Introduction
Splenic hemangiomas, although infrequent, represent
the most common benign neoplasms of the spleen with an incidence of
0.03–14% found in large autopsy series (1,2). In
the vast majority of cases, splenic hemangiomas are incidental
surgical, radiologic, or autopsy findings, made during evaluation
for other disorders (1–3). Arber et al (3) reported 7 localized splenic
hemangiomas all of which were discovered incidentally in surgical
patients. In a large series of 32 patients with splenic hemangioma,
only 6 presented with abdominal symptoms and only 4 had a palpable
spleen (1).
Because of the general absence of presenting
symptoms and the typically incidental nature of splenic
hemangiomas, the age at presentation varies considerably. Several
non-autopsy, surgical series revealed an average age at
detection/presentation of between 51 and 63 years (1,3).
However, examination of autopsy data reveals a much younger average
patient age (4) indicating that
these benign lesions are likely to be present but remain undetected
for long periods of time. No gender or race predilection has been
reported (1).
Splenic hemangiomas are thought to be congenital in
origin, arising from sinusoidal epithelium (5). Whether they are neoplastic or
represent some other type of misgrowth, remains uncertain. They
typically appear as circumscribed, non-encapsulated,
honeycomb-like, red-purple masses that frequently blend
imperceptibly into the surrounding splenic parenchyma (4). The spaces are composed of sponge-like
tissue filled with blood and separated by fibrous septa. Occasional
calcification may be seen, often in association with an organized
infarct (6). Microscopically, the
majority of hemangiomas are cavernous in nature, consisting of
large interconnected, dilated, blood-filled spaces lined by a
monolayer of cytologically bland endothelial cells separated by
thin fibrous septa or splenic pulp tissue. Pure capillary
architecture is less common. Instead, many lesions contain varying
proportions of both cavernous and capillary components (4). Immunophenotypically, splenic
hemangiomas show reactivity of endothelial lining cells for CD31,
von Willebrand factor, Ulex europeaus, lectin I, and CD34. This
pattern raises the possibility that splenic hemangioma may derive
from a combination of splenic venous structures as well as from
splenic sinusoidal cells (4).
Most splenic hemangiomas tend to be small in size
(<4 cm) although lesions ≤36 cm have been reported (4). They need not be entirely without
complications as larger lesions may rupture with resulting
intra-abdominal hemorrhage (7–11).
In some patients, they cause the Kasabach-Meritt syndrome (12).
The etiology and pathogenesis of splenic hemangiomas
are unclear and no cytogenetic or molecular genetic information
about the disease has been published. We here describe the
cytogenetic analysis of a splenic hemangioma and the fusion gene
corresponding to the chromosomal translocation thus found.
Materials and methods
Ethical approval
The study was approved by the Regional Committee for
Medical and Health Research Ethics, SouthEast Norway (REK Sør)
http://helseforskning.etikkom.no).
Written informed consent was obtained from the patient. The consent
included acceptance that the clinical details be published. The
ethics committee's approval included a review of the consent
procedure. All patient information has been anonymized and
de-identified.
Patient
A twenty-nine-year-old woman was incidentally
diagnosed with a splenic hemangioma during an ultrasound
examination for cholecystitits. She had been without symptoms
attributable to the splenic lesion, possibly except some pressure
in the upper left abdomen. Because of continuous growth of the
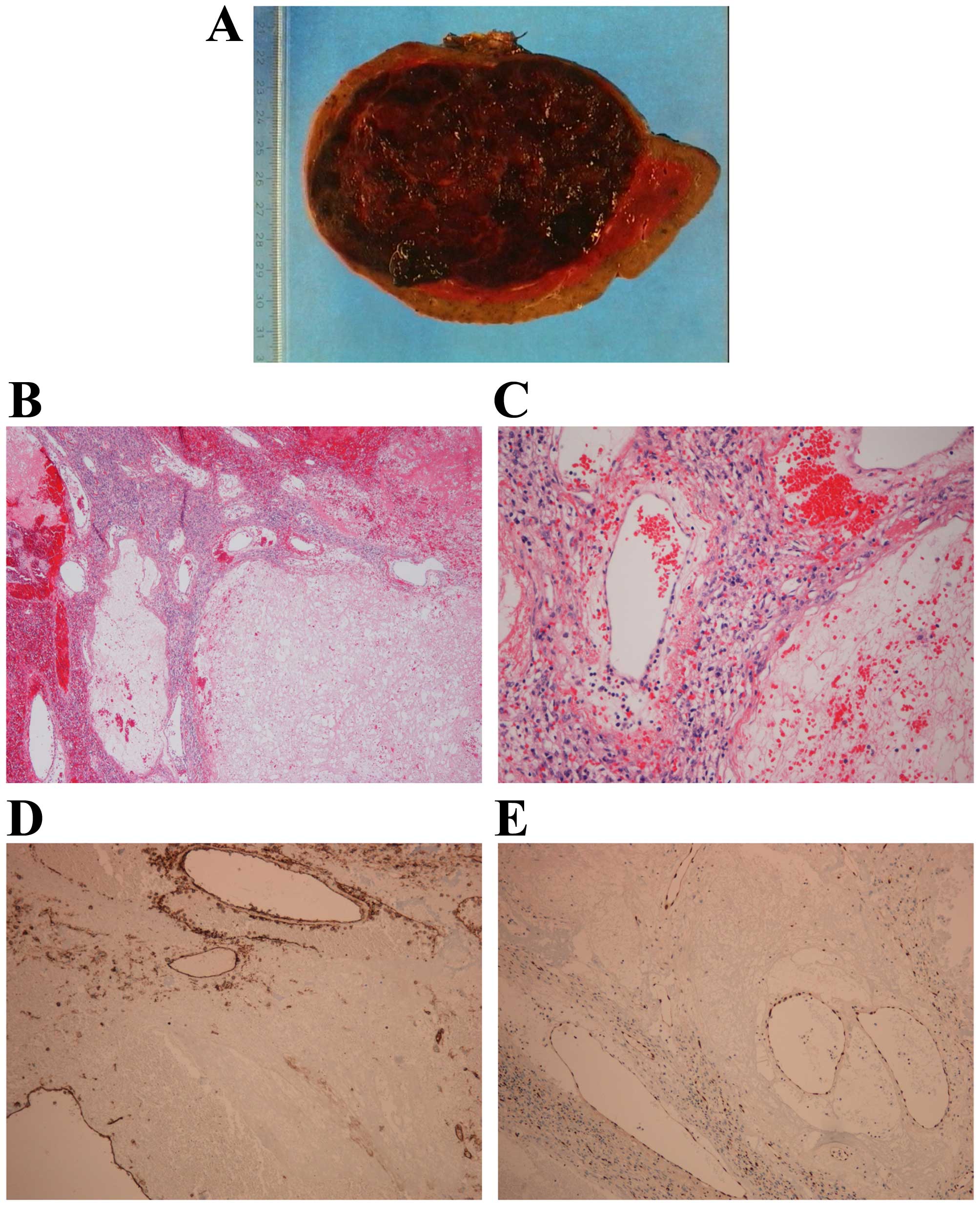
hemangioma, it was decided to do a splenectomy. Histological
examination (Fig. 1) showed that
the lesion was composed of large, blood-filled vessels lined by
flat endothelium and separated by thin fibrous septa or splenic
pulpa. Immunohistochemical analysis showed positivity for CD31 and
ERG.
