Introduction
Lung cancer is one of the primary causes of
cancer-related death worldwide and >80% of lung cancer patients
have non-small cell lung cancer (NSCLC) (1,2).
Despite improvements in treatment modalities (such as surgical
resection, chemo-therapy, radiotherapy, and targeted therapy), the
long-term survival of NSCLC is still dismal with 5-year survival
rate >10% due to cancer relapse and metastasis (3,4).
Hence, good understanding of the mechanisms underlying NSCLC
metastasis is very important.
The transforming growth factor β (TGF-β) superfamily
plays crucial roles in cell proliferation, apoptosis,
differentiation, and angiogenesis (5,6). In
particular, its alterations significantly associated with the
tumorigenic and metastatic processes of NSCLC (7,8). In
the canonical signaling, upon ligand stimulation, TGF-β receptor
type II (TGF-βR2) recruits and transphosphorylates TGF-β receptor
type I (TGF-β R1), which in turn phosphorylate Smad2 and Smad3
proteins, and these phosphorylated proteins in turn form tight
protein complexes with Smad4 (also known as DPC4), and subsequently
translocate to the nucleus where they interact with other
transcription activator or repressors to signal downstream pathways
(9). Notably, there is more and
more evidence showing that TGF-β signaling is an important inducer
of epithelial-mesenchymal transition (EMT) in various cancers,
including NSCLC (10–12). EMT is a critical step for
morphogenesis during embryonic development and the conversion of
early-stage tumors into invasive malignancies (13,14),
which is marked by repression of E-cadherin and induction of
N-cadherin, and Vimentin (12,15).
Importantly, such tumor-suppressing function of TGF-β signaling
pathway is strongly dependent on the status of Smad4 (16). Despite such functional importance,
how Smad4 is regulated at transcriptional level is unclear.
MicroRNAs (miRNAs) are non-coding RNA molecules
~19–24 nucleotides long that regulate gene expression at the
post-transcriptional level (17,18).
Approximately 30% of messenger RNAs are regulated by miRNAs
(19). Thus, miRNAs represent a
group of important players in diverse biological and pathological
processes, including tumor cell proliferation, differentiation, and
survival (20–23). Recently a study showed that the
expression of microRNAs appear to be tissue or tumor type-specific
(24), and there is accumulating
evidence showing that it could be a candidate biomarker for
clinical diagnosis, including identification of cancer type or
tumor subtype (25,26). miR-205, which is located at lung
cancer associated genomic amplification region 1q32.2.
Dysregualtion of miR-205 was observed in many types of tumors,
including lung cancer (27),
moreover, miR-205 was reported to be expressed at higher level in
squamous cell lung carcinoma than other types of NSCLC (25).
In this study, based on experiments in vitro
and biochemical analyses, we explored the functional roles of
miR-205 and its molecular link to Smad4. Our data indicate that
miR-205 moderates TGF-β-induced epithelial-mesenchymal transition
in NSCLC by directly targeting Smad4. Moreover, this study provides
crucial molecular and cellular bases of miR-205 as one of potential
clinical therapeutic targets for highly invasive NSCLC.
Materials and methods
Cell culture
Human NSCLC cells A549, obtained from the Cell Bank
of the Chinese Academy of Sciences (Shanghai, China), were seeded
and grown in RPMI-1640 medium (Hyclone, South Logan, UT, USA)
supplemented with 10% heat-inactivated fetal bovine serum (Gibco,
Carlsbad, CA, USA) and antibiotics (Invitrogen, Carlsbad, CA, USA)
at the condition of 37°C in a humidified incubator containing 5%
CO2.
RNA extraction, cDNA synthesis and
quantitative real-time PCR (qRT-PCR)
Total RNA of cells and tissues was extracted by
adding 1.0 ml RNAiso Plus (Takara, Osaka, Japan) according to the
manufacturer's protocol. The concentration of RNA was measured
using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA).
Synthesis of cDNA was carried out with reverse transcriptase M-MLV
(Takara). The primers for reverse transcription and amplification
of miR-205 and U6 were designed and synthesized by Guangzhou
RiboBio Co. (Guangzhou, China). The sequences of qRT-PCR primers
for Smad4, Snail, MMP-9 and β-actin were as follows:
Smad4, forward, 5′-CAGCCATCTTG TCCACT-3′; reverse,
5′-GCTGGGGTGCTGTATGTC-3′, Snail, forward,
5′-CGAAAGGCCTTCAACTGCAAAT-3′; reverse, 5′-ACTGGTACTTCTTGACATCTG-3′.
MMP-9, forward, GACGATGACGAGTTGTGG; reverse, GAAGGGG
AAGTGGCAG. β-actin, forward, 5′-CACAGAGCCTCGC CTTTGCC-3′;
reverse, 5′-ACCCATGCCCACCATCACG-3′. qRT-PCR was performed using
SYBR Premix ExTaq™ (Takara) according to the manufacturer's
instructions on an ABI Step One Plus Real-Time PCR system (Applied
Biosystems, Foster City, CA, USA). β-actin and U6 were
respectively used as the internal controls for Smad4 and
miR-205. The relative mRNA expression was measured using the ΔΔCT
method.
Western blot assay
Cells were harvested and then lysed in RIPA buffer
(Cell Signaling Technology, Danvers, MA, USA) containing protease
inhibitor and phosphatase inhibitor cocktail (Sigma-Aldrich, St.
Louis, MO, USA). After incubation in 4°C for 15 min, the total
lysates were centrifuged at 12,000 rpm for 15 min at 4°C. Then the
cell lysate per well was separated by 10% SDS-PAGE electrophoresis,
transferred to nitrocellulose membranes (Millipore, Billerica, MA,
USA), blocked with 5% BSA in TBST buffer for 1 h and then
immunoblotted with primary antibodies overnight at 4°C. After 4
times washing with TBST, the membranes were further incubated with
the HRP-conjugated secondary antibodies for 2 h at room
temperature. Signal detection was performed using the ECL kit
(Pierce, Rockford, IL, USA). All antibodies for western blotting
including anti-Smad4; anti-β-actin and secondary antibodies were
purchased from Cell Signaling Technology.
