Introduction
Phosphatidylinositol 3-kinase (PI3K) represents a
family of closely related enzymes that serve to transduce
downstream signaling and have a profound role in multiple critical
cellular processes, including growth, differentiation, metabolism,
survival and cellular proliferation (1,2).
PI3K are divided into three distinct classes on the basis of
primary structure, function and lipid substrate specificity
(3). Class I PI3Ks, the most
widely implicated as aberrant in cancers, are heterodimers composed
of a regulatory and catalytic subunit and subdivided into 1A and 1B
based on their mode of activation. Class 1A is composed of p110α,
p110β and p110δ (catalytic subunits), bound by p85, p50 or p55
(regulatory subunits). Class 1B consists of a single catalytic
domain, p110γ bound by the regulatory domain p101 (4). Class II PI3Ks are monomeric and three
catalytic isoforms have been identified: the ubiquitously expressed
PI3K-C2α and PI3K-C2β and liver-specific PI3K-C2γ. Class III PI3Ks
are heterodimeric enzymes consisting of a catalytic (Vps34) and a
regulatory (Vps15/p150) subunit (5).
PI3K binds to and is activated by several upstream
receptors and non-receptor protein tyrosine kinases (6,7).
Once activated, PI3K phosphorylates its lipid substrate
phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) to
PtdIns(3,4,5) P3, a critical intra-cellular lipid second messenger.
This process is opposed by the tumor suppressor phosphatase and
tension homolog (PTEN) which is frequently deleted or mutated in
human cancers that results in constitutive PI3K activation
(8). It in turn activates Akt, an
important downstream effector through interacting with
PtdIns(3,4,5)P3 via its pleckstrin homology (PH) domain. Activated
Akt inhibits the tuberous sclerosis complexes 1 and 2 (TSC1 and
TSC2), permitting activation of the mTOR complex 1 (mTORC1) and
subsequent phosphorylation of proteins 70S6K1, S6 and the
eukaryotic translation initiation factor 4E-binding protein 1
(4EBP1), resulting in dysregulation of protein synthesis and cell
survival (9). A second complex of
mTOR, known as mTORC2, appears to act as a feedback loop via Akt.
Activation of mTORC2 via phosphorylated 70S6K1 induces
phosphorylation of Akt at its serine 473 residue and facilitates
its complete activation (10).
The oncogenic potential of the PI3K/Akt/mTOR pathway
is well documented. Forced expression of PI3K is shown to induce
cell line transformation (11),
tumor formation (12) and
angiogenesis in vivo (13).
It has also been shown that Akt is involved in malignant
transformation (14). PI3K
signaling activation frequently occurs in numerous human tumors due
to multiple molecular alterations, such as mutations (PIK3CA, Akt1
and PTEN), gene amplifications (PIK3CA, Akt1 and Akt2), loss of
expression of the tumor suppressors PTEN and inositol
polyphosphate-4-phosphatase type II and other mechanisms (15). In addition, deregulation of the
PI3K/Akt/mTOR axis is also observed when upstream oncogenes are
mutated or amplified and tumor suppressor genes are deleted
(16). Indeed, numerous inhibitors
targeting this signaling pathway have been developed and have been
shown to possess preliminary clinical activity (16).
Aggressive B-cell non-Hodgkin's lymphomas (B-NHL)
such as diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma
(MCL), Burkitt's lymphoma (BL) and follicular lymphoma (FL) are
incurable with current chemoimmunotherapy regimens and development
of novel and effective treatments based on biologically validated
targets is urgently needed (17).
Same as in solid tumors, activation of the PI3K/Akt/mTOR signaling
pathway occurs commonly in B-NHL (18). The mechanism by which the pathway
is activated differs by lymphoma subtype. In MCL, most patients
have an increased copy number of PIK3CA, resulting in increased
transcription and pathway activation (19). PTEN loss has been found in GCB
subtype of DLBCL, which results in increased PI3K/Akt signaling and
in vitro PI3K inhibitor sensitivity (20). Moreover, phosphorylation of Akt and
p70S6K occurs in most DLBCL cell lines and patient samples
(21,22). The B-cell receptor (BCR), a
critical signaling pathway for B-cell survival, also induces PI3K
pathway activation (23,24). Importantly, inhibition of
PI3K/Akt/mTOR pathway have shown anti-lymphoma activity associated
with cell cycle arrest and apoptosis both in vivo and in
vitro (25–28).
PF-04691502 is a potent, selective and orally active
ATP-competitive PI3K/mTOR inhibitor with Ki values of 1.6, 1.8,
1.9, 2.1 and 16 nM for human PI3K δ, α, γ, β and mTOR, respectively
(29). Many in vitro and
in vivo studies have shown that PF-04691502 can inhibit cell
proliferation, induce apoptosis and has antitumor activity in solid
tumors (30–34). However, PF-04691502 has not been
tested in hematologic malignancies. In the present study, we
demonstrated that PF-04691502 inhibited cell proliferation, induced
cell cycle arrest and apoptosis in aggressive B-cell NHL cell lines
associated with inhibition of PI3K/Akt/mTOR signaling activity.
Materials and methods
Cells and reagents
U-2932 (DLBC) and Granta-519 (MCL) cell lines were
purchased from Deutsche Sammlung von Mikroorganismen und
Zellkulturen GmbH (DSMZ; Braunschweig, Germany). SUDHL-10 (DLBC)
cell line was from the American Type Culture Collection (ATCC;
Rockville, MD, USA). Cells were maintained in RPMI-1640 medium
(Mediatech, Herndon, VA, USA) supplemented with 10% fetal bovine
serum (FBS), 2 mM sodium pyruvate and 100 units/ml
penicillin/streptomycin at 37°C in a humidified atmosphere
containing 5% CO2. The doubling time of each of these
three cell lines is ~48 h. PF-04691502 (PI3K(α/β/δ/γ)/mTOR dual
inhibitor), CAL-101 (p110δ inhibitor) and INK-128 (mTORC1/2
inhibitor) were purchased from Selleckchem (Houston, TX, USA). The
compounds were dissolved at 50 mM in DMSO as a stock solution, and
then further diluted to desired concentrations for in vitro
experiments. Rituximab was a kind donation by the affiliated
hospital clinic. Anti-phospho-Akt (Ser473 and Thr308; 1:500
dilution), anti-Akt (1:500 dilution), anti-phospho-S6 (1:1,000
dilution) and anti-GAPDH (14C10; 1:1,000 dilution) antibodies were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Anti-PARP (H-250; 1:500 dilution) and anti-cyclin D1 (1:500
dilution) were from Santa Cruz Biotechnology (Santa Cruz, CA,
USA).