Control sample
The control sample was FirstChoice human spleen
total RNA (Life Tehnologies, Carlsbad, CA, USA).
G-banding and karyotyping
Fresh tissue from a representative area of the tumor
was received and analyzed cytogenetically as part of our diagnostic
routine. The sample was disaggregated mechanically and
enzymatically with collagenase II (Worthington, Freehold, NJ, USA).
The resulting cells were cultured and harvested using standard
techniques. Chromosome preparations were G-banded with Wright stain
and examined. Peripheral blood lymphocytes stimulated with
phytohemagglutinin (PHA) for 72 h were also karyotyped. The
karyotypes were written according to the International System for
Human Cytogenetic Nomenclature (ISCN) 2009 guidelines (13).
RNA and DNA extraction
Tumor tissue adjacent to that used for cytogenetic
analysis and histologic examination had been frozen and stored at
−80°C. Total RNA was extracted using miRNeasy Mini kit according to
the manufacturer's instructions (Qiagen Nordic, Oslo, Norway).
Tumor tissue was disrupted and homogenized in Qiazol Lysis Reagent
(Qiagen) using a 5-mm stainless steel bead and TissueLyser II
(Qiagen). Subsequently, total RNA was purified using QIAcube
(Qiagen). The RNA quality was evaluated using the Experion
Automated Electrophoresis system (Bio-Rad Laboratories, Oslo,
Norway). The RNA quality indicator (RQI) was 9.0. Genomic DNA was
extracted using the Maxwell 16 Instrument System and the Maxwell 16
Tissue DNA Purification kit (Promega, Madison, WI, USA), and the
concentration and purity of DNA were measured using NanoVue Plus
Spectrophotometer (GE Healthcare Life Sciences, Oslo, Norway).
High-throughput paired-end
RNA-sequencing
Three micrograms of total RNA were sent for
high-throughput paired-end RNA-sequencing at the Norwegian
Sequencing Centre, Ullevål Hospital (http://www.sequencing.uio.no/). The RNA was sequenced
using an Illumina HiSeq 2000 instrument and the Illumina software
pipeline was used to process image data into raw sequencing data.
The regular TruSeq library preparation protocol (http://support.illumina.com/downloads/truseq_rna_sample_preparation_guide_15008136.ilmn)
was used. The reads obtained had a length of 100 base pairs (bp). A
total of 74 million reads were obtained. The quality of the raw
sequence data was assessed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
The software TopHat-Fusion was used for the discovery of fusion
transcripts (14,15). To verify further the fusion gene
which was found by TopHat-Fusion, the ‘grep’ command (http://en.wikipedia.org/wiki/Grep) was used to
search the fastq files of sequence data (http://en.wikipedia.org/wiki/FASTQ_format). Our
‘specific expression’ was a sequence of 20 nucleotides at the
fusion point, 10 bases upstream (5′-end gene), and 10 bases
downstream from the junction (3′-end gene). The expression was
‘CGACCAATAGGTCCCCAAGT’. The sequences obtained by ‘grep’ were
blasted against the human genomic plus transcript database
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) as well as
the sequences with accession numbers NM_024665.4 (TBL1XR1)
and NM_002131.3 (HMGA1) and DB051170.1 (TESTI2 Homo
sapiens cDNA clone TESTI2040990, 5′, mRNA sequence).
RT-PCR and genomic PCR analyses
The primers used for PCR amplification and Sanger
sequencing are listed in Table I.
For RT-PCR, 1 μg of total RNA was reverse-transcribed in a 20-μl
reaction volume using iScript Advanced cDNA Synthesis kit for
RT-qPCR according to the manufacturer's instructions (Bio-Rad
Laboratories). The 25 μl PCR volume contained 12.5 μl Premix Ex
Taq™ DNA Polymerase Hot Start Version (Takara Bio Europe/SAS,
Saint-Germain-en-Laye, France), 1 μl of cDNA, and 0.4 μM of each of
the forward and reverse primer. The primer sets
TBL1XR1-229F1/DB051170-Intr2R1 and TBL1XR1-229F1/HMGA1-324R1 were
used to detect possible TBL1XR1-HMGA1 fusion transcripts.
The quality of the cDNA synthesis was examined by amplification of
a cDNA fragment of the ABL1 gene using the primers ABL1-91F1
and ABL1-404R1 (16).
 | Table IPrimers used for PCR amplification
and Sanger sequencing analyses. |
Table I
Primers used for PCR amplification
and Sanger sequencing analyses.
| Name | Sequence
(5′→3′) | Direction in
PCR | Position on Human
Feb. 2009 (GRCh37/hg19) Assembly |
|---|
| TBL1XR1-229F1 |
TGTTGTGACCTCATGGTTTAAGTGG | Forward |
chr3:176,782,774-176,782,798 |
|
TBL1XR1-intron4-F1 |
AAGCAGTCATTTCCAGTTGCTGC | Forward |
chr3:176,769,680-176,769,702 |
| HMGA1-324R1 |
GGACTTCGAGCTCGACTCACTCA | Reverse |
chr6:34,208,559-34,208,581 |
|
DB051170-Intr2R1 |
GTACACCCAAGGGAGGCTTCATAC | Reverse |
chr6:34,203,951-34,203,974 |
|
DB051170-Intr2R2 |
TGATCAAGGTGGAGCCTTCCAGC | Reverse |
chr6:34,204,001-34,204,023 |
| ABL1-91F1 |
CAGCGGCCAGTAGCATCTGACTTTG | Forward |
chr9:133,729,459-133,729,483 |
| ABL1-404R1 |
CTCAGCAGATACTCAGCGGCATTGC | Reverse |
chr9:133,730,335-133,730,359 |
For genomic PCR, the 25 μl PCR volume contained 12.5
μl Premix Ex Taq™ DNA Polymerase Hot Start Version, 100 ng DNA, and
0.4 μM of each of the forward and reverse primers
TBL1XR1-intron4-F1 and DB051170-intr2R1.