Luciferase reporter assays
Constructing a plasmid containing the Smad4 3′-UTR
fused to the 3′-end of a luciferase reporter, we used psiCHECK2
dual luciferase vector (Promega, Madison, WI, USA). Briefly, a
215-bp fragment containing predicted miR-205 target site (positions
262–269) was chosen for the luciferase assay. The wild-type and
mutanted fragment were directly synthesized (Genewiz, Suzhou,
China), and then each subcloned into psiCHECK2 vector to generate a
psiCHECK2-Smad4-3′-UTR wild-type and a
psiCHECK2-Smad4-3′-UTR-mutant. Subsequently, A549 cells were plated
in a 24-well plate and cotransfected with wild-type or mutanted
plasmid with either miR-205 mimics or miR-negative control (miR-NC)
using Lipofectamine 2000 (Life Technologies). After 48 h, cells
were all harvested, and luciferase activities were measured by the
Dual-Luciferase Reporter Assay kit (Promega). Each experiment was
done in triplicate.
The −960 to +124 promoter region of PAI-1 was
amplified by PCR with primers (forward, CGGGGTACCGCACACCC
TGCAAACCTGCC and reverse, CCGCTCGAGCGATTGG CGGTTCGTCCTG). Digested
with KpnI and XhoI, the acquired fragments were
directly ligated into the pGL3 basic vector (Promega). pGL3 basic
vector (800 ng), 800 ng pGL3-PAI-1, 32 ng pRL-TK were
co-transfected with miR-205 mimics or miR-NC using Lipofectamine
2000 (Life Technologies). Twenty-four hours later, the cells
treated with/ without 5 ng/ml TGF-β1. At last, luciferase activity
of the transfected cells was determined using the Dual-Luciferase
Reporter Assay kit (Promega). Each experiment was done in
triplicate.
miR-205 mimics, plasmid, siRNA and cell
transfection
miR-205 mimics, and matched miR-NC were synthesized
by GenePharma company (Suzhou, China), The control (Si-NC) and
Smad4 siRNA (Si-Smad4) were prepared as previously described
(28). The target sequences of
siRNA were as follows: 5′-GTACTTCATACCATGCCGA-3′. Cell
transfections were performed using Lipofectamine 2000 (Invitrogen)
according to the manufacturer's instructions. After 48-h
transfection, the cells were collected for further experiments.
Immunocytochemistry staining
Cells were seeded into 24-well plates at a
concentration of 1×104 cells/well. After treatment
with/without 5 ng/ml TGF-β1 for 48 h, discarding the culture
medium, cells were fixed with 95% alcohol and permeabilized with
0.5% Triton X-100. Five minutes later, cells were incubated with
primary antibodies against E-cadherin, N-cadherin and Vimentin
(1:100, BD Biosciences, San Jose, CA, USA) for 2 h, negative
controls were made with PBS instead of the primary antibodies. The
second antibodies were diluted in PBS and incubated for 1 h at room
temperature. Diaminobenzidine (DAB) was used to visualize the
immunoreactivity and a light microscope was used for photography of
the sections.
Wound healing assay
After 24 h of transfection with miR-NC and miR-205,
A549 cells were seeded into a 6-well culture plate at a density of
25×104/well. When cells reached ~80–90% confluence as a
monolayer, the medium was changed with 2 ml PBS, the monolayer was
gently scratched with a 10-μl pipette tip, ensuring that the tip
was always vertical to the bottom of the well. The obtained gap
distance equals the diameter of the end of the 10-μl pipette tip.
After scratching, the well were washed with 1X PBS gently two times
to remove the detached cells. Fresh medium was added to the well
and the cells were cultured for an additional 24 h. Photos were
taken of the gap distance using a microscope and the gap distance
was quantitatively evaluated using Photoshop.
Migration and invasion assays
Transwell inserts 8.0-μm pores in size (Corning,
NewYork, NY, USA) were used for performing cell migration and
invasion assays. For migration assay, 800 μl RPMI-1640 medium with
10% FBS was added into each lower champer of a Transwell insert.
Briefly, cells were transfected with 50 nM miR-NC or miR-205
mimics. Forty-eight hours post-transfection, cells were
trypsinized, and then cells (5×104) with medium
containing 1% FBS were seeded into the upper champer and incubated
at 37°C for 24 h in a humidified incubator. Furtherly, the cells
migrated onto the lower surface of the insert were fixed with 100%
methanol for 30 min, air-dried for 10 min, stained with 0.1%
crystal violet overnight and washed with 1X PBS two times. Lastly,
the cells were photographed and counted. For invasion assay, the
inserts were coated with Matrigel matrix (BD Science, Sparks, MD,
USA) diluted in serum-free medium, then incubated at 37°C for 2 h,
remaining procedures were conducted similarly to migration assay.
Each experiment was performed in triplicate.
Statistical analysis
Results are presented as mean ± standard deviation
(SD). Statistical significance was tested using Student's t-test
and a P-value <0.05 was considered significant. All statistical
analyses were performed using GraphPad Prism 5.0 (GraphPad, San
Diego, CA, USA) and SPSS 7.0 software (SPSS, Chicago, IL, USA).
