Introduction
Glioblastoma, classified as a grade IV astrocytoma
according to the World Health Organization prognostic system, is
the most common and lethal type of intracranial tumor,
characterized by high angiogenic and infiltrative capacities
(1,2). The current standard treatment for
glioblastoma consists of maximal surgical resection, radiotherapy,
and concomitant and adjuvant chemotherapy with temozolomide
(1,3). Despite advances in the understanding
of the molecular pathogenesis of glioblastoma, diagnosis, and more
promising and tailored therapeutic approaches, the median survival
time of patients with glioblastoma remains <1 year (4,5). The
requirement for novel therapeutic strategies remains paramount
given the sustained development of drug resistance and tumor
recurrence.
Bufadienolides are the major effective constituents
of cinobufacini (also known as Huachansu), a well-known Chinese
medicine that comes from the dried skin of Bufo bufo gargarizans
Cantor, and cinobufacini has been used to treat patients with
various types of cancer, including hepatoma, gallbladder carcinoma
and lung cancer in China (6-8). The
authors of the present study recently demonstrated that active
bufadienolide compounds, including gamabufotalin and arenobufagin,
exhibit selective cytocidal effects against intractable cancer
cells, including in glioblastoma, but minimal effects on normal
peripheral blood mononuclear cells (PBMCs) prepared from healthy
volunteers (9). Notably, it was
demonstrated that nearly non-toxic gamabufotalin concentrations on
PBMCs efficiently downregulated the percentages of
CD4+CD25+Fop3+ regulatory T (Treg)
cells (9), which have been
considered to serve an important role in limiting antitumor immune
response in the body and promoting immunological ignorance of
cancer cells (10-12). These results suggest that
bufadienolides, including gamabufotalin and arenobufagin, may
provide a therapeutic benefit to patients with glioblastoma via
their cytocidal effects against tumor cells and antitumor
immunity-promoting characteristics. Despite the selective cytocidal
effects of bufadienolides against glioblastoma and their
possibility for further clinical applications, the detailed
molecular mechanisms of action of these active bufadienolides in
glioblastoma cells remain unexplored.
Cell cycle arrest, necrosis and apoptosis are
considered the major mechanisms underlying the cytocidal effects of
the majority of chemotherapeutic drugs (13-15).
A number of previous studies have demonstrated that active
bufadienolides, including bufalin, arenobufagin and hellebrigenin,
induce cytotoxicity associated with cell cycle arrest (16-18).
They have also been implicated in the inhibition of the
phosphatidylinositol 3-kinase (PI3K)/AKT serine/threonine kinase
(Akt) signaling pathway, and downregulation of essential regulators
of the cell cycle transition, such as Cdc25C and Cyclin B1, in
human hepatocellular carcinoma cells (16-18).
Survivin, a well-known cancer-associated protein that is highly
expressed in the majority of human tumor cells, has been associated
with an increase in the malignant potential of human glioma, and is
considered to serve an important role in mediating radiation
resistance in primary glioblastoma cells (19). In addition, members of the
mitogen-activated protein kinase (MAPK) family are involved in cell
cycle arrest, cell death signaling and the regulation of growth.
The MAPKs include p38 kinase, c-Jun NH2-terminal protein kinase
(JNK) and extra-cellular signal-regulated kinase (Erk) (20). Similar to Akt, Erk is usually
referred to as a survival mediator involved in cytoprotection
(21,22). Although p38 MAPK and JNK are
generally considered to be required for the induction of cell death
by diverse stimuli (21,23,24),
a novel prosurvival role for p38 MAPK has been reported in human
cancer cells, including in glioblastoma cells (25-27).
However, whether and how these molecules contribute to the
potential cytotoxic effects induced by active bufadienolides
against glioblastoma cells, and the association between these
molecules and cellular responses, including cell cycle arrest,
remain to be elucidated.