The PCR amplifications were run on a C-1000 Thermal
cycler (Bio-Rad Laboratories) with an initial denaturation at 94°C
for 30 sec, followed by 35 cycles of 7 sec at 98°C, 30 sec at 55°C
(58°C for genomic PCR), 1 min at 72°C, and a final extension for 5
min at 72°C. For amplification of the ABL1 cDNA fragment, the PCR
cycling was an initial denaturation at 94°C for 30 sec followed by
35 cycles of 7 sec at 98°C and 2 min at 68°C, and a final extension
for 5 min at 68°C. Three microliters of the PCR products were
stained with GelRed (Biotium, Hayward, CA, USA), analysed by
electrophoresis through 1.0% agarose gel, and photographed. The
remaining 22 μl PCR products were purified using the MinElute PCR
purification kit (Qiagen Nordic) and sequenced at GATC Biotech
(Germany, http://www.gatc-biotech.com/en/home.html). The BLAST
software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for
computer analysis of sequence data.
Results
G-banding
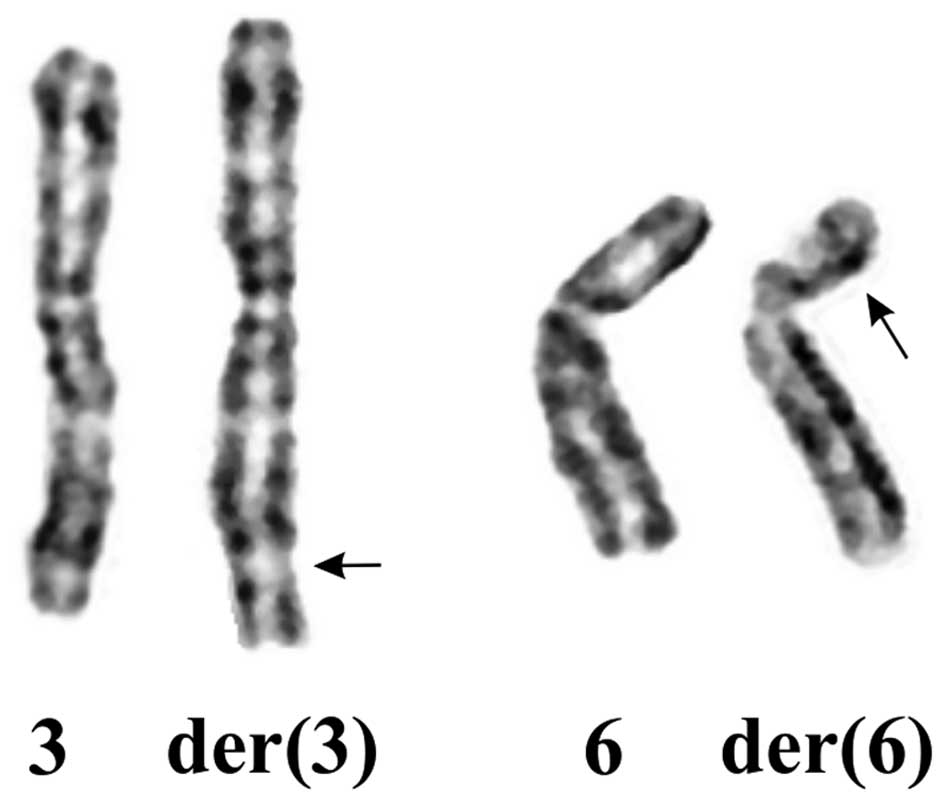
The G-banding analysis of the splenic hemangioma
yielded a karyotype with a single clonal chromosome abnormality:
45~47,XX,t(3;6)(q26;p21)[cp22] (Fig.
2). The G-banding analysis of PHA-stimulated peripheral blood
cells yielded a normal 46,XX karyotype. Thus, the translocation
t(3;6)(q26;p21) was found in cells of the splenic hemangioma
only.
High-throughput paired-end RNA-sequencing
analysis
Using the TopHat-Fusion on the raw sequencing data
obtained by Norwegian Sequencing Centre, four fusions were found
between chromosome bands 3q26 and 6p21 (Table II). Among them was a fusion
between TBL1XR1 (from 3q26) and the sequence with accession
number DB051170 which is an alternative splicing transcript of
HMGA1 found in testis (17).
| Table IIFusions between chromosome bands 3q26
and 6p21 which were found with the TopHat-Fusion program.a |
Table II
Fusions between chromosome bands 3q26
and 6p21 which were found with the TopHat-Fusion program.a
| 3q26-fusion
point | 6p21-fusion
point | Fifty bases on the
left side of the fusion (3p21) | Fifty bases on the
right side of the fusion (6p21) |
|---|
| 166506603 | 30593594 |
GGGATTACAACTCCTCCTGGCATTA
CAGGAGTGAGCCACCATGCCTGGCC |
TCTCTTCCCAGTCTACAGCTCCTACAGAGCACTCTGAGGGCCTGTCTGCC |
|
176769483 |
34203899 |
CACAATTTTCCAAATCCAGGATGG
TACCTTGTTTGATGGTCGACCAATAG |
GTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAA |
| 176799652 | 35441544 |
TCTTTAGTCAGTATAATTACAGT
ATTGTATATATAATATACAATATAAAA |
CCTAGGTGGGGTTTGGAGGTGGCCTGAGCGATATGCAAACAGTGAGGACC |
| 161838267 | 39157355 |
TTGGGTTTGAGACTATAGCTAAA
ATATAATAAAACAAAAAATAACAAATA |
CATTCAGGACACAGTGGGCCAGCCCCAGGGTGAACATCATGGAGAACCCA |
In order to verify this fusion, we used the ‘grep’
command utility to search for expressions composed of 10 nt of
TBL1XR1 and 10 nt of HMGA1 upstream and downstream of
the fusion point (Table III).