Results
TGF-β induces EMT in NSCLC cells
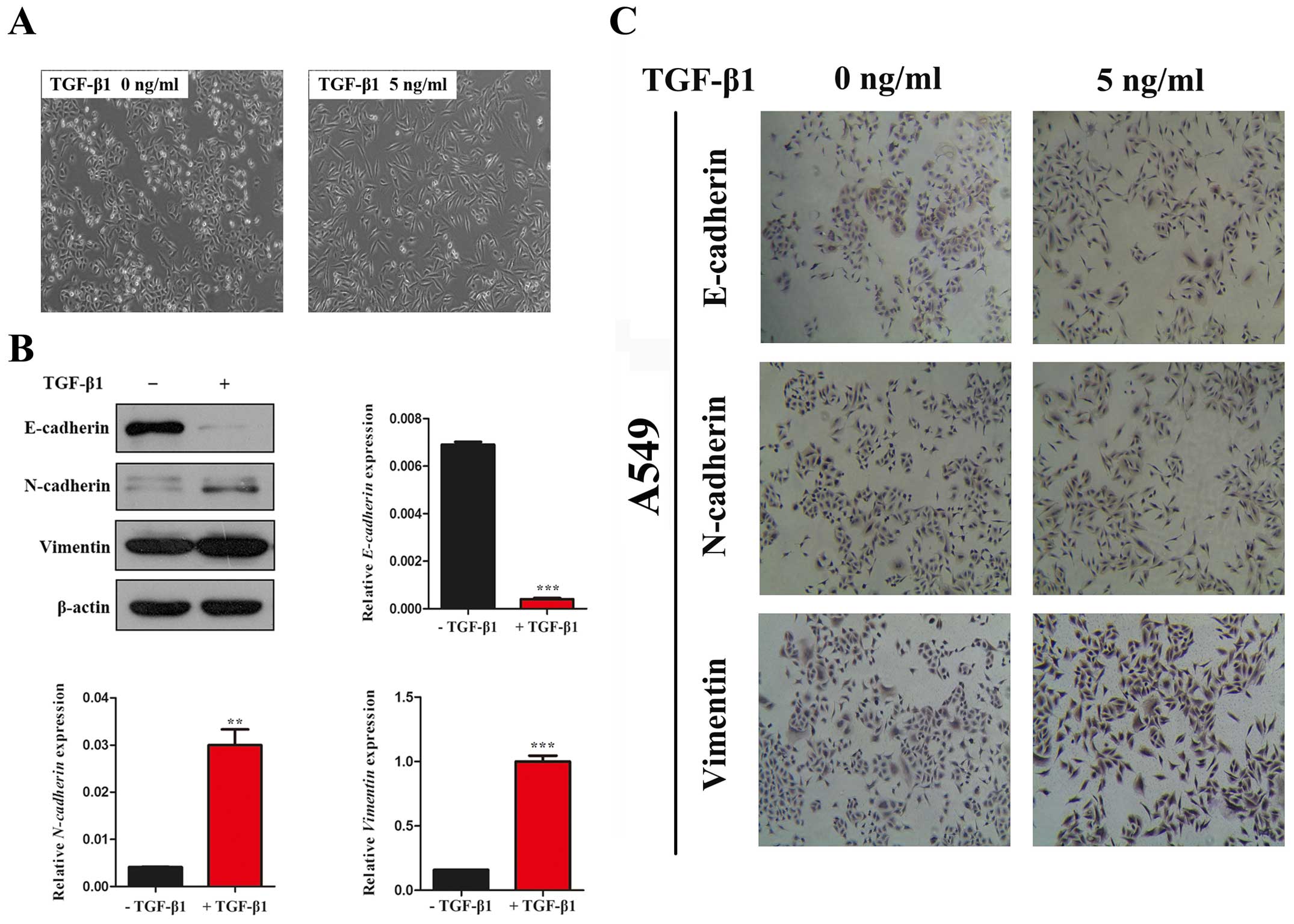
Firstly, we observed the morphological changes in
A549 cells which have been widely used as a model system to study
the mechanisms of carcinogenesis and tumor progression in lung
cancer. In the absence of TGF-β1, A549 cells maintained a classic
epithelial morphology. On the contrary, A549 cells displayed a
spindle-shape, fibroblast-like morphology after treatment with
TGF-β1 for 24 h (Fig. 1A). Then,
we investigated the expression of epithelial marker, E-cadherin,
the mesenchymal marker, N-cadherin and Vimentin by western blot
assay. In line with the morphological changes, the results showed
that the expression of E-cadherin was significantly decreased and
N-cadherin and Vimentin was increased with TGF-β1 treatment for 24
h in A549 cells (Fig. 1B and C).
Taken together, our data indicated that TGF-β1 was able to induce
EMT in A549 cell lines.
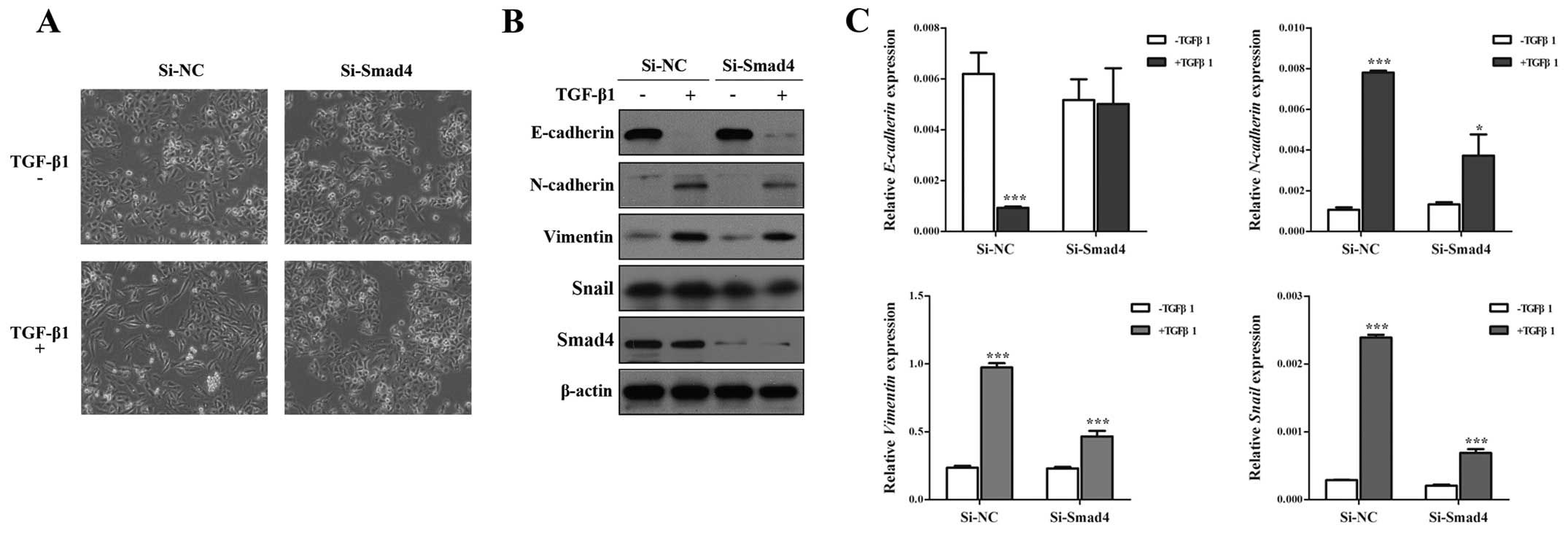
Knockdown of Smad4 inhibits TGF-β-induced
EMT, invasion and migration in NSCLC cells
Although Smad4 is the key molecule to TGF-β-induced