Inhibition analysis of cell proliferation
(MTS assay)
Cells were seeded at 10,000/well in 96-well culture
plates and allowed to grow for 24 h followed by the desired
treatment with increasing concentrations of the indicated agents
for 4 days. The studies were conducted in three separate
experiments with triplicate on one plate. Viable cell densities
were determined using a CellTiter 96 Cell Proliferation assay
(Promega, Madison, WI, USA). Absorbance readings at 490 nm were
analyzed against the control group for each drug treatment to
determine cell viability. The IC50 values were estimated
by Calcusyn software (Biosoft, Cambridge, UK).
Cell cycle analysis
Cells were treated with different concentrations of
PF-04691502 for 48 h and then the cells were centrifuged at 1,500 ×
g for 5 min at 4°C and resuspended in PBS, fixed by drop wise
addition of ice-cold ethanol (100%) to a final concentration of
70%, and incubated for 30 min on ice. Fixed cells were pelleted and
treated with 100 μl of RNase A (0.2 mg/ml in PBS) for 5 min at room
temperature, then suspended in 0.5 ml of ddH2O. After
staining with 4 μg/ml propidium iodide, the DNA content was
determined using a BD Biosciences LSR II flow cytometer and the
cell cycle profile was analyzed by ModFit software. Cell aggregates
were gated out of the analysis, based on the width of the propidium
iodide fluorescence signal. Each profile was compiled from 10,000
gated events.
Apoptosis assay
Using Annexin V staining to detect apoptosis,
treated cells were harvested and rinsed with cold PBS once. After
centrifugation for 5 min, cells were resuspended in 500 μl of 1X
Annexin V binding buffer (Annexin V-FITC reagent kit,
cat.#1001-1000; BioVision, Inc., Milpitas, CA, USA) and then 5 μl
of Annexin V-FITC and 5 μl of propidium iodide (PI) (Annexin V-FITC
reagent kit; BioVision) were added. After incubation for 5 min at
room temperature in the dark, the samples were analyzed by a BD
Biosciences FACSCalibur flow cytometer.
Immunoblotting
The cells were lysed in NP-40 lysis buffer
containing 50 mM Tris-Cl (pH 7.4), 0.15 M NaCl, 0.5% NP-40, 1 mM
DTT, 50 mM sodium fluoride, and 2 μl/ml protease inhibitor cocktail
(Sigma, St. Louis, MO, USA). Protein concentrations were determined
using the Bio-Rad protein assay kit (Bio-Rad Laboratories,
Hercules, CA, USA) and 50 μg of protein was resolved by
electrophoresis on a 10% SDS-PAGE gel. The proteins were then
transferred onto a nitrocellulose membrane and non-specific binding
was blocked by incubating with 5% non-fat milk in TBST buffer (0.01
M Tris-Cl, 0.15 M NaCl, 0.5% Tween-20, pH 8.0) at room temperature
for 1 h. The membrane was subjected to the indicated antibodies and
the proteins were detected by a LI-COR Odyssey Infrared Imaging
System.
Statistical analysis
All in vitro experiments were performed in
triplicate. The results were expressed as mean ± SD. The difference
between two mean values were measured by the Student's t-test
(Excel) and considered to be statistically significant at
P≤0.05.
Results
Inhibition of PI3K/mTOR suppresses cell
proliferation in aggressive B-NHL cells
The PI3K/mTOR pathway plays a central role in cancer
cell survival and proliferation. PF-04691502, which is against all
class I isoforms of PI3K and mTOR kinases, has been reported to
have antitumor activity in several types of solid cancers including
glioblastoma, breast, colorectal, nasopharyngeal, hepatocellular
and non-small cell lung carcinoma cells (30,31,34–36).
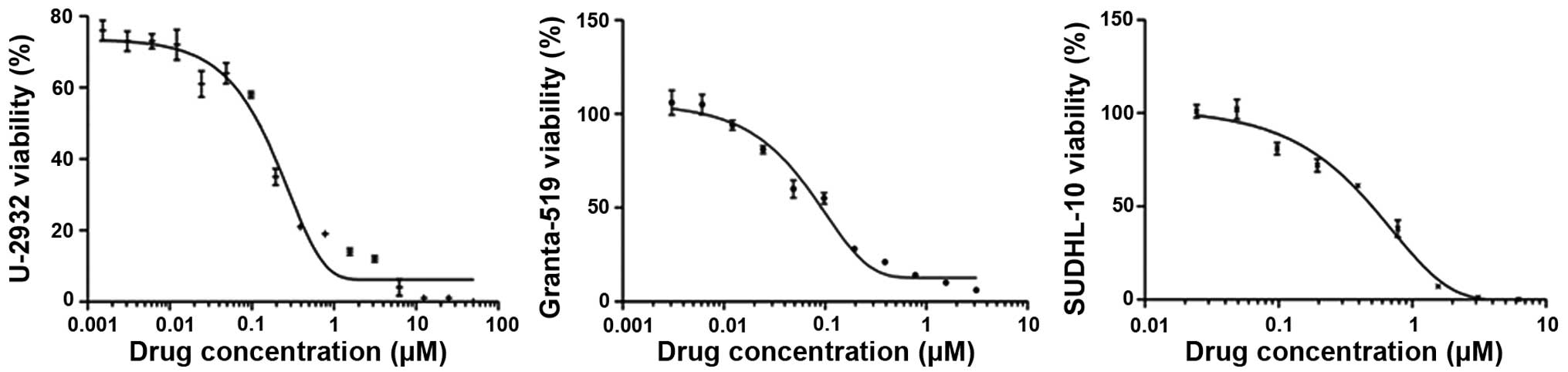
To examine whether PF-04691502 inhibits B-NHL cells, MTS assays
were performed to evaluate the growth including U-2932, SUDHL-10
and Granta-519 which represents DLBCL and MCL subtypes,
respectively. Consistent with previous studies in solid tumors,
PF-04691502 effectively inhibited the growth of these cell lines
with IC50 values ranging from 0.121 to 0.545 μM
(Fig. 1 and Table I). CAL-101, a p110δ selective PI3K
inhibitor which has been shown to inhibit multiple myeloma
(37), B-cell acute lymphoblastic
leukemia (38) and chronic
lymphocyte leukemia (39) and
INK-128, a selective mTOR inhibitor which has antitumor activity in
breast, pancreatic and colorectal cancers (40–42)
were also used in this experiment. Notably, the IC50 of
CAL-101 in these cells was higher than that of INK-128 (ranging
from 3.15 to 15.37 μM and from 0.014 to 0.170 μM, respectively)
(Table I). Together, these data
demonstrate that inhibition of PI3K/mTOR signaling effectively
suppresses cell proliferation in B-cell NHL cell lines.