In the current study, the cytotoxicity of two active
bufadienolide compounds, arenobufagin and hellebrigenin, was
investigated in the human glioblastoma cell line U-87 by focusing
on proliferation inhibition associated with cell cycle arrest,
necrosis as well as apoptosis, in order to provide novel insight
into therapeutic approaches against glioblastoma. Primary mouse
astrocytes were also used to evaluate whether the two drugs
exhibited cytotoxic selectivity for cancer cells, rather than
normal cells. Key regulatory molecules involved in the cell cycle
and cell death were investigated to further elucidate the cytotoxic
mechanisms. Considering the controversial role of p38 MAPK in cell
death/survival in human cancer cells, the details of whether and
how p38 MAPK is implicated in active bufadienolides-mediated
cytocidal effects in U-87 cells were also investigated using its
specific inhibitor, SB203580.
Materials and methods
Materials
Arenobufagin was purchased from Baoji Herbest
Bio-Tech Co., Ltd. (Baoji, China). XTT, propidium iodide (PI),
proteinase K, Hoechst 33258 and ribonuclease A (RNaseA) were
purchased from Merck KGaA (Darmstadt, Germany). Dulbecco's modified
Eagle's medium (DMEM), phenazine methosulfate, agarose X and 25%
glutaraldehyde solution were purchased from Wako Pure Chemical
Industries, Ltd. (Osaka, Japan). Crystal violet (C.I. 42555)
Certistain® was purchased from Merck KGaA. SB203580, a
specific inhibitor of p38 MAPK, and its negative control SB202474
were purchased from Merck KGaA. Fetal bovine serum (FBS) was
obtained from Nichirei Biosciences, Inc. (Tokyo, Japan). High
performance liquid chromatography (HPLC)-grade acetonitrile and
methanol were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Mass spectrometry (MS)-grade formic acid was
obtained from ROE Scientific, Inc. (Newark, DE, USA).
Isolation and identification of
hellebrigenin from the dried skin of Bufo bufo gargarizans
Cantor
Dried skin of Bufo bufo gargarizans Cantor
was purchased from Anhui Jinchan Biochemistry Co., Ltd. (Huaibei,
China), and further identified by Professor Hongjie Wang (Institute
of Chinese Materia Medica, China Academy of Chinese Medical
Sciences, Beijing, China). The dried toad skin (10 kg) was cut into
pieces, and then extracted under reflux with 95% ethanol into 20
liters. The extracting solution was dried with rotary evaporation
at 45°C under reduced pressure (vacuum drying) to yield ~150 g
residue. Following separation through a silica gel (2,000 g;
160-200 mesh; Qingdao Haiyang Chemical Co., Ltd., Qingdao, China)
column chromatography with chloroform-methanol solution (50:1-1:1)
with gradient elution, a total of eight fractions were obtained
(Fr. 1-8). Fr. 4 (8 g) was further separated by C18 (320 ml;
SP-120-50-ODS-RPS; DAISO Co., Ltd., Osaka, Japan) column
chromatography using 30% (500 ml, Fr. 4.1-4.5), 40% (500 ml, Fr.
4.6-4.10), 50% (500 ml, Fr. 4.11-4.15) and 60% (400 ml, Fr.
4.16-4.19) methanol. Hellebrigenin was purified from Fr. 4.10-4.16
by preparative HPLC. It was obtained as a white powder with
molecular formula of C24H32O6
based on high-resolution electrospray ionization MS (HR-ESI-MS).
The compound was identified as hellebrigenin with >96% purity
according to previously reported values (28).
Cell culture and treatment
U-87, a human glioblastoma cell line, was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
cultured in DMEM supplemented with 10% heat-inactivated FBS, 100
U/ml of penicillin and 100 µg/ml of streptomycin (both from
Wako Pure Chemical Industries, Ltd.) in a humidified atmosphere
with 5% CO2 at 37°C. Primary mouse glial cells were
prepared according to a method previously reported with
modifications (29). Briefly, the
cerebral cortex of 4-5 newborn C57BL/6J mice [2-4 days old (sex
unconfirmed), body weight 2-3 g, housed at 23±1°C in a room with a
constant humidity of 55±5% and a regular 12-h light/12-h dark cycle
together with dams] was removed and digested by trituration in PBS
(Wako Pure Chemical Industries, Inc.) containing 0.25% trypsin.