EMT (29,30), the role of Smad4 in TGF-β-induced
EMT has not been studied fully. To identify this issue, we silenced
Smad4 by specific SiRNA against Smad4 (Si-Smad4), and found that
Si-Smad4 could remarkably decrease TGF-β-induced EMT, including
morphology (Fig. 2A), and EMT
markers (Fig. 2B), Si-Smad4
significantly restored E-cadherin expression and impaired
N-cadherin, Vimentin and Snail expression in the presence of TGF-β1
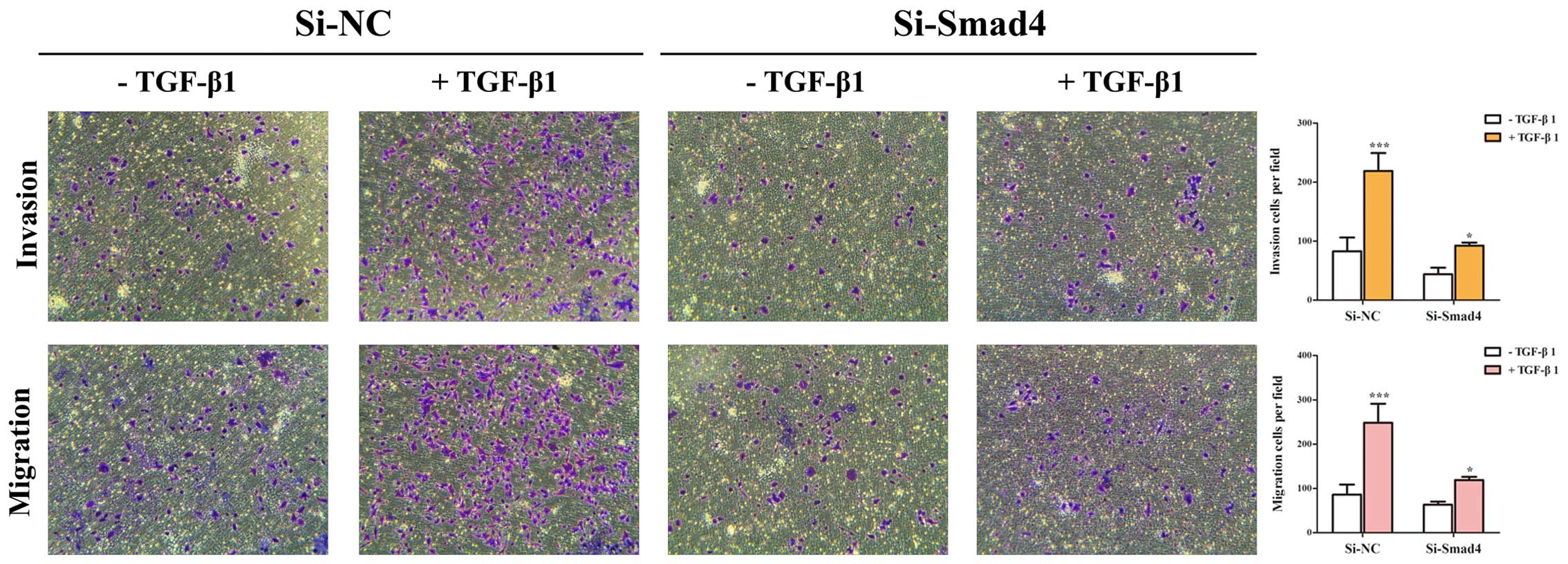
(Fig. 2B and C). Then, Transwell
assays showed that TGF-β-induced migratory and invasive abilities
could be inhibited by knockdown of Smad4 in A549 cells (Fig. 3). These results suggested that
TGF-β-induced EMT, invasion and migration depends on Smad4 in lung
cancer cells, and reduced expression of Smad4 inhibits TGF-β
induced EMT, invasion and migration of NSCLC cells.
miR-205 directly targets Smad4 in NSCLC
cells
Given the fact miRNAs can regulate various
biological processes including cell proliferation by targeting
proliferation-related genes (23),
and Huang et al (31)
predicted in silico that Smad4 was a direct target of
miR-205, we consider the possibility that miR-205 can inhibit Smad4