 | Table IIC50 (μM) of the different
PI3K/mTOR inhibitors in B-NHL cells. |
Table I
IC50 (μM) of the different
PI3K/mTOR inhibitors in B-NHL cells.
| Cell
lines/drugs | U-2932 | Granta-519 | SUDHL-10 |
|---|
| CAL-101 | 12.58±0.62 | 3.15±0.26 | 15.37±0.73 |
| PF-04691502 | 0.175±0.064 | 0.121±0.013 | 0.545±0.089 |
| INK-128 | 0.014±0.003 | 0.022±0.006 | 0.170±0.015 |
PF-04691502 inhibits downstream signaling
of PI3K in B-cell NHL cell lines
Receptor tyrosine kinase or chronic B-cell receptor
signaling results in activation of the PI3K pathway, and
subsequently leads to trigger the downstream targets such as Akt
and S6 ribosomal protein which promote cell proliferation and
survival. In the present study, we evaluated the effect of
PF-04691502 on the activity of Akt and S6 in B-NHL cells. U-2932,
Granta-519 and SUDHL-10 cells were treated with PF-04691502 at
different concentrations for 1 and 24 h. As expected,
phosphorylation of Akt at both Ser473 and Thr308 sites was
completely decreased with PF-04691502. However, the total Akt
protein level was unchanged upon PF-04691502 treatment, indicating
that the decreased pSer473 and pThr308 were due to inhibition of
phosphorylation and not Akt protein degradation or translational