Dissociated glial cells were cultured in 75 cm2 flasks
(BD Biosciences, Franklin Lakes, NJ, USA) in DMEM containing 10%
FBS in a humidified atmosphere with 5% CO2 at 37°C.
After 2 days of cultivation, the medium was changed
to remove unattached cells. Glial cells were allowed to form a
confluent monolayer for 7-9 days and harvested from the flask by
treatment with 0.125% trypsin. Subsequently, cells were seeded at a
density of 5×105 cells/ml in 96-well plates and allowed
to grow to ~80% confluency. The glial cell preparation was highly
enriched in astrocytes (>90%) according our previous report
(29). The U-87 and primary glial
cells were treated with SB203580, a specific inhibitor of p38 MAPK,
and its negative control SB202474 at various concentrations (1, 5
or 10 µM) for 30 min prior to treatment with arenobufagin
and hellebrigenin [20 ng/ml, almost equal to their half maximal
inhibitory concentration (IC50) values at 48 h], in the
presence or absence of these reagents. The present study was
approved by the Committee of Animal Care and Welfare of Tokyo
University of Pharmacy and Life Sciences (Tokyo, Japan;
registration no. P17-60).
Qualitative assessment of arenobufagin
and hellebrigenin in the cerebrospinal fluid of rats receiving a
single oral dose of arenobufagin or hellebrigenin
As mice are too small to successfully collect clear
cerebrospinal fluid without blood contamination, and there is no
clear difference in the structure and function of the blood-brain
barrier (BBB) in rats and mice, rats were used instead of mice for
the qualitative assessment. Nine male Sprague Dawley rats (220-240
g) were purchased from Shanghai Shibei Biotechnology Co., Ltd.
(Shanghai, China), and housed at 23±1°C in a room with a constant
humidity of 55±5% and a regular 12/12-h light/dark cycle for one
week prior to experimentation. Throughout the experiment, the rats
had free access to food and water. After an overnight fasting with
water not limited, all animals were randomly divided into three
groups (n=3) and administered the following treatments: Vehicle
control (saline), arenobufagin (50 mg/kg) and hellebrigenin (50
mg/kg). All animals received a single oral dose of 50 mg
arenobufagin/hellebrigenin or the same volume of saline/kg of body
weight. At 30 min after the oral administration, cerebrospinal
fluids were collected according to the method previously described
with slight modifications (30).
Briefly, the fur on the head and neck regions of rats were
carefully removed, and animals were placed in an induction chamber
and anesthetized. Following disinfection with ethanol, an ~2-cm
skin incision was made along the midline. The spinous process and
lamina were clamped with hemostatic forceps in order to make the
arachnoid membrane more visible. Then, the cerebrospinal fluid was
carefully extracted from the subarachnoid space using a 1-ml
syringe with a 23G needle without blood contamination. Following
centrifugation at 9,730 × g for 15 min at 4°C, the cerebrospinal
fluid supernatants were collected and stored at −20°C for later
analysis.
The analysis of cerebrospinal fluids was performed
using the ultimate 3000 hyperbaric LC system coupled with a Velos
Pro LTQ Orbitrap Mass Spectrometer via an ESI interface from Thermo
Fisher Scientific, Inc. Eclipse XDB-C18 (4.6×150 mm, 3.5 µm;
Agilent Technologies, Inc., Santa Clara, CA, USA) was used as the
separation column in the ultra HPLC. The mobile phase consisted of
0.1% formic acid in water (solvent A) and acetonitrile (solvent B),
with the gradient elution as follows: 0-5 min, 20-40% B; 5-20 min,
40-50% B; 20-21 min, 50-95% B; 21-26 min, held at 95% B; 26-33 min,
balanced to 20% B. The flow rate was 0.2 ml/min and the
temperature-controlled column was maintained at 30°C. Column
effluents were monitored at 296 nm following the injection of 4
µl cerebrospinal fluid obtained from the control and
treatment groups. MS detection was performed in the positive ion
mode. The ESI source parameters were as follows: Ion spray voltage,
+5.0 kV; sheath gas flow rate, 40 arb; aux gas flow rate, 10 arb;
capillary temperature, 350°C; and S-lens RF level, 60%. The mass
resolution of Fourier transform was 30,000 with a full scan in the
range of m/z 200-800. The tandem MS (MS/MS) experiments were
set as a data-dependent scan. The experimental procedures complied
with the Animal Ethics Committee Guidelines of Beijing Animals
Science Biology Technology Co., Ltd. (Beijing, China; registration
no. 170703002).