expression by directly binding to Smad4 3′-UTR region. To confirm
that Smad4 is directly regulated by miR-205, we subcloned Smad4
3′-UTR containing miR-205 binding site (wild-type/mutant type) into
psiCHECK-2 vector (Fig. 4). As
miR-205 is significantly downregulated in NSCLC cell lines
(32,33), we only transiently cotransfected
the reporter construct with miR-205 mimics/miR-NC into A549 cells.
Experimental results showed that miR-205 significantly inhibited
the luciferase activities in cells transfected with the Smad4
3′-UTR wild-type but did not repress the luciferase activities in
cells with the mutant construct (Fig.
4), moreover, the cells trans-fected with miR-205 mimics showed
lower expression of Smad4 than miR-NC (Fig. 5A). The results suggested that
miR-205 can directly target the 3′-UTR of Smad4 in NSCLC cells.
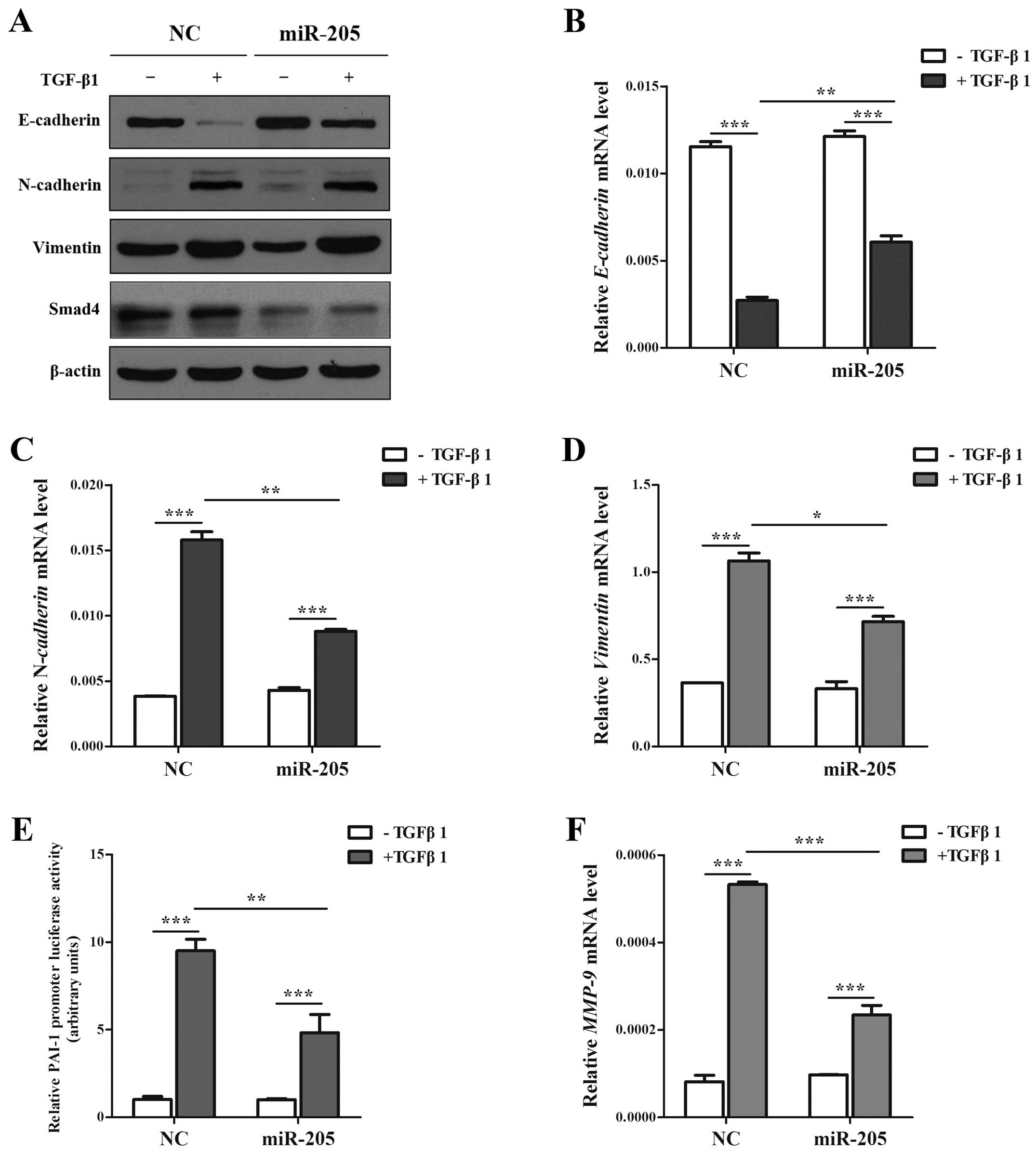
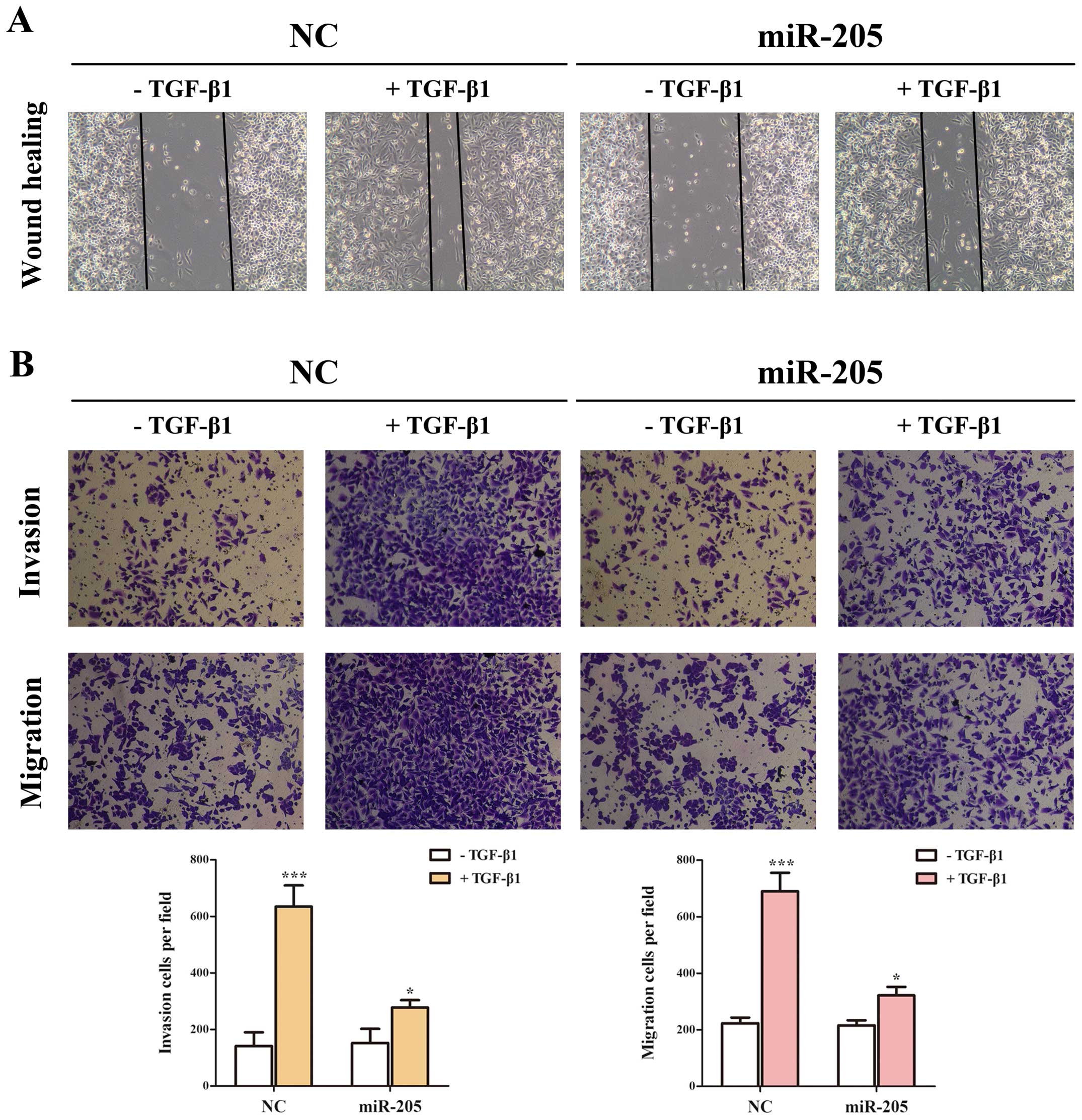
miR-205 moderates the EMT, invasion and
migration induced by TGF-β/Smad4 signal in NSCLC cell lines
As is known, the production of TGF-β has been found
increased in NSCLC cells and tissues (34,35).