downregulation (Fig. 2A).
Phospho-S6 ribosomal protein was also reduced markedly by
PF-04691502 even at a low concentration of 0.05 μM (Fig. 2B). Hence, PF-04691502 effectively
inhibits the PI3K/Akt/mTOR pathway in B-NHL cells.
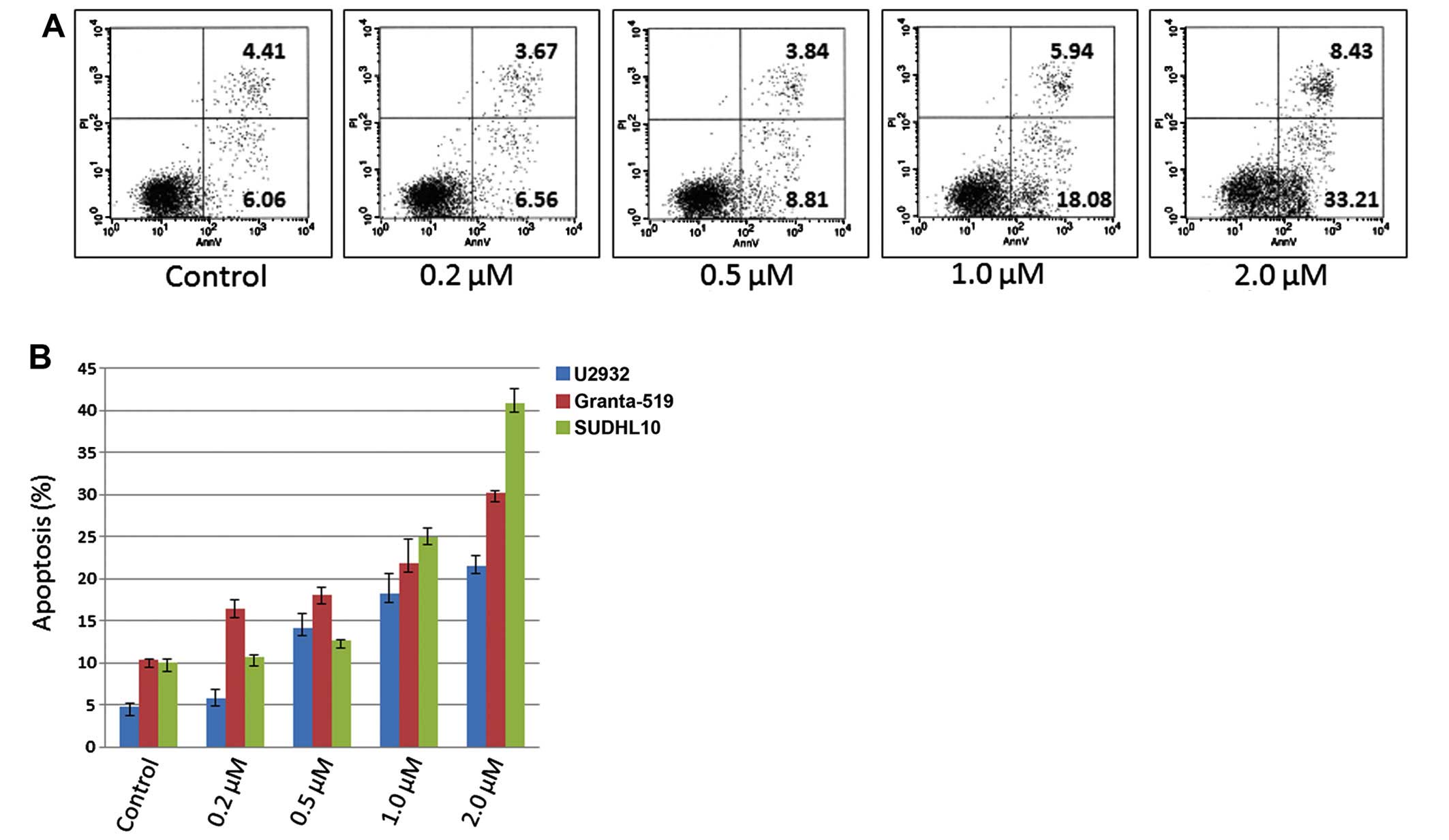
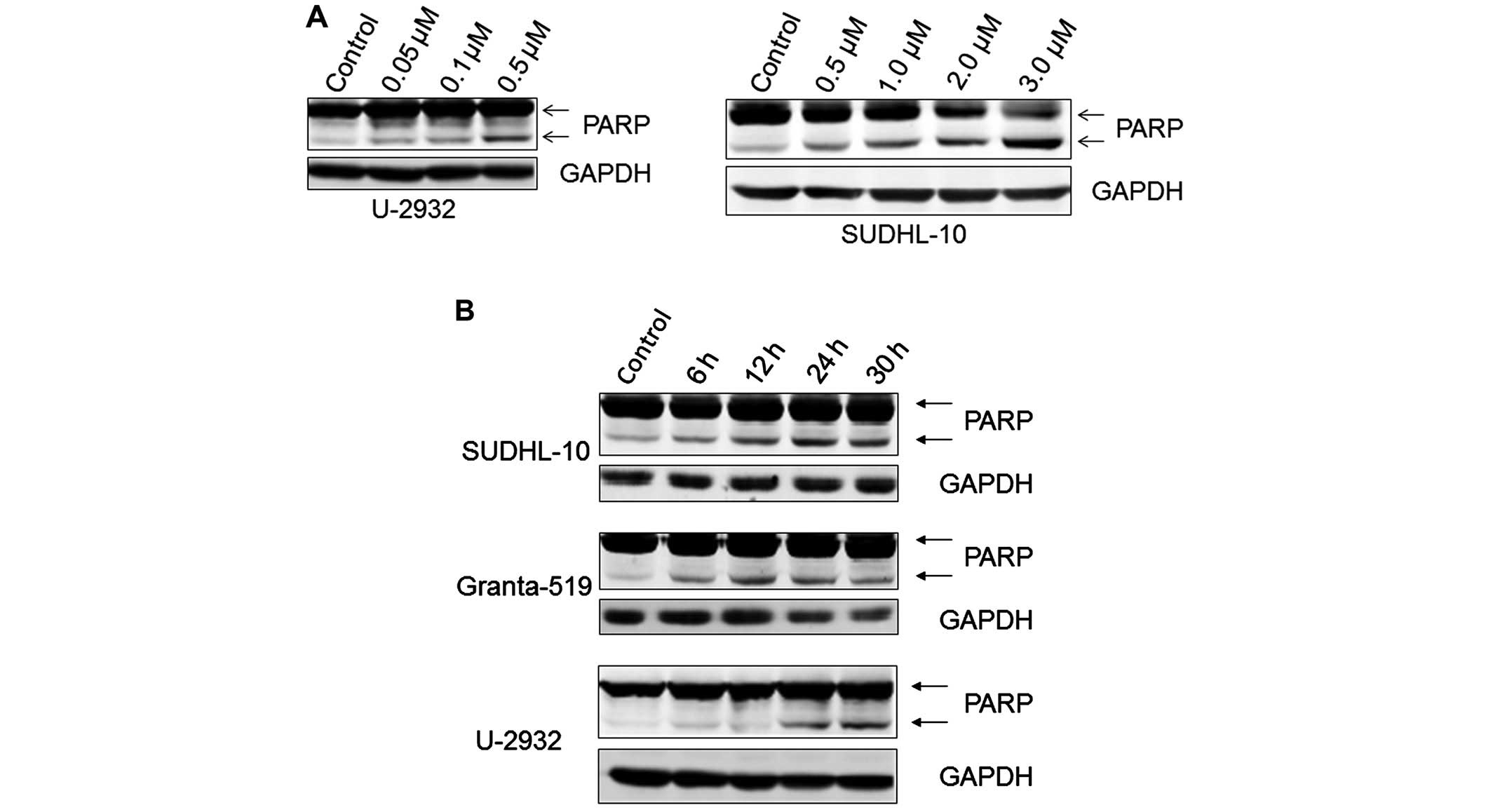
PF-04691502 induces apoptosis in B-NHL
cell lines
It is known that apoptosis is induced when
suppressing the PI3K pathway. To examine apoptosis, U-2932,
Granta-519 and SUDHL-10 cell lines were treated with PF-04691502 at
varying doses of 0.2, 0.5, 1.0 and 2.0 μM for 48 h, stained with
Annexin V and PI and evaluated by flow cytometry assays. As
expected, PF-04691502 induced apoptosis in all three cells in a
dose-dependent manner (Fig. 3A and
B). These results were confirmed by demonstrating an increased
level of cleaved PARP in treated B-NHL cells (Fig. 4A). In addition, apoptosis occurred
as early as 6 h in Granta-519 and SUDHL10 and 24 h in U-2932 cells
(Fig. 4B) indicating apoptosis
induction is in a time-dependent manner in different B-NHL
subtypes. Together, the data demonstrate that inhibition of
PI3K/mTOR by PF-04691502 leads to apoptosis in aggressive B-NHL
cells.