Using the expression ‘CGACCAATAGGTCCCCAAGT’, which is composed of
10 nt, ‘CGACCAATAG’, from TBL1XR1 and 10 nt, ‘GTCCCCAAGT’,
from HMGA1, 16 sequences were retrieved. BLAT of these
sequences on the human genome browser-hg19 assembly (http://genome-euro.ucsc.edu/cgi-bin/hgGateway) showed
that they were chimeric cDNA fragments composed of nucleotides
which mapped on 3q26 in the coding region of TBL1XR1 and
nucleotides mapped on chromosome band 6p21, circa 700 bp upstream
of the HMGA1 sequence with accession number NM_002131 and
within the sequence with accession number DB051170 which is an
alternative splicing transcript of HMGA1 found in testis
(17). The fusion had occurred
between nt 496 of TBL1XR1 mRNA reference sequence
NM_024665.4 and nt 215 of the sequence with accession number
DB051170.1 (Table III). We
therefore decided to investigate the tumor further for the presence
of the TBL1XR1-HMGA1 fusion transcript using molecular
techniques. No other fusions were examined.
| Table IIISequences retrieved with the ‘grep’
command using the expression ‘CGACCAATAGGTCCCCAAGT’.a |
Table III
Sequences retrieved with the ‘grep’
command using the expression ‘CGACCAATAGGTCCCCAAGT’.a
| 1 |
CTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTATGAAGCCTCCCTTGGGTGTACCTG |
| 2 |
ATTTTCCAAATCCAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTAT |
| 3 |
CTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTATGAAGCCTCCCTTGGGTGTACCTG |
| 4 |
GGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTATGAAGCCTCCCTTGGGTGT |
| 5 |
AGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCAC |
| 6 |
ATTTTCCAAATCCAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTAT |
| 7 |
GTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTA |
| 8 |
CAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATC |
| 9 |
CTATCATCCAGAAAGGTCTACAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTG |
| 10 |
CTATCATCCAGAAAGGTCTACAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTG |
| 11 |
CTATCATCCAGAAAGGTCTACAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTG |
| 12 |
TTCTATCATCCAGAAAGGTCTACAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCC |
| 13 |
TTTATTTCTATCATCCAGAAAGGTCTACAGTATGTAGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGT |
| 14 |
AGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCAC |
| 15 |
AGAAGCAGAAGTTAGTATTAATGAGGATGGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCAC |
| 16 |
GGTACCTTGTTTGATGGTCGACCAATAGGTCCCCAAGTGGGCCTGCGTATCTCCAGAACACCATCTAAGTCACCTCAAGGTATGAAGCCTCCCTTGGGTGT |
Molecular genetic confirmation of the
TBL1XR1-HMGA1 fusion
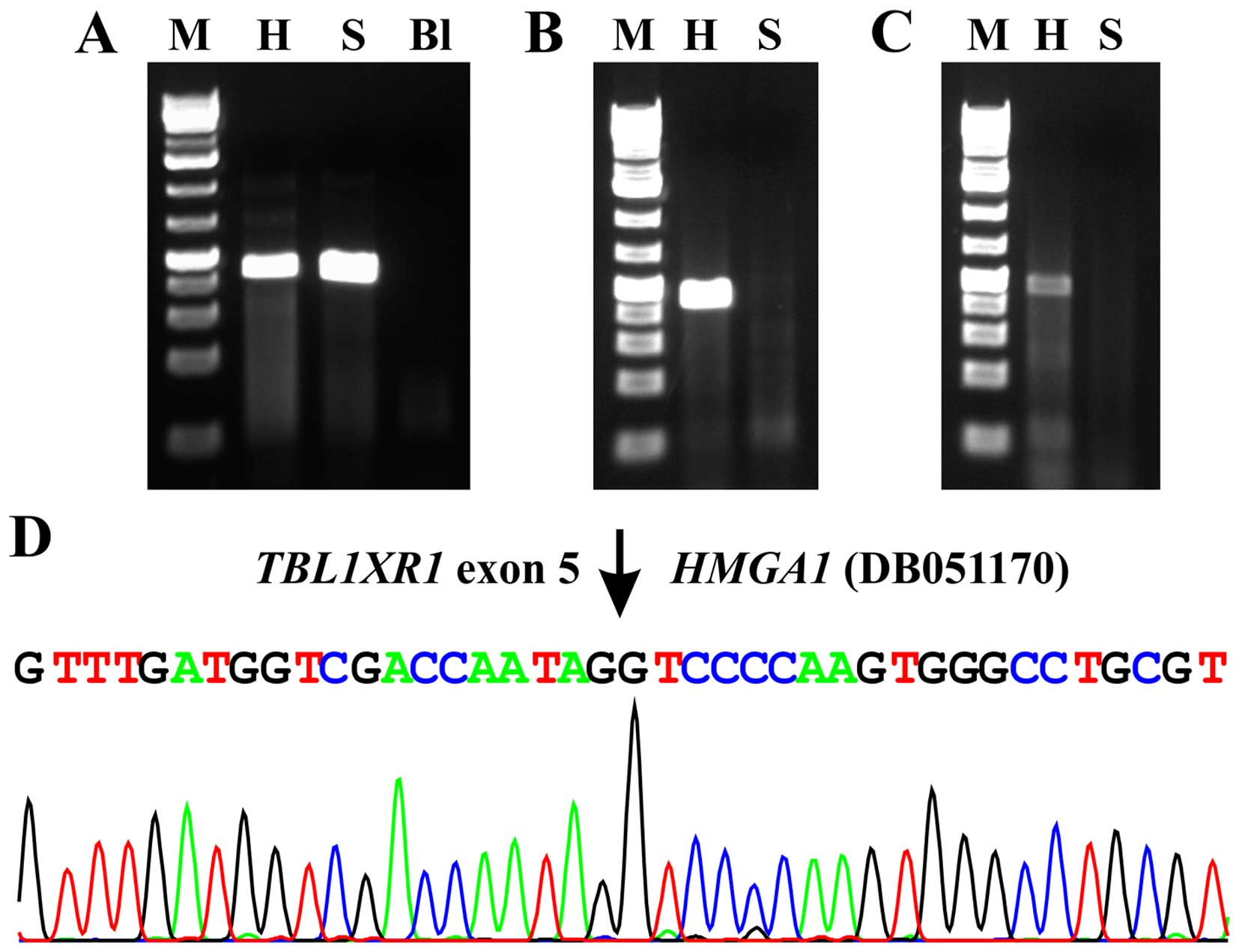
A 338-bp ABL1 cDNA fragment was amplified
indicating the good quality of the synthesized cDNA (Fig. 3A).
To verify the data obtained with the
RNA-sequencing/TopHat-Fusion software and ‘grep’ command, PCR
amplifications were performed using forward TBL1XR1 and
reverse HMGA1 primers corresponding to sequences located
upstream and downstream of the putative breakpoint, respectively.
