. Urol Int.
70:167–171. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bochner BH, Cote RJ, Weidner N, Groshen S,
Chen SC, Skinner DG and Nichols PW: Angiogenesis in bladder cancer:
Relationship between microvessel density and tumor prognosis. J
Natl Cancer Inst. 87:1603–1612. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nicolini A, Ferrari P, Masoni MC, Fini M,
Pagani S, Giampietro O and Carpi A: Malnutrition, anorexia and
cachexia in cancer patients: A mini-review on pathogenesis and
treatment. Biomed Pharmacother. 67:807–817. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Höglund P, Eriksson T and Björkman S: A
double-blind study of the sedative effects of the thalidomide
enantiomers in humans. J Pharmacokinet Biopharm. 26:363–383. 1998.
View Article : Google Scholar
|
|
47
|
Ridoux O and Drancourt M: Lack of in vitro
antimicrosporidian activity of thalidomide. Antimicrob Agents
Chemother. 43:2305–2306. 1999.PubMed/NCBI
|
|
48
|
Moreira AL, Wang J, Sarno EN and Kaplan G:
Thalidomide protects mice against LPS-induced shock. Braz J Med
Biol Res. 30:1199–1207. 1997. View Article : Google Scholar
|
|
49
|
Shannon EJ, Sandoval FG and Morales MJ: In
vitro thalidomide does not interfere with the activation of
complement by M. leprae. J Drugs Dermatol. 10:274–278.
2011.PubMed/NCBI
|
|
50
|
Mall JW, Schwenk W, Philipp AW, Müller JM
and Pollmann C: Thalidomide given intraperitoneally reduces the
number of postoperative adhesions after large bowel resection in
rabbits. Eur J Surg. 168:641–645. 2002. View Article : Google Scholar
|
|
51
|
D'Amato RJ, Loughnan MS, Flynn E and
Folkman J: Thalidomide is an inhibitor of angiogenesis. Proc Natl
Acad Sci USA. 91:4082–4085. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lentzsch S, LeBlanc R, Podar K, Davies F,
Lin B, Hideshima T, Catley L, Stirling DI and Anderson KC:
Immunomodulatory analogs of thalidomide inhibit growth of Hs Sultan
cells and angiogenesis in vivo. Leukemia. 17:41–44. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Oz ES, Aydemir E and Fışkın K: DMSO
exhibits similar cytotoxicity effects to thalidomide in mouse
breast cancer cells. Oncol Lett. 3:927–929. 2012.PubMed/NCBI
|
|
54
|
Eter N and Spitznas M: DMSO mimics
inhibitory effect of thalidomide on choriocapillary endothelial
cell proliferation in culture. Br J Ophthalmol. 86:1303–1305. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Loftsson T and Duchêne D: Cyclodextrins
and their pharmaceutical applications. Int J Pharm. 329:1–11. 2007.
View Article : Google Scholar
|
|
56
|
Kratz JM, Teixeira MR, Ferronato K,
Teixeira HF, Koester LS and Simões CM: Preparation,
characterization, and in vitro intestinal permeability evaluation
of thalidomide-hydroxypropyl-β-cyclodextrin complexes. AAPS
PharmSciTech. 13:118–124. 2012. View Article : Google Scholar
|
|
57
|
Alvarez C, Calero J, Menéndez JC, Torrado
S and Torrado JJ: Effects of hydroxypropyl-beta-cyclodextrin on the
chemical stability and the aqueous solubility of thalidomide
enantiomers. Pharmazie. 63:511–513. 2008.PubMed/NCBI
|
|
58
|
Kale R, Tayade P, Saraf M and Juvekar A:
Molecular encapsulation of thalidomide with sulfobutyl ether-7
beta-cyclodextrin for immediate release property: Enhanced in vivo
antitumor and antiangiogenesis efficacy in mice. Drug Dev Ind
Pharm. 34:149–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stella VJ, Rao VM, Zannou EA and Zia V:
Mechanisms of drug release from cyclodextrin complexes. Adv Drug
Deliv Rev. 36:3–16. 1999. View Article : Google Scholar
|
|
60
|
Kirsh EJ, Baunoch DA and Stadler WM:
Expression of bcl-2 and bcl-X in bladder cancer. J Urol.
159:1348–1353. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sedlakova O, Svastova E, Takacova M,
Kopacek J, Pastorek J and Pastorekova S: Carbonic anhydrase IX, a
hypoxia-induced catalytic component of the pH regulating machinery
in tumors. Front Physiol. 4:4002014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen MC, Lee CF, Huang WH and Chou TC:
Magnolol suppresses hypoxia-induced angiogenesis via inhibition of
HIF-1α/VEGF signaling pathway in human bladder cancer cells.