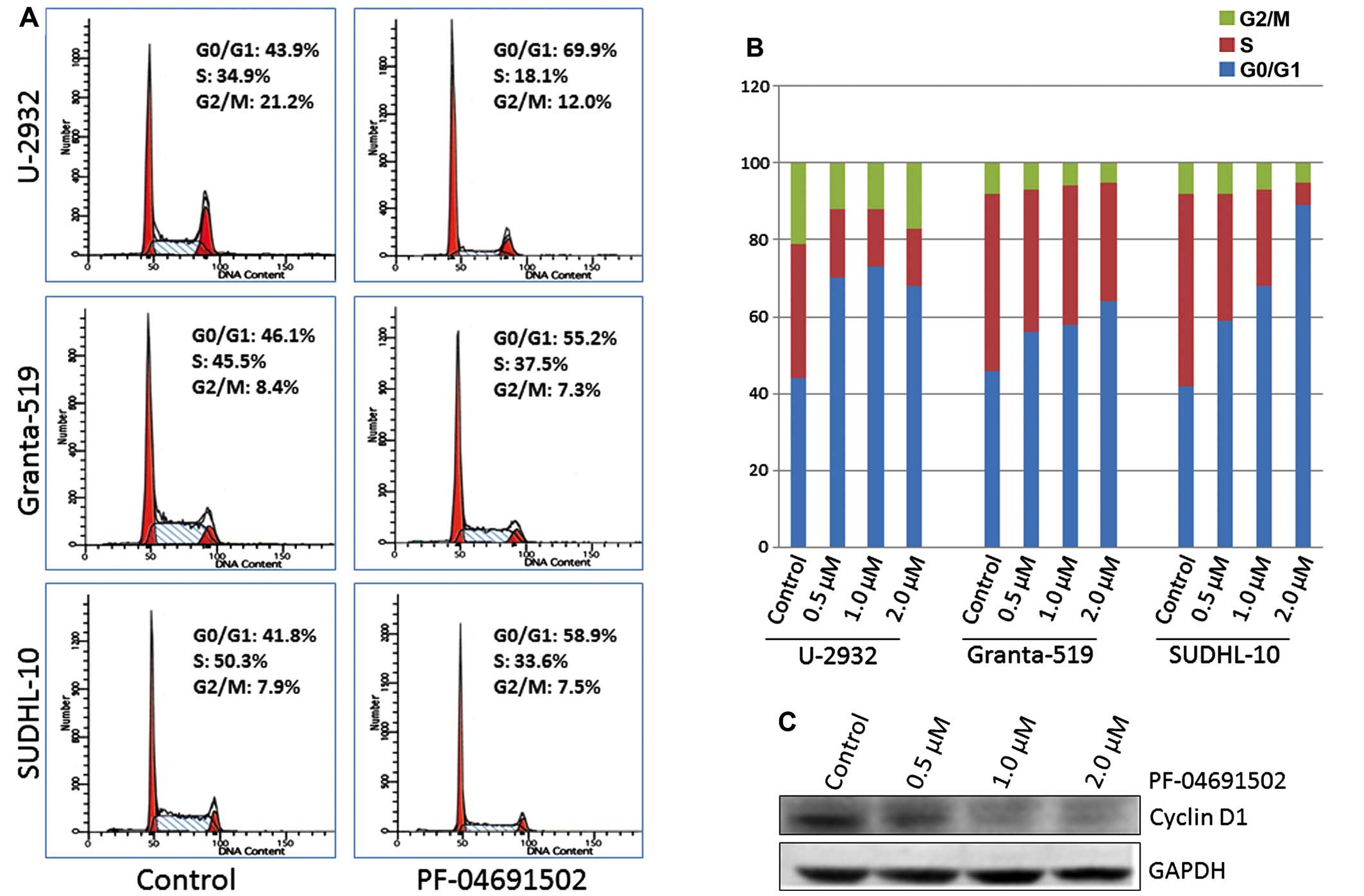
PF-04691502 inhibits cell cycle
progression by downregulating cyclin D1 in aggressive B-NHL
cells
The PI3K/Akt/mTOR pathway plays an important role in
cell cycle progression. PF-04691502 has been shown to induce G1
phase arrest in glioblastoma and hepatoma cells (30,36).
To examine cell cycle progression in B-NHL, we treated U-2932,
Granta-519 and SUDHL-10 cells with PF-04691502 at 0.5, 1.0 and 2.0
μM for 48 h and DNA content was evaluated using flow cytometry
(Fig. 5A and B). Treatment of
B-NHL cells with PF-04691502 strongly increased G0/G1 and decreased
S phase populations (Fig. 5A and
B). Since cyclin D1 is a critical player in the G1/S cell cycle
progression and is regulated by the PI3K pathway, we evaluated it
in a representative cell line Granta-519 which highly expresses
cyclin D1. Fig. 5C clearly showed
that PF-04691502 inhibited cyclin D1 at the protein level which is
consistent with the G1 arrest observed in B-NHL cells.
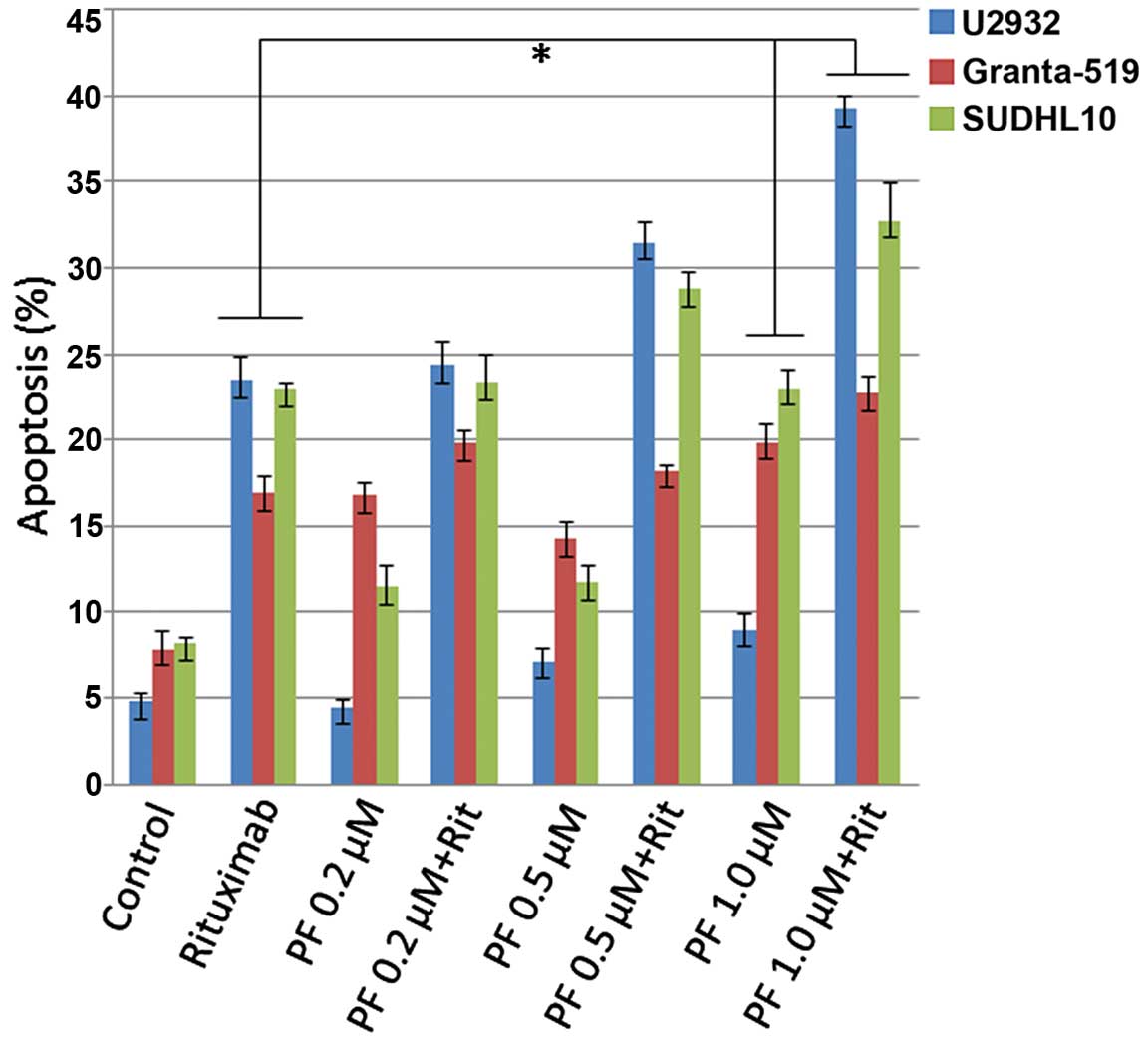
PF-04691502 in combination with rituximab
enhances apoptosis in B-NHL cells
Rituximab is often used as part of the initial
treatment or as part of a second-line regimen for treating
non-Hodgkin lymphoma patients either by itself or along with
chemotherapy. It has been reported that rituximab diminishes the
constitutive activity of the PI3K/Akt signaling pathway and
increases chemo-sensitization to drug-induced apoptosis (43). Here we treated U-2932, Granta-519
and SUDHL-10 cells with PF-04691502 at 0.2, 0.5 and 1.0 μM alone or
in combination with 10 μg/ml of rituximab for 48 h. Apoptosis was
evaluated after Annexin V and PI staining and the results clearly
showed rituximab plus PF-04691502 enhanced apoptosis significantly
(P<0.05) compared with rituximab or PF-04691502 alone (Fig. 6), suggesting rituximab increases
the antitumor activity of PI3K/mTOR inhibitor in B-cell NHL
cells.
Discussion
Most aggressive B-cell non-Hodgkin lymphomas (B-NHL)
are not curable with current chemo-immunotherapy combi- nations
(17). The PI3K/Akt/mTORC
signaling pathway is frequently dysregulated in B-NHL (44), promoting the evaluation of novel
small molecule inhibitors as an alternative treatment strategy.
Indeed, disruption of PI3K and mTOR activity is now accepted as a
therapeutic concept in hematologic malignancies and multiple agents
are currently being investigated in various stages of clinical
trials (44). In the present
study, we demonstrate that PF-04691502, a novel selective dual