Based on the facts that TGF-β is one of the primary inducers of EMT
in NSCLC (36,37), and accumulating results showed that
miR-205 can regulate key molecules and influence the signal
transduction in tumor development (27), notably in EMT (12). However, Smad4 acts as the only
Co-Smad of TGF/Smad signaling pathway and plays the key role in
TGF-β-mediated EMT. Based on the results above, we determined
whether miR-205 inhibited TGF-β/Smad4 signal-induced EMT in A549
cell lines treated with TGF-β1 with/without miR-205 by targeting
Smad4. In our study, the cells treated with TGF-β1 in combination
with miR-205 showed higher expression of E-cadherin and lower
expression of N-cadherin and Vimentin only in the mRNA level than
miR-NC (Fig. 5A–D). Then cells
transfected with Smad-mediated PAI-1 reporter plasmid were
stimulated with TGF-β in the presence and absence of miR-205. As
illustrated (Fig. 5E) both the
basal and TGF-β-induced PAI-1 promoter activation could been
attenuated significantly by miR-205. Accordingly, miR-205 can
diminish TGF-β-induced enhancement in MMP-9 (Fig. 5F), which has been widely reported
to promote cancer metastasis. Given the facts that TGF-β signaling
can stimulate the migration and invasion of NSCLC cells (13), we used Transwell and wounding
healing assays to examine whether miR-205 could effect
TGF-β-induced migration and invasion in A549 cell lines. Taken
together, the results indicated that the invasion and migration
function induced by TGF-β/Smad4 signal of NSCLC cell lines
(Fig. 6) could be repressed by
miR-205.
Discussion
Metastasis and relapse is the major cause of death
for lung cancer patients (4). EMT
is a critical step in cancer metastasis, which can invert
early-stage tumor into invasive malignancy (14). TGF-β-mediated signaling functions
as a potent inducer of EMT which is marked by repression of
E-cadherin and induction of N-cadherin and Vimentin in cancer cells
(16), contribute to progression
of various cancers, including lung cancer (13).
As a Co-Smad of the Smad family, Smad4 was
identified as a tumor suppressor gene in pancreatic carcinomas and
initially known as ‘deleted in pancreatic carcinoma locus 4 (DPC4)’
(38). Although the TGF-β pathway
has been extensively studied for more than two decades, many
efforts have focused on R-Smads regulation and less is known about
Smad4. Recently studies showed TGF-β-responsive tumors are involved
in TGF-β/ Smad4 signaling pathway showing poor differentiation and
increased EMT (39). Another study
showed the Smad3/Smad4 complex could bind directly to regulatory
promoter region of Snail, increasing its transcription and
subsequently repressing the E-cadherin expression (40). Interesting, Yang et al found
that canonical TGF-β/Smad4 pathway could activate CDH2 promoter to
increase N-cadherin expression in NSCLC (28). These above mentioned activities
improve the understanding of Smad4 serving as a tumor suppressor
through its ability to inhibit epithelial cell proliferation.
However, more mechanisms on TGF-β-induced EMT and its
tumor-suppressive role should be elaborated and verified (41).
Since miRNAs are generally involved in the
pathogenesis of cancer by directly regulating the expression of
their targets at a post-transcriptional level. miR-205, which has a
relationship with lung cancer associated genomic amplification
region 1q32.2. Dysregualtion of miR-205 was observed in many types
of tumors, including lung cancer, and it was shown to be
overexpressed in different non-small cell lung carcinomas tissues,
resulting in increased cell proliferation and activated
angiogenesis both in vitro and in vivo through
directly targeting PTEN and PHLPP2 tumor suppressor genes and
subsequently activating AKT/FOXO3a and AKT/ mTOR pathways (27). Recent studies have shown altered
miRNAs could affect EMT by regulated key molecules (12), including lung cancer (42). miR-205 has been found to effect EMT
specially by targeting ZEB1/ZEB2 in breast cancer cells along with
a decrease in E-cadherin and an increase in N-cadherin (12). On the contrary, ectopic expression
of miR-205 in mesenchymal cell-initiated MET (mesenchymal to
epithelial transition) with the upregulation of E-cadherin and
decrease of cell migration and invasion. A recent study even found
that miR-34a inhibits EMT in human cholangio-carcinoma by targeting
Smad4 through TGF-β/Smad pathway (43). Our study shows that miR-205
suppresses the activation of TGF-β/Smad4 signal-induced EMT,
invasion and migration in NSCLC cell lines by targeting Smad4.
Accumulating evidence has demonstrated that
dual-functional miR-205 may contribute to normal physiological
processes, including wound healing and in development of
pathological events such as cancers. The tissue-type origin of the
cancer and target genes therein is thus far the only logical
determinant for the dual roles of miR-205. As a tumor suppressor or
oncogene, miR-205 could effectively impair or promote cancer
progression through a wide variety of cellular and molecular
signaling pathways; cell proliferation, programmed cell death, EMT
and angiogenesis. Downregulation of miR-205 has been reported in a
number of cancers, including lung cancer, Larzabal et al
have also reported significant downregulation of miR-205 in
non-small cell lung carcinoma compared with non-cancerous lung
epithelial cells, describing it as a tumor suppressor miRNA
(33). For clarification of this
contradictory finding, they investigated a novel molecular
signaling pathway in which miR-205 has an ability to regulate
cancer cell migration and metastases by making connection between
TMPRSS4 (trans-membrane protease, serine 4) and integrin α5.
TMPRSS4 is been known as a membrane-anchored proteases implicated
in cell invasion and motility. Upon knockdown of TMPRSS4, they
found miR-205 was upregulated, resulting in increased E-cadherin
expression, reduction of fibronectin and inhibition of
epithelial-mesenchymal transition. They further found that integrin
α5 which is involved in cell motility and invasion and also is a
direct target for miR-205, was downregulated due to upregulation of
miR-205. Eventually, they found a hindrance in cell migration and
reduction in cell proliferation. On the other hand, miR-205 has
also been detected as upregulated in lung cancer, it was shown to
be overexpressed in different non-small cell lung carcinomas
tissues, resulting in increased cell proliferation and activated
angiogenesis both in vitro and in vivo through
directly targeting PTEN and PHLPP2 tumor suppressor genes and
subsequently activating AKT/FOXO3a and AKT/mTOR pathways (27). The variation of miR-205 expression
may act as a diagnostic and/or prognostic biomarker tool in human
cancers, with different relationships in various cancers and their
subtypes. More importantly, appreciation of molecular function of
miR-205 in initiation and progression of cancer via targeting
numerous tumor suppressor genes and oncogenes could help us to
address a cancer therapeutic issue and open new avenues for gene
therapy in those cancers where its tumor suppressive functions are
dominant.