Biochem Pharmacol. 85:1278–1287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Borst P: Cancer drug pan-resistance:
Pumps, cancer stem cells, quiescence, epithelial to mesenchymal
transition, blocked cell death pathways, persisters or what? Open
Biol. 2:1200662012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Goss PE and Chambers AF: Does tumour
dormancy offer a therapeutic target? Nat Rev Cancer. 10:871–877.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Podhorecka M, Halicka HD, Klimek P, Kowal
M and Dmoszynska A: Thalidomide induces phosphorylation of histone
H2AX and increases rate of apoptosis caused by fludarabine in
malignant lymphocytes of chronic lymphocytic leukemia in short-term
cell cultures. Leuk Res. 33:997–1000. 2009. View Article : Google Scholar
|
|
66
|
Yu J, Liu F, Sun Z, Sun M and Sun S: The
enhancement of radio-sensitivity in human esophageal carcinoma
cells by thalidomide and its potential mechanism. Cancer Biother
Radiopharm. 26:219–227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Margulis V, Lotan Y and Shariat SF:
Survivin: A promising biomarker for detection and prognosis of
bladder cancer. World J Urol. 26:59–65. 2008. View Article : Google Scholar
|
|
68
|
Lai PC, Fang TC, Chiu TH and Huang YT:
Overexpression of securin in human transitional cell carcinoma
specimens. Tzu Chi Med J. 22:171–176. 2010. View Article : Google Scholar
|
|
69
|
Jian W, Levitt JM, Lerner SP and Sonpavde
G: The preclinical activity of lenalidomide in indolent urothelial
carcinoma. Anticancer Res. 34:3383–3389. 2014.PubMed/NCBI
|
|
70
|
Ren S, Xu C, Cui Z, Yu Y, Xu W, Wang F, Lu
J, Wei M, Lu X, Gao X, et al: Oncogenic CUL4A determines the
response to thalidomide treatment in prostate cancer. J Mol Med
Berl. 90:1121–1132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lopez-Girona A, Mendy D, Ito T, Miller K,
Gandhi AK, Kang J, Karasawa S, Carmel G, Jackson P, Abbasian M, et
al: Cereblon is a direct protein target for immunomodulatory and
antiproliferative activities of lenalidomide and pomalidomide.
Leukemia. 26:2326–2335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Greenberg AJ, Walters DK, Kumar SK,
Vincent Rajkumar S and Jelinek DF: Responsiveness of
cytogenetically discrete human myeloma cell lines to lenalidomide:
Lack of correlation with cereblon and interferon regulatory factor
4 expression levels. Eur J Haematol. 91:504–513. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Puskás LG, Fehér LZ, Vizler C, Ayaydin F,
Rásó E, Molnár E, Magyary I, Kanizsai I, Gyuris M, Madácsi R, et
al: Polyunsaturated fatty acids synergize with lipid droplet
binding thalidomide analogs to induce oxidative stress in cancer
cells. Lipids Health Dis. 9:562010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lv P, Luo HS, Zhou XP, Xiao YJ, Paul SC,
Si XM and Zhou YH: Reversal effect of thalidomide on established
hepatic cirrhosis in rats via inhibition of nuclear
factor-kappaB/inhibitor of nuclear factor-kappaB pathway. Arch Med
Res. 38:15–27. 2007. View Article : Google Scholar
|
|
75
|
Piura B, Medina L, Rabinovich A, Dyomin V
and Huleihel M: Thalidomide distinctly affected TNF-α, IL-6 and MMP
secretion by an ovarian cancer cell line (SKOV-3) and primary
ovarian cancer cells. Eur Cytokine Netw. 24:122–129.
2013.PubMed/NCBI
|
|
76
|
Zhang S, Li M, Gu Y, Liu Z, Xu S, Cui Y
and Sun B: Thalidomide influences growth and vasculogenic mimicry
channel formation in melanoma. J Exp Clin Cancer Res. 27:602008.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kotoh T, Dhar DK, Masunaga R, Tabara H,
Tachibana M, Kubota H, Kohno H and Nagasue N: Antiangiogenic
therapy of human esophageal cancers with thalidomide in nude mice.
Surgery. 125:536–544. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sasano H and Suzuki T: Pathological
evaluation of angiogenesis in human tumor. Biomed Pharmacother.
59(Suppl 2): S334–S336. 2005. View Article : Google Scholar
|
|
79
|
Gordon JN, Trebble TM, Ellis RD, Duncan
HD, Johns T and Goggin PM: Thalidomide in the treatment of cancer
cachexia: A randomised placebo controlled trial. Gut. 54:540–545.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Grover JK, Uppal G and Raina V: The
adverse effects of thalidomide in relapsed and refractory patients
of multiple myeloma. Ann Oncol. 13:1636–1640. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhu D, Corral LG, Fleming YW and Stein B:
Immunomodulatory drugs Revlimid (lenalidomide) and CC-4047 induce
apoptosis of both hematological and solid tumor cells through NK
cell activation. Cancer Immunol Immunother. 57:1849–1859. 2008.
View Article : Google Scholar : PubMed/NCBI
|