PI3K/mTOR inhibitor, potently suppresses cell proliferation,
induces G1 cell cycle arrest by inhibiting cyclin D1 protein level,
and promotes apoptosis associated with suppression of Akt and S6
ribosomal protein activity in aggressive B-NHL cells including
DLBCL and MCL. PF-04691502 has been shown to have potent antitumor
activity in hepatocellular carcinoma cells (30), nasopharyngeal carcinoma (31) and colorectal cancer (34). A phase I trial evaluation of
PF-04691502 has also been done in patients with advanced solid
tumors which demonstrated that PF-04691502 was tolerable at 8 mg
orally once a day, with a safety profile similar to other PI3K/mTOR
inhibitors (35). However, no
studies have been performed to evaluate PF-04691502 activity in
aggressive B-NHL. Our results here support the inhibition of
PI3K/mTOR by PF-04691502 may represent a novel therapeutic strategy
that warrants clinical trial evaluation in aggressive B-NHL.
CAL-101, a potent and selective p110δ inhibitor, has
been extensively studied and shown to have pre-clinical and
clinical activity in lymphoid malignancies, such as CLL (44). Compared with PF-04691502, CAL-101
is less effective in suppressing cell proliferation in B-NHL
(Table I), suggesting a pan-PI3K
inhibitor is likely to be more active than an isoform selective
inhibitor for the treatment of B-NHL. The most likely explanation
is that other PI3K class I isoforms such as p110α, p110β and p110γ
exist in an activated form leading to resistance. In fact, all of
the PI3K class I isoforms are expressed and active in a variety of
aggressive B-NHL (4,44). Additionally PF-04691502 also
inhibits mTOR which is also a therapeutic target in B-NHL (44). Notably, INK-128, a potent and
selective mTOR inhibitor has more activity than PF-04691502 in
B-NHL cells (Table I). Further
preclinical and clinical studies are needed to determine whether
selective mTOR inhibitors have a therapeutic advantage over dual
PI3K/mTOR or selective PI3K inhibitors.
The PI3K/Akt/mTOR signaling has been implicated to
regulate both G1/S and G2/M transition. Inhibition of PI3K by
LY294002 or other small molecular inhibitors and mTOR by rapamycin
has been demonstrated to induce G1 cell cycle arrest in human
malignances (42,45). However, recently LY294002 has been
shown to block G2/M transition in retinal cells (46). The novel PI3K/mTOR inhibitors,
GDC-0980, GDC-0941 and PF-04691502 have been reported to induce G1
cell cycle arrest in breast, lung, glioblastoma and hepatocellular
carcinoma cells (30,36,47,48).