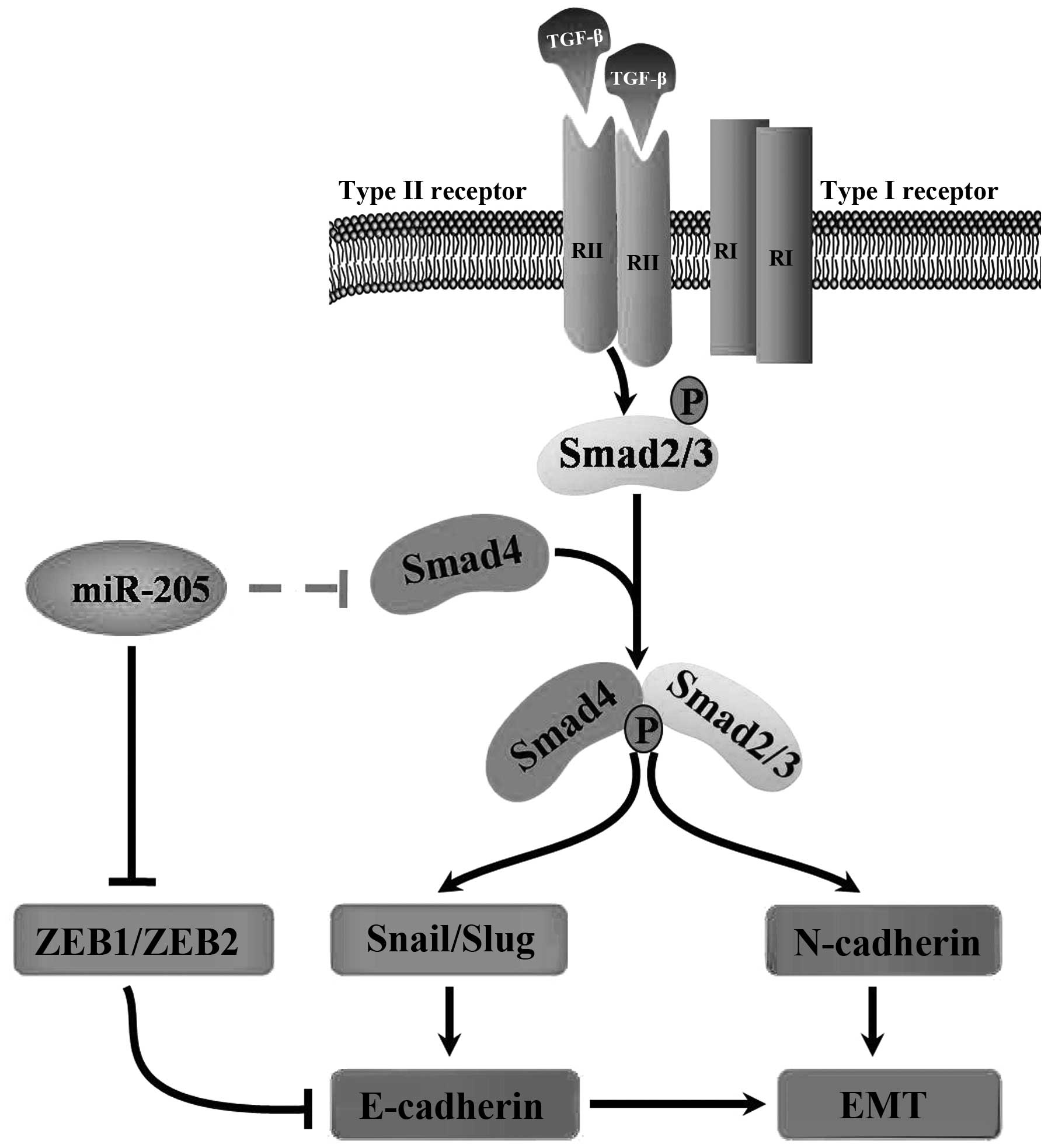
In conclusion, our findings demonstrate that miR-205
regulates the expression of Smad4 and impairs its functions in
cells, therefore miR-205 is important for TGF-β-induced EMT,
invasion and migration in NSCLC. The promising therapeutic role of
miRNAs in the prevention of advanced NSCLC remains a major clinical
challenge. In this study, our results strongly indicated the
importance of miR-205 as a potential target in clinical therapy and
demonstated that this miRNA merits further investigation as a
promising gene therapy target for the treatment of NSCLC (Fig. 7). Notably, our data provide
theoretical basis for study on the relationship between the miRNAs
and EMT in other diseases.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81201575 to Z-Y. Liu; 31270940
to J-A. Huang), Jiangsu Province Colleges and Universities Natural
Science Research Foundation (12KJB310016 to Z-Y. Liu; 14KJB0017 to
Z.L), Clinical Medical Center of Suzhou (Szzx201502) and Clinical
Key Speciality Project of China.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang T, Nelson RA, Bogardus A and Grannis
FW Jr: Five-year lung cancer survival: Which advanced stage
nonsmall cell lung cancer patients attain long-term survival?
Cancer. 116:1518–1525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gupta GP and Massagué J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Massagué J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kitisin K, Saha T, Blake T, Golestaneh N,
Deng M, Kim C, Tang Y, Shetty K, Mishra B and Mishra L: TGF-Beta
signaling in development. Sci STKE. 2007.cm1:2007.
|
|
7
|
Kim WS, Park C, Jung YS, Kim HS, Han J,
Park CH, Kim K, Kim J, Shim YM and Park K: Reduced transforming
growth factor-beta type II receptor (TGF-beta RII) expression in
adeno-carcinoma of the lung. Anticancer Res. 19A:301–306. 1999.
|
|
8
|
Park C, Kim WS, Choi Y, Kim H and Park K:
Effects of transforming growth factor beta (TGF-beta) receptor on
lung carcinogenesis. Lung Cancer. 38:143–147. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
David CJ, Huang YH, Chen M, Su J, Zou Y,
Bardeesy N, Iacobuzio-Donahue CA and Massagué J: TGF-β tumor
suppression through a lethal EMT. Cell. 164:1015–1030. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gregory PA, Bracken CP, Smith E, Bert AG,
Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, et al:
An autocrine TGF-beta/ZEB/miR-200 signaling network regulates
establishment and maintenance of epithelial-mesenchymal transition.