PCR with the two primer combinations, TBL1XR1-229F1/DB01170-intr2R1
and TBL1XR1-229F1 and HMGA1-324R1, amplified fragments from the
cDNA of splenic hemangioma but not from cDNA of the normal spleen.
The results strongly suggested the presence of TBL1XR1-HMGA1
chimeric transcript in splenic hemangioma (Fig. 3A). Direct Sanger sequencing of the
amplified fragments showed that both were TBL1XR1-HMGA1
chimeric cDNA fragments with the fusion point identical to that
found with TopHat-Fusion and the ‘grep’ command, i.e., the fusion
had occurred between nt 496 of TBL1XR1 mRNA reference
sequence NM_024665.4 and nt 215 of the sequence with accession
number DB051170.1 (Fig. 3B).
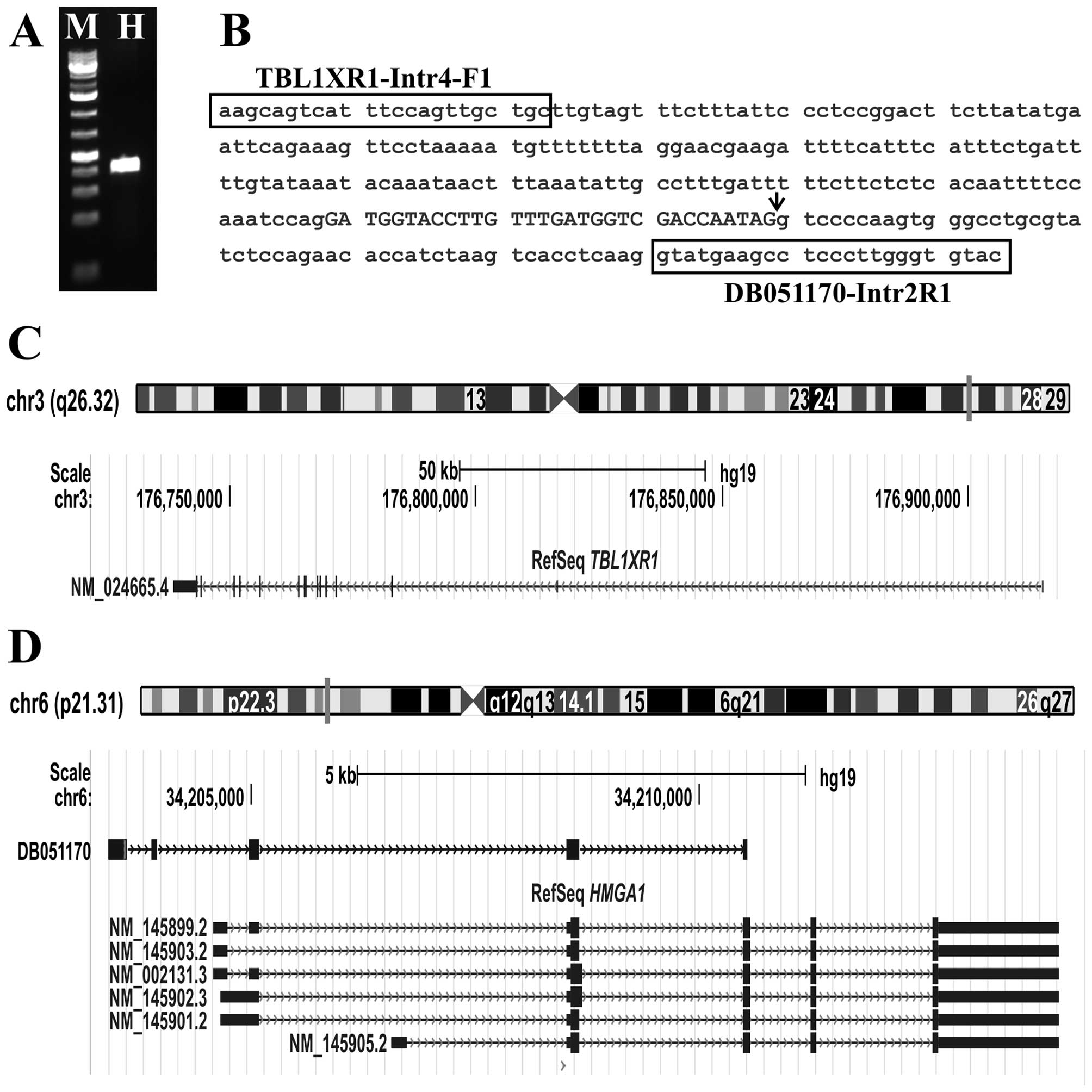
Genomic PCR with the primer combination
TBL1XR1-intron4-F1/DB051170-intr2R1 amplified a fragment from the
DNA of the splenic hemangioma. Direct Sanger sequencing showed that
it was a genomic hybrid DNA fragment with sequences from the
TBL1XR1 and HMGA1 genes (Fig. 4B). The junction point was identical
to the fusion point found with TopHat-Fusion and the ‘grep’ command
and with RT-PCR.
Discussion
We describe the first cytogenetic and molecular
genetic analysis of a splenic hemangioma. The tumor had an acquired
chromosomal translocation, t(3;6)(q26;p21), which resulted in
fusion of the TBL1XR1 (from 3q26) and HMGA1 (from
6p21) genes. The fact that an acquired genetic aberration was found
in the splenic hemangioma cells argues strongly that the disease is
neoplastic.
The protein encoded by the TBL1XR1 gene has
sequence similarity with members of the WD40 repeat-containing
protein family (18). The WD40
group is a large family of proteins which appear to have a
regulatory function (19–21). WD40 repeats mediate protein-protein
interactions and members of the family are involved in signal
transduction, RNA processing, gene regulation, vesicular
trafficking, cytoskeletal assembly, and they may also play a role
in the control of cytotypic differentiation (19–21).
TBL1XR1 is a core component of NCoR (nuclear receptor corepressor)
and SMRT (silencing mediator of retinoic acid and thyroid hormone
receptors) repressor complexes and is essential in targeting
SMRT/NCoR corepressor complexes to the promoter of target genes. It
is also required for transcriptional activation by nuclear
receptors and other regulated transcription factors (18). TBL1XR1 is further essential in the
activation of Wnt-β-catenin and NF-κB signaling pathways (18). De novo deletions and
recurrent mutations have been identified in the TBL1XR1 gene
and are linked to intellectual disability (18). TBL1XR1 plays an important
role in tumorigenesis, invasion, metastasis, and the development of
therapy resistance (18).