Cell viability, morphological alterations
and clonogenic survival
Following treatment with various concentrations (10,
20, 30, 40, 100 and 150 ng/ml) of arenobufagin and hellebrigenin
for 48 h, cell viability was measured using the XTT assay as
described previously (31).
Relative cell viability was expressed as the ratio of the
absorbance at 450 nm of each treatment group against those of the
corresponding untreated control group. The IC50 values
of each drug were calculated using GraphPad Prism® 6.0
software (GraphPad Software, Inc., La Jolla, CA, USA). With respect
to the morphological alterations of U-87 cells, the cells were
imaged using an inverted microscope (CKX53; Olympus Corporation,
Tokyo, Japan) fitted with a digital camera following treatment with
various concentrations (20, 40, 100, 150 and 200 ng/ml) of
arenobufagin and hellebrigenin for 48 h. Untreated cells were used
as the control. Clonogenic survival assays were performed according
to a method previously described, with slight modifications
(14). Briefly, U-87 cells were
seeded at a density of 5×103 cells/well in 6-well
plates, and treated with various concentrations (5, 10, 20, 30 and
40 ng/ml) of arenobufagin and hellebrigenin for 24 h. Untreated
cells were used as the control. The medium was then replaced with
fresh DMEM (supplemented with 10% heat-inactivated FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin) and the cells were
allowed to grow for 7-10 days in a humidified 5% CO2
atmosphere at 37°C. Following washing gently with PBS twice, the
cells were fixed with 0.25% glutaraldehyde/PBS for 15 min prior to
staining with 0.2% crystal violet/PBS for 10 min at room
temperature. Following a washout of extra crystal violet with water
to get an adequate staining pattern, the images of crystal
violet-stained cells were scanned into a computer, followed by
dissolution of the violet-stained cells in 1% SDS. The absorbance
of cell lysates was determined at 550 nm. The relative colony
formation rate was expressed as the ratio of the absorbance at 550
nm of each treatment group against those of the corresponding
untreated control group.
Cell cycle analysis
Following treatment with various concentrations (10,
20, 30 and 40 ng/ml) of arenobufagin or hellebrigenin for 48 h, and
20 ng/ml arenobufagin or hellebrigenin for 24, 48 or 72 h, cell
cycle analysis was performed using a FACSCanto™ flow cytometer (BD
Biosciences), according to a previous method (14,21).
Briefly, cells were washed twice with cold PBS, fixed with 1%
paraformaldehyde/PBS on ice for 30 min, washed twice again with
cold PBS, permeabilized in 70% (v/v) cold ethanol at −20°C for at
least 4 h. Cell pellets were then washed twice with cold PBS
following centrifugation (430 × g for 5 min at 4°C) and incubated
with 0.25% Triton X-100 for 5 min on ice. Following centrifugation
(430 × g for 5 min at 4°C) and washing with PBS, cells were
resuspended in 500 µl PI/RNase A/PBS (5 µg/ml PI and
0.1% RNase A in PBS) and incubated for 30 min in the dark at room
temperature. A total of 10,000 events were recorded, and FACSDiva™
software (v6.0; BD Biosciences) and ModFit LT™ v3.0 (Verity
Software House, Inc., Topsham, ME, USA) were used to calculate the
number of cells in each G0/G1, S and
G2/M phase fraction.