Mol Biol Cell. 22:1686–1698. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014.PubMed/NCBI
|
|
14
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Principe DR, Doll JA, Bauer J, Jung B,
Munshi HG, Bartholin L, Pasche B, Lee C and Grippo PJ: TGF-β:
Duality of function between tumor prevention and carcinogenesis. J
Natl Cancer Inst. 106:djt3692014. View Article : Google Scholar
|
|
17
|
Mlcochova J, Faltejskova P, Nemecek R,
Svoboda M and Slaby O: MicroRNAs targeting EGFR signalling pathway
in colorectal cancer. J Cancer Res Clin Oncol. 139:1615–1624. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmidt A and Küppers R: Role of microRNAs
in B cell leukemias and lymphomas. Curr Mol Med. 14:580–597. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi X, Zhan L, Xiao C, Lei Z, Yang H, Wang
L, Zhao J and Zhang HT: miR-1238 inhibits cell proliferation by
targeting LHX2 in non-small cell lung cancer. Oncotarget.
6:19043–19054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mirzamohammadi F, Papaioannou G and
Kobayashi T: MicroRNAs in cartilage development, homeostasis, and
disease. Curr Osteoporos Rep. 12:410–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu J, Zeng Y, Xu C, Qin H, Lei Z, Shen D,
Liu Z and Huang JA: Expression profile analysis of microRNAs and
downregulated miR-486-5p and miR-30a-5p in non-small cell lung
cancer. Oncol Rep. 34:1779–1786. 2015.PubMed/NCBI
|
|
24
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lebanony D, Benjamin H, Gilad S, Ezagouri
M, Dov A, Ashkenazi K, Gefen N, Izraeli S, Rechavi G, Pass H, et
al: Diagnostic assay based on hsa-miR-205 expression distinguishes
squamous from nonsquamous non-small-cell lung carcinoma. J Clin
Oncol. 27:2030–2037. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Markou A, Tsaroucha EG, Kaklamanis L,
Fotinou M, Georgoulias V and Lianidou ES: Prognostic value of
mature microRNA-21 and microRNA-205 overexpression in non-small
cell lung cancer by quantitative real-time RT-PCR. Clin Chem.
54:1696–1704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai J, Fang L, Huang Y, Li R, Yuan J, Yang
Y, Zhu X, Chen B, Wu J and Li M: miR-205 targets PTEN and PHLPP2 to
augment AKT signaling and drive malignant phenotypes in non-small
cell lung cancer. Cancer Res. 73:5402–5415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang H, Wang L, Zhao J, Chen Y, Lei Z, Liu
X, Xia W, Guo L and Zhang HT: TGF-β-activated SMAD3/4 complex
transcriptionally upregulates N-cadherin expression in non-small
cell lung cancer. Lung Cancer. 87:249–257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hesling C, Fattet L, Teyre G, Jury D,
Gonzalo P, Lopez J, Vanbelle C, Morel AP, Gillet G, Mikaelian I, et
al: Antagonistic regulation of EMT by TIF1γ and Smad4 in mammary
epithelial cells. EMBO Rep. 12:665–672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng H, Fertig EJ, Ozawa H, Hatakeyama H,
Howard JD, Perez J, Considine M, Thakar M, Ranaweera R, Krigsfeld
G, et al: Decreased SMAD4 expression is associated with induction
of epithelial-to-mesenchymal transition and cetuximab resistance in
head and neck squamous cell carcinoma. Cancer Biol Ther.
16:1252–1258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang W, Jin Y, Yuan Y, Bai C, Wu Y, Zhu H
and Lu S: Validation and target gene screening of hsa-miR-205 in
lung squamous cell carcinoma. Chin Med J (Engl). 127:272–278.
2014.
|
|
32
|
Du L, Schageman JJ, Irnov, Girard L,
Hammond SM, Minna JD, Gazdar AF and Pertsemlidis A: MicroRNA
expression distinguishes SCLC from NSCLC lung tumor cells and
suggests a possible pathological relationship between SCLCs and
NSCLCs. J Exp Clin Cancer Res. 29:752010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Larzabal L, de Aberasturi AL, Redrado M,
Rueda P, Rodriguez MJ, Bodegas ME, Montuenga LM and Calvo A:
TMPRSS4 regulates levels of integrin α5 in NSCLC through miR-205
activity to promote metastasis. Br J Cancer. 110:764–774. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Asselin-Paturel C, Echchakir H, Carayol G,
Gay F, Opolon P, Grunenwald D, Chouaib S and Mami-Chouaib F:
Quantitative analysis of Th1, Th2 and TGF-beta1 cytokine expression
in tumor, TIL and PBL of non-small cell lung cancer patients. Int J
Cancer. 77:7–12. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bennett WP, el-Deiry WS, Rush WL, Guinee
DG Jr, Freedman AN, Caporaso NE, Welsh JA, Jones RT, Borkowski A,
Travis WD, et al: p21waf1/cip1 and transforming growth factor beta
1 protein expression correlate with survival in non-small cell lung
cancer. Clin Cancer Res. 4:1499–1506. 1998.PubMed/NCBI
|
|
36
|
Akhurst RJ and Balmain A: Genetic events
and the role of TGF beta in epithelial tumour progression. J
Pathol. 187:82–90. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han G, Lu SL, Li AG, He W, Corless CL,
Kulesz-Martin M and Wang XJ: Distinct mechanisms of
TGF-beta1-mediated epithelial-to-mesenchymal transition and
metastasis during skin carcinogenesis. J Clin Invest.
115:1714–1723. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hahn SA, Schutte M, Hoque AT, Moskaluk CA,
da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban
RH, et al: DPC4, a candidate tumor suppressor gene at human
chromosome 18q21.1. Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bardeesy N, Cheng KH, Berger JH, Chu GC,
Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, et
al: Smad4 is dispensable for normal pancreas development yet
critical in progression and tumor biology of pancreas cancer. Genes
Dev. 20:3130–3146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Katsuno Y, Lamouille S and Derynck R:
TGF-β signaling and epithelial-mesenchymal transition in cancer
progression. Curr Opin Oncol. 25:76–84. 2013. View Article : Google Scholar
|
|
42
|
Kumarswamy R, Mudduluru G, Ceppi P,
Muppala S, Kozlowski M, Niklinski J, Papotti M and Allgayer H:
MicroRNA-30a inhibits epithelial-to-mesenchymal transition by
targeting Snai1 and is downregulated in non-small cell lung cancer.
Int J Cancer. 130:2044–2053. 2012. View Article : Google Scholar
|
|
43
|
Qiao P, Li G, Bi W, Yang L, Yao L and Wu
D: microRNA-34a inhibits epithelial mesenchymal transition in human
cholangio-carcinoma by targeting Smad4 through transforming growth
factor-beta/Smad pathway. BMC Cancer. 15:4692015. View Article : Google Scholar
|