TBL1XR1 is overexpressed in primary lung squamous cell
carcinoma, breast cancer, cervical cancer, nasopharyngeal
carcinoma, esophageal squamous cell carcinoma, and invasive
prostate cancer (reviewed in ref. 18). Mutation of TBL1XR1 was found
in primary central nervous system lymphomas (22,23).
Deletions of TBL1XR1 were described in 15% of
ETV6-RUNX1 positive acute lymphoblastic leukemias (24). In frame TBL1XR1 chimeric
transcripts which code for chimeric proteins, were found in
different neoplasias. A recurrent TBL1XR1-TP63 fusion gene
was reported in diffuse large B-cell lymphoma, peripheral T-cell
lymphoma, and follicular lymphoma which was the result of a
chromosomal rearrangement between 3q26 (TBL1XR1) and 3q28
(TP63) (25,26). In most of the cases, the exons 1–7
of TBL1XR1 were fused in frame to exons 4–8 or 4–10 of
TP63. Exon 14 or exon 4 of TBL1XR1 was involved in
the remaining cases (25,26).
TBL1XR1 is fused to RARA in an acute
promyelocytic leukemia carrying a variant t(3;17)(q26;q21)
translocation (27). The
TBL1XR1-RARA fusion protein was predominantly localized in the
nucleus, formed homodimers or heterodimers with retinoid X receptor
α, and acted as transcriptional activator in the presence of
ligand. In the presence of pharmacologic doses of ATRA, TBLR1-RARA
protein could be degraded, and its homodimerization was abrogated
(27).
Fusions of TBL1XR1 resulting in promoter
swapping were also found (28,29).
In the breast carcinoma MCB7 cell line, the untranslated (5′-UTR)
exon 1 of TBL1XR1 is fused with the start codon-bearing exon
2 of RGS17, generating a fusion gene that encodes the
full-length RGS17 protein under the control of TBL1XR1 gene
promoter (28). The same 5′-UTR
exon 1 of TBL1XR1 was also found to be fused with the
PIK3CA gene in breast cancer and prostate adenocarcinomas.
The result was again the expression of PIK3CA which came
under the control of the TBL1XR1 promoter (29).
The HMGA1 gene encodes a non-histone protein
involved in many cellular processes, including regulation of
inducible gene transcription, integration of retroviruses into
chromosomes, and the metastatic progression of cancer cells
(30–32). The encoded protein preferentially
binds to the minor groove of A+T-rich regions in double-stranded
DNA. It has little secondary structure in solution but assumes
distinct conformations when bound to substrates such as DNA or
other proteins. The encoded protein is frequently acetylated and is
found in the nucleus. Six transcript variants were identified
encoding two different isoforms (http://www.ncbi.nlm.nih.gov/gene/3159). However,
expressed sequence tags (EST) indicate that additional alternative
splicing exists with various 5′-UTR sequences occurring in
different tissues. For example, the EST with accession numbers
DB051170 and DB03591 are found in testis, EST BM845429 in lymph
node, CN350278 in embryonic stem cells, DC338100 in brain, and
DA921912 is found in the small intestine. These splicing variants
may be regulated by alternative promoters of HMGA1 (17).
HMGA1 is overexpressed in poorly
differentiated cancers originating from all three germ layers,
suggesting that it plays a fundamental role in tumorigenesis
regardless of the cell type from which the tumor begins. High
expression of the HMGA1 gene or high levels of HMGA1 protein
were found in cancers of the thyroid, lung, breast, bladder,
prostate, colon, pancreas, uterine corpus, uterine cervix, kidney,
head and neck, nervous system, stomach, liver, and hematopoietic
system. Expression studies reveal that HMGA1 overexpression
correlates with adverse clinical outcomes in cancers (33–35).
The basis for HMGA1 overexpression is not
well understood but it seems that HMGA1 expression is
induced through different pathways. For example, HMGA1
expression was shown to be induced by cMYC and AP1 transcription
factors (36,37). Recent studies suggest that
microRNAs regulate HMGA1 in some types of cancer (38–40).
Another recent study showed that HMGA1 was an immediate target of
the β-catenin/TCF-4 signaling pathway in colon cancer (41).
Dysregulation of HMGA1 as a result of
specific chromosomal rearrangements involving chromosomal region
6p21.3 has also been identified in a variety of common benign
mesenchymal tumors such as lipomas, uterine leiomyomas, endometrial
polyps, and pulmonary hamartomas (42–51).
The breakpoints in most tumors are located 3′ of HMGA1, but
5′ or intragenic breakpoints have also been reported (42,43,46,47).
The findings we present indicate that a similar pathogenetic
mechanism may be operative also in splenic hemangioma. Whether any
systematic tumorigenic differences exist between this and the other
benign neoplasias is a question that can be answered only after
many more splenic hemangiomas are genetically analyzed.
So far, only one HMGA1-fusion transcript has
been described in the literature: an HMGA1-LAMA4 found in a
pulmonary hamartoma as a result of inv(6)(p21q21) (46). Fusion of HMGA1 to the
intergenic region between LPP and TPRG1 in a lipoma
carrying a t(3;6)(q27;p21) has also been reported (52).
The present t(3;6)(q26;p21) chromosomal
rearrangement would have molecular consequences for both
TBL1XR1 and HMGA1. The TBL1XR1 gene would have
only a single functional copy of the gene left in the cell, while
the other, rearranged allele would produce a putative truncated
form of TBL1XR1 protein containing the LiSH and F-box-like domains
(NP_078941; LiSH domain: 4–32, F-box-like domain: 41–86). Thus, the
truncated form of TBL1XR1, through the LisH and F-box-like domains,
could participate in protein dimerization, affect protein
half-life, and could influence specific cellular localizations
(53,54).
With regard to HMGA1, the
TBL1XR1-HMGA1 fusion transcript leads untranslated exons of
HMGA1 to be replaced by the first 5 exons of the
TBL1XR1 gene. The result is that the entire coding region of
HMGA1 comes under the control of the TBL1XR1 promoter
leading to dysregulation of HMGA1.
Acknowledgements
This study was supported by grants from the
Norwegian Radium Hospital Foundation.
References
|
1
|
Willcox TM, Speer RW, Schlinkert RT and
Sarr MG: Hemangioma of the spleen: Presentation, diagnosis, and
management. J Gastrointest Surg. 4:611–613. 2000. View Article : Google Scholar
|
|
2
|
Husni EA: The clinical course of splenic
hemangioma with emphasis on spontaneous rupture. Arch Surg.