Similarly, our data in this study show inhibition of PI3K/mTOR by
PF-04691502 markedly blocked B-NHL cells from entering S phase
(Fig. 5A and B). Cyclin D1, is an
oncogene that regulates the G1-S cell cycle transition in B-NHL,
particularly in MCL where it is overexpressed due to the t(11;14)
(q13;q32) chromosomal translocation. Overexpression of
constitutively active Akt extends the half-life of cyclin D1
protein whereas treatment with the PI3K inhibitor accelerated its
degradation (45,49). Consistent with these findings,
PF-04691502 inhibits Akt and decreases cyclin D1 protein in B-NHL
(Fig. 5C).
Rituximab is a chimeric monoclonal antibody against
the protein CD20 and is often used for treating leukemias and
lymphomas either by itself or along with chemotherapy. Rituximab
has been demonstrated to inhibit the constitutively activated Akt
pathway in B-NHL cell lines, and this inhibition contributes to
sensitization of drug-resistant cells to apoptosis by
chemotherapeutic drugs (43). In
this study, we found combination of PF-04691502 and rituximab
significantly increases apoptosis in all tested B-NHL cell lines
compared to a single agent treatments (Fig. 6), implying this combination may
have better efficacy to treat lymphoma patients. In fact, several
PI3K inhibitors including Idelalisib and XL765 (SAR245409) plus
rituximab are entering clinical trials in NHL or CLL (44). Results from these trials are
eagerly awaited.
In conclusion, our findings indicate that inhibition
of the PI3K/mTOR pathway is an excellent therapeutic strategy for
aggressive B-NHL. PF-04691502, a dual PI3K/mTOR inhibitor had
strong activity in reducing tumor cell proliferation, enhancing
apoptosis and induction of G1 cell cycle arrest in aggressive
B-NHL. Inhibition of PI3K/mTOR pathway by PF-04691502 was confirmed
by decreased phosphorylation of Akt and S6 ribosomal protein.
Finally, combining PF-04691502 with rituximab increased apoptosis
and may be a synthetic lethal interaction. These results suggest
that PF-04691502 with and without rituximab should be examined
further as a potential therapeutic strategy for patients with
aggressive B-cell NHL.
Acknowledgements
We wish to thank the Health Administration of the
Jiangsu Province for funding the present project.
References
|
1
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vanhaesebroeck B, Stephens L and Hawkins
P: PI3K signalling: The path to discovery and understanding. Nat
Rev Mol Cell Biol. 13:195–203. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blachly JS and Baiocchi RA: Targeting
PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br J Haematol.
167:19–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Foster FM, Traer CJ, Abraham SM and Fry
MJ: The phosphoinositide (PI) 3-kinase family. J Cell Sci.
116:3037–3040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen HC and Guan JL: Association of focal
adhesion kinase with its potential substrate phosphatidylinositol
3-kinase. Proc Natl Acad Sci USA. 91:10148–10152. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Izuhara K, Feldman RA, Greer P and Harada
N: Interleukin-4 induces association of the c-fes proto-oncogene
product with phosphatidylinositol-3 kinase. Blood. 88:3910–3918.
1996.PubMed/NCBI
|
|
8
|
Leslie NR and Downes CP: PTEN: The down
side of PI 3-kinase signalling. Cell Signal. 14:285–295. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang S, Denley A, Vanhaesebroeck B and
Vogt PK: Oncogenic transformation induced by the p110beta, -gamma,
and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl
Acad Sci USA. 103:1289–1294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang HW, Aoki M, Fruman D, Auger KR,
Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM and Vogt PK:
Transformation of chicken cells by the gene encoding the catalytic
subunit of PI 3-kinase. Science. 276:1848–1850. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang BH, Zheng JZ, Aoki M and Vogt PK:
Phosphatidylinositol 3-kinase signaling mediates angiogenesis and
expression of vascular endothelial growth factor in endothelial
cells. Proc Natl Acad Sci USA. 97:1749–1753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: A target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodon J, Dienstmann R, Serra V and
Tabernero J: Development of PI3K inhibitors: Lessons learned from
early clinical trials. Nat Rev Clin Oncol. 10:143–153. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mahadevan D and Fisher RI: Novel
therapeutics for aggressive non-Hodgkin's lymphoma. J Clin Oncol.
29:1876–1884. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fang X, Zhou X and Wang X: Clinical
development of phosphatidylinositol 3-kinase inhibitors for
non-Hodgkin lymphoma. Biomark Res. 1:302013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Psyrri A, Papageorgiou S, Liakata E,
Scorilas A, Rontogianni D, Kontos CK, Argyriou P, Pectasides D,
Harhalakis N, Pappa V, et al: Phosphatidylinositol 3′-kinase
catalytic subunit alpha gene amplification contributes to the
pathogenesis of mantle cell lymphoma. Clin Cancer Res.
15:5724–5732. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pfeifer M, Grau M, Lenze D, Wenzel SS,
Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et
al: PTEN loss defines a PI3K/AKT pathway-dependent germinal center
subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA.
110:12420–12425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baohua Y, Xiaoyan Z, Tiecheng Z, Tao Q and
Daren S: Mutations of the PIK3CA gene in diffuse large B cell
lymphoma. Diagn Mol Pathol. 17:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao MY, Auerbach A, D'Costa AM, Rapoport
AP, Burger AM, Sausville EA, Stass SA, Jiang F, Sands AM, Aguilera
N, et al: Phospho-p70S6K/p85S6K and cdc2/cdk1 are novel targets for
diffuse large B-cell lymphoma combination therapy. Clin Cancer Res.
15:1708–1720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi MY and Kipps TJ: Inhibitors of B-cell
receptor signaling for patients with B-cell malignancies. Cancer J.
18:404–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Inabe K and Kurosaki T: Tyrosine
phosphorylation of B-cell adaptor for phosphoinositide 3-kinase is
required for Akt activation in response to CD19 engagement. Blood.
99:584–589. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gupta M, Hendrickson AE, Yun SS, Han JJ,
Schneider PA, Koh BD, Stenson MJ, Wellik LE, Shing JC, Peterson KL,
et al: Dual mTORC1/mTORC2 inhibition diminishes Akt activation and
induces Puma-dependent apoptosis in lymphoid malignancies. Blood.