83:681–688. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arber DA, Strickler JG, Chen YY and Weiss
LM: Splenic vascular tumors: A histologic, immunophenotypic, and
virologic study. Am J Surg Pathol. 21:827–835. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kutok JL and Fletcher CD: Splenic vascular
tumors. Semin Diagn Pathol. 20:128–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abbott RM, Levy AD, Aguilera NS, Gorospe L
and Thompson WM: From the archives of the AFIP: primary vascular
neoplasms of the spleen: radiologic-pathologic correlation.
Radiographics. 24:1137–1163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Halgrimson CG, Rustad DG and Zeligman BE:
Calcified hemangioma of the spleen. JAMA. 252:2959–2960. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaplan J and McIntosh GS: Spontaneous
rupture of a splenic vascular malformation. A report of three cases
and review of the literature. J R Coll Surg Edinb. 32:346–347.
1987.PubMed/NCBI
|
|
8
|
Rao RC, Ghose R, Sawhney S and Berry M:
Hemangioma of spleen with spontaneous, extra-peritoneal rupture,
with associated splenic tuberculosis - an unusual presentation.
Australas Radiol. 37:100–101. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pachl M, Elmalik K, Cohen M, Kamupira S,
Walker J and Murthi G: Ruptured splenic cavernous hemangioma in a
neonate. J Pediatr Surg. 43:407–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carta G, D'Alfonso A, Nallbani A, Palermo
P, Franchi V and Patacchiola F: Spontaneous rupture of splenic
hemangioma in puerperium. Clin Exp Obstet Gynecol. 39:407–408.
2012.PubMed/NCBI
|
|
11
|
Kang LY, Huang FD and Liu YY: Blunt
abdominal injury with rupture of giant hepatic cavernous hemangioma
and laceration of the spleen. Hepatobiliary Pancreat Dis Int.
14:109–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hoeger PH, Helmke K and Winkler K: Chronic
consumption coagulopathy due to an occult splenic haemangioma:
Kasabach-Merritt syndrome. Eur J Pediatr. 154:365–368. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schaffer LG, Slovak ML and Campbell LJ:
ISCN 2009: an International System for Human Cytogenetic
Nomenclature. Karger; Basel: 2009
|
|
14
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim D and Salzberg SL: TopHat-Fusion: An
algorithm for discovery of novel fusion transcripts. Genome Biol.
12:R722011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Panagopoulos I, Gorunova L, Bjerkehagen B
and Heim S: Fusion of the genes EWSR1 and PBX3 in retroperitoneal
leiomyoma with t(9;22)(q33;q12). PLoS One. 10:e01242882015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kimura K, Wakamatsu A, Suzuki Y, Ota T,
Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K,
Wakaguri H, et al: Diversification of transcriptional modulation:
Large-scale identification and characterization of putative
alternative promoters of human genes. Genome Res. 16:55–65. 2006.
View Article : Google Scholar :
|
|
18
|
Li JY, Daniels G, Wang J and Zhang X:
TBL1XR1 in physiological and pathological states. Am J Clin Exp
Urol. 3:13–23. 2015.PubMed/NCBI
|
|
19
|
Xu C and Min J: Structure and function of
WD40 domain proteins. Protein Cell. 2:202–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li D and Roberts R: WD-repeat proteins:
Structure characteristics, biological function, and their
involvement in human diseases. Cell Mol Life Sci. 58:2085–2097.
2001. View Article : Google Scholar
|
|
21
|
Neer EJ, Schmidt CJ, Nambudripad R and
Smith TF: The ancient regulatory-protein family of WD-repeat
proteins. Nature. 371:297–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura T, Tateishi K, Niwa T, Matsushita
Y, Tamura K, Kinoshita M, Tanaka K, Fukushima S, Takami H, Arita H,
et al: Recurrent mutations of CD79B and MYD88 are the hallmark of
primary central nervous system lymphomas. Neuropathol Appl
Neurobiol. Jun 25–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gonzalez-Aguilar A, Idbaih A, Boisselier
B, Habbita N, Rossetto M, Laurenge A, Bruno A, Jouvet A, Polivka M,
Adam C, et al: Recurrent mutations of MYD88 and TBL1XR1 in primary
central nervous system lymphomas. Clin Cancer Res. 18:5203–5211.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parker H, An Q, Barber K, Case M, Davies
T, Konn Z, Stewart A, Wright S, Griffiths M, Ross FM, et al: The
complex genomic profile of ETV6-RUNX1 positive acute lymphoblastic
leukemia highlights a recurrent deletion of TBL1XR1. Genes
Chromosomes Cancer. 47:1118–1125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Scott DW, Mungall KL, Ben-Neriah S, Rogic
S, Morin RD, Slack GW, Tan KL, Chan FC, Lim RS, Connors JM, et al:
TBL1XR1/TP63: A novel recurrent gene fusion in B-cell non-Hodgkin
lymphoma. Blood. 119:4949–4952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vasmatzis G, Johnson SH, Knudson RA,
Ketterling RP, Braggio E, Fonseca R, Viswanatha DS, Law ME, Kip NS,
Ozsan N, et al: Genome-wide analysis reveals recurrent structural
abnormalities of TP63 and other p53-related genes in peripheral
T-cell lymphomas. Blood. 120:2280–2289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Y, Li S, Zhou C, Li C, Ru K, Rao Q,
Xing H, Tian Z, Tang K, Mi Y, et al: TBLR1 fuses to retinoid acid
receptor α in a variant t(3;17)(q26;q21) translocation of acute
promyelocytic leukemia. Blood. 124:936–945. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hahn Y, Bera TK, Gehlhaus K, Kirsch IR,
Pastan IH and Lee B: Finding fusion genes resulting from chromosome
rearrangement by analyzing the expressed sequence databases. Proc
Natl Acad Sci USA. 101:13257–13261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stransky N, Cerami E, Schalm S, Kim JL and
Lengauer C: The landscape of kinase fusions in cancer. Nat Commun.
5:48462014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Benecke AG and Eilebrecht S, Benecke A and
Eilebrecht S: RNA-mediated regulation of HMGA1 function.
Biomolecules. 5:943–957. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reeves R and Beckerbauer L: HMGI/Y
proteins: Flexible regulators of transcription and chromatin
structure. Biochim Biophys Acta. 1519:13–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Resar LM: The high mobility group A1 gene:
Transforming inflammatory signals into cancer? Cancer Res.