119:476–487. 2012. View Article : Google Scholar :
|
|
26
|
Bhende PM, Park SI, Lim MS, Dittmer DP and
Damania B: The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious
against follicular lymphoma. Leukemia. 24:1781–1784. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uddin S, Hussain AR, Siraj AK, Manogaran
PS, Al-Jomah NA, Moorji A, Atizado V, Al-Dayel F, Belgaumi A,
El-Solh H, et al: Role of phosphatidylinositol 3′-kinase/AKT
pathway in diffuse large B-cell lymphoma survival. Blood.
108:4178–4186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mahadevan D, Chiorean EG, Harris WB, Von
Hoff DD, Stejskal-Barnett A, Qi W, Anthony SP, Younger AE, Rensvold
DM, Cordova F, et al: Phase I pharmacokinetic and pharmacodynamic
study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in
patients with advanced solid tumours and B-cell malignancies. Eur J
Cancer. 48:3319–3327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng H, Li C, Bailey S, Baxi SM, Goulet
L, Guo L, Hoffman J, Jiang Y, Johnson TO, Johnson TW, et al:
Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04979064
through structure-based drug design. ACS Med Chem Lett. 4:91–97.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang FZ, Peng-Jiao, Yang NN, Chuang-Yuan,
Zhao YL, Liu QQ, Fei HR and Zhang JG: PF-04691502 triggers cell
cycle arrest, apoptosis and inhibits the angiogenesis in
hepatocellular carcinoma cells. Toxicol Lett. 220:150–156. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wong CH, Loong HH, Hui CW, Lau CP, Hui EP,
Ma BB and Chan AT: Preclinical evaluation of the PI3K-mTOR dual
inhibitor PF-04691502 as a novel therapeutic drug in nasopharyngeal
carcinoma. Invest New Drugs. 31:1399–1408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Devereaux K, Dall'Armi C, Alcazar-Roman A,
Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P and Di Paolo
G: Regulation of mammalian autophagy by class II and III PI
3-kinases through PI3P synthesis. PLoS One. 8:e764052013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carver BS, Chapinski C, Wongvipat J,
Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J,
Scher H, et al: Reciprocal feedback regulation of PI3K and androgen
receptor signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fang DD, Zhang CC, Gu Y, Jani JP, Cao J,
Tsaparikos K, Yuan J, Thiel M, Jackson-Fisher A, Zong Q, et al:
Antitumor efficacy of the dual PI3K/mTOR inhibitor PF-04691502 in a
human xenograft tumor model derived from colorectal cancer stem
cells harboring a PIK3CA mutation. PLoS One. 8:e672582013.
View Article : Google Scholar :
|
|
35
|
Britten CD, Adjei AA, Millham R, Houk BE,
Borzillo G, Pierce K, Wainberg ZA and LoRusso PM: Phase I study of
PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and
mTOR, in patients with advanced cancer. Invest New Drugs.
32:510–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan J, Mehta PP, Yin MJ, Sun S, Zou A,
Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, et al:
PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR
kinases with antitumor activity. Mol Cancer Ther. 10:2189–2199.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ikeda H, Hideshima T, Fulciniti M, Perrone
G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, et
al: PI3K/p110{delta} is a novel therapeutic target in multiple
myeloma. Blood. 116:1460–1468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lannutti BJ, Meadows SA, Herman SE,
Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM,
Deininger M, et al: CAL-101, a p110delta selective
phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell
malignancies, inhibits PI3K signaling and cellular viability.
Blood. 117:591–594. 2011. View Article : Google Scholar
|
|
39
|
Herman SE, Gordon AL, Wagner AJ, Heerema
NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ,
et al: Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows
promising preclinical activity in chronic lymphocytic leukemia by
antagonizing intrinsic and extrinsic cellular survival signals.
Blood. 116:2078–2088. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
García-García C, Ibrahim YH, Serra V,
Calvo MT, Guzmán M, Grueso J, Aura C, Pérez J, Jessen K, Liu Y, et
al: Dual mTORC1/2 and HER2 blockade results in antitumor activity
in preclinical models of breast cancer resistant to anti-HER2
therapy. Clin Cancer Res. 18:2603–2612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lou HZ, Weng XC, Pan HM, Pan Q, Sun P, Liu
LL and Chen B: The novel mTORC1/2 dual inhibitor INK-128 suppresses
survival and proliferation of primary and transformed human
pancreatic cancer cells. Biochem Biophys Res Commun. 450:973–978.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kampa-Schittenhelm KM, Heinrich MC, Akmut
F, Rasp KH, Illing B, Döhner H, Döhner K and Schittenhelm MM: Cell
cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor
NVP-BGT226 in acute leukemia. Mol Cancer. 12:462013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suzuki E, Umezawa K and Bonavida B:
Rituximab inhibits the constitutively activated PI3K-Akt pathway in
B-NHL cell lines: Involvement in chemosensitization to drug-induced
apoptosis. Oncogene. 26:6184–6193. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jabbour E, Ottmann OG, Deininger M and
Hochhaus A: Targeting the phosphoinositide 3-kinase pathway in
hematologic malignancies. Haematologica. 99:7–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gao N, Zhang Z, Jiang BH and Shi X: Role
of PI3K/AKT/mTOR signaling in the cell cycle progression of human
prostate cancer. Biochem Biophys Res Commun. 310:1124–1132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ornelas IM, Silva TM, Fragel-Madeira L and
Ventura AL: Inhibition of PI3K/Akt pathway impairs G2/M transition
of cell cycle in late developing progenitors of the avian embryo
retina. PLoS One. 8:e535172013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wallin JJ, Edgar KA, Guan J, Berry M,
Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, et al:
GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust
activity in cancer models driven by the PI3K pathway. Mol Cancer
Ther. 10:2426–2436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zou ZQ, Zhang LN, Wang F, Bellenger J,
Shen YZ and Zhang XH: The novel dual PI3K/mTOR inhibitor GDC-0941
synergizes with the MEK inhibitor U0126 in non-small cell lung
cancer cells. Mol Med Rep. 5:503–508. 2012.
|
|
49
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|