70:436–439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Z, Wang Q, Chen F and Liu J:
Elevated expression of HMGA1 correlates with the malignant status
and prognosis of non-small cell lung cancer. Tumour Biol.
36:1213–1219. 2015. View Article : Google Scholar
|
|
34
|
Huang R, Huang D, Dai W and Yang F:
Overexpression of HMGA1 correlates with the malignant status and
prognosis of breast cancer. Mol Cell Biochem. 404:251–257. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liau SS, Rocha F, Matros E, Redston M and
Whang E: High mobility group AT-hook 1 (HMGA1) is an independent
prognostic factor and novel therapeutic target in pancreatic
adenocarcinoma. Cancer. 113:302–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wood LJ, Mukherjee M, Dolde CE, Xu Y,
Maher JF, Bunton TE, Williams JB and Resar LM: HMG-I/Y, a new c-Myc
target gene and potential oncogene. Mol Cell Biol. 20:5490–5502.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dhar A, Hu J, Reeves R, Resar LM and
Colburn NH: Dominant-negative c-Jun (TAM67) target genes: HMGA1 is
required for tumor promoter-induced transformation. Oncogene.
23:4466–4476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao XX, Yuan QZ, Mu DP, Sun DW, Bo QA,
Pan GZ, Li GQ, Cui T, Ding PP, You FP, et al: MicroRNA-26a inhibits
proliferation by targeting high mobility group AT-hook 1 in breast
cancer. Int J Clin Exp Pathol. 8:368–373. 2015.PubMed/NCBI
|
|
39
|
Liu K, Zhang C, Li T, Ding Y, Tu T, Zhou
F, Qi W, Chen H and Sun X: Let-7a inhibits growth and migration of
breast cancer cells by targeting HMGA1. Int J Oncol. 46:2526–2534.
2015.PubMed/NCBI
|
|
40
|
Xu G, Wang J, Jia Y, Shen F, Han W and
Kang Y: MiR-142-3p functions as a potential tumor suppressor in
human osteosarcoma by targeting HMGA1. Cell Physiol Biochem.
33:1329–1339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bush BM, Brock AT, Deng JA, Nelson RA Jr
and Sumter TF: The Wnt/β-catenin/T-cell factor 4 pathway
up-regulates high-mobility group A1 expression in colon cancer.
Cell Biochem Funct. 31:228–236. 2013. View Article : Google Scholar :
|
|
42
|
Kazmierczak B, Bol S, Wanschura S,
Bartnitzke S and Bullerdiek J: PAC clone containing the HMGI(Y)
gene spans the breakpoint of a 6p21 translocation in a uterine
leiomyoma cell line. Genes Chromosomes Cancer. 17:191–193. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kazmierczak B, Wanschura S, Rommel B,
Bartnitzke S and Bullerdiek J: Ten pulmonary chondroid hamartomas
with chromosome 6p21 breakpoints within the HMG-I(Y) gene or its
immediate surroundings. J Natl Cancer Inst. 88:1234–1236. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dal Cin P, Wanschura S, Christiaens MR,
Van den Berghe I, Moerman P, Polito P, Kazmierczak B, Bullerdiek J
and Van den Berghe H: Hamartoma of the breast with involvement of
6p21 and rearrangement of HMGIY. Genes Chromosomes Cancer.
20:90–92. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams AJ, Powell WL, Collins T and
Morton CC: HMGI(Y) expression in human uterine leiomyomata.
Involvement of another high-mobility group architectural factor in
a benign neoplasm. Am J Pathol. 150:911–918. 1997.PubMed/NCBI
|
|
46
|
Xiao S, Lux ML, Reeves R, Hudson TJ and
Fletcher JA: HMGI(Y) activation by chromosome 6p21 rearrangements
in multilineage mesenchymal cells from pulmonary hamartoma. Am J
Pathol. 150:901–910. 1997.PubMed/NCBI
|
|
47
|
Kazmierczak B, Dal Cin P, Wanschura S,
Borrmann L, Fusco A, Van den Berghe H and Bullerdiek J: HMGIY is
the target of 6p21.3 rearrangements in various benign mesenchymal
tumors. Genes Chromosomes Cancer. 23:279–285. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kazmierczak B, Meyer-Bolte K, Tran KH,
Wöckel W, Breightman I, Rosigkeit J, Bartnitzke S and Bullerdiek J:
A high frequency of tumors with rearrangements of genes of the
HMGI(Y) family in a series of 191 pulmonary chondroid hamartomas.
Genes Chromosomes Cancer. 26:125–133. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tallini G, Vanni R, Manfioletti G,
Kazmierczak B, Faa G, Pauwels P, Bullerdiek J, Giancotti V, Van Den
Berghe H and Dal Cin P: HMGI-C and HMGI(Y) immunoreactivity
correlates with cytogenetic abnormalities in lipomas, pulmonary
chondroid hamartomas, endometrial polyps, and uterine leiomyomas
and is compatible with rearrangement of the HMGI-C and HMGI(Y)
genes. Lab Invest. 80:359–369. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Medeiros F, Araujo AR, Erickson-Johnson
MR, Kashyap PC, Dal Cin P, Nucci M, Wang X, Bell DA and Oliveira
AM: HMGA1 and HMGA2 rearrangements in mass-forming endometriosis.
Genes Chromosomes Cancer. 49:630–634. 2010.PubMed/NCBI
|
|
51
|
Nezhad MH, Drieschner N, Helms S, Meyer A,
Tadayyon M, Klemke M, Belge G, Bartnitzke S, Burchardt K, Frantzen
C, et al: 6p21 rearrangements in uterine leiomyomas targeting
HMGA1. Cancer Genet Cytogenet. 203:247–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang X, Zamolyi RQ, Zhang H, Pannain VL,
Medeiros F, Erickson-Johnson M, Jenkins RB and Oliveira AM: Fusion
of HMGA1 to the LPP/TPRG1 intergenic region in a lipoma identified
by mapping paraffin-embedded tissues. Cancer Genet Cytogenet.
196:64–67. 2010. View Article : Google Scholar
|
|
53
|
Gerlitz G, Darhin E, Giorgio G, Franco B
and Reiner O: Novel functional features of the Lis-H domain: Role
in protein dimerization, half-life and cellular localization. Cell
Cycle. 4:1632–1640. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mateja A, Cierpicki T, Paduch M, Derewenda
ZS and Otlewski J: The dimerization mechanism of LIS1 and its
implication for proteins containing the LisH motif. J Mol Biol.
357:621–631. 2006. View Article : Google Scholar : PubMed/NCBI
|