Western blot analysis
For preparation of the protein samples, cell pellets
(1-2×106 per 110 µl buffer) were suspended in
Laemmli buffer containing 100 mM DTT, 2 µg/ml leupeptin, 2
µg/ml aprotinin, 1 µg/ml pepstatin, 1 mM PMSF. Cell
suspensions were sonicated (Qsonica, LLC, Newtown, CT, USA) with 10
short bursts of 2 sec followed by intervals of 2 sec for cooling.
The suspensions were then kept in an ice bath. Sonicated cells were
heated in 95°C for 5 min, and then centrifuged at 13,000 × g for 15
min at 4°C. Protein concentrations of the supernatant were
determined according to Bradford's method using the Protein Assay
Dye Reagent Concentrate (Bio-Rad Laboratories, Inc.), according to
the manufacturer's instructions, using BSA as the standard. Western
blot analysis was carried out according to methods previously
described (31). Briefly, after
separation of proteins (10-20 µg protein/lane) via SDS-PAGE
(8, 10 or 12.5% according to the molecular weight of the target
protein), followed by transference to a polyvinylidene difluoride
(PVDF) membrane, which was then blocked with 5% skim milk/PBST (PBS
containing 0.5% Tween-20) for 1 h at room temperature. Protein
bands were detected using the following primary antibodies: Mouse
anti-human β-actin (1:5,000 dilution; cat. no. A-5441;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), rabbit anti-human
Cdc25c (1:1,000 dilution; cat. no. 4688), rabbit anti-human p27
(1:1,000 dilution; cat. no. 2552), mouse anti-human Cyclin B1
(1:2,000 dilution; cat. no. 4135), mouse anti-human survivin
(1:1,000 dilution; cat. no. 2802), rabbit anti-human phospho-Akt
(Ser473) (1:2,000 dilution; cat. no. 4060) and Akt (1:1,000
dilution; cat. no. 4691), rabbit anti-human phospho-p38
(Thr180/Tyr182, 1:1,000 dilution; cat. no. 9211) and p38 (1:1,000
dilution; cat. no. 9212), rabbit anti-human phospho-MK2 (Thr334)
(1:1,000 dilution; cat. no. 3041), rabbit anti-human phospho-ERK
(Thr202/Tyr2042) (1:2,000 dilution; cat. no. 4370) and ERK (1:1,000
dilution; cat. no. 4695; all from Cell Signaling Technology, Inc.,
Danvers, MA, USA). PVDF membranes containing blotted protein bands
were incubated overnight with the respective primary antibody at
4°C, followed by the appropriate horseradish peroxidase-conjugated
secondary antibody (anti-mouse IgG, 1:3,000 dilution, cat. no.
A5906; anti-rabbit IgG, 1:3,000 dilution, cat. no. A0545; both from
Sigma-Aldrich; Merck KGaA) for 1 h at room temperature, and then
detected with an enhanced chemiluminescence (ECL) analysis system
(Amersham; GE Healthcare Life Sciences, Chalfont, UK).
Lactate dehydrogenase (LDH) assay
Following treatment with various concentrations (10,
20, 40, 100, 150 and 200 ng/ml) of arenobufagin or hellebrigenin
for 48 h as described in the previous section of the cell viability
assay, LDH leakage from U-87 cells was measured using a LDH
cytotoxicity detection kit (Wako Pure Chemical Industries, Ltd.)
according to the method previously described with slight
modifications (31). Briefly,
culture medium served as the negative control (NC). Culture
supernatants (S) were collected by centrifugation at 450 × g for 5
min at 4°C and stored at −80°C until use. Cultured cells without
treatment were lysed in the culture medium containing 0.2%
Tween-20, and the cell lysate was used as the non-damaged positive
control (PC) following centrifugation at 12,000 × g for 5 min at
4°C. In order to avoid the influence of Tween-20, culture medium
containing 0.2% Tween-20 served as the negative control for PC and
was referred to as NCT. Samples were diluted 16-fold with PBS and
50 µl of the diluted solution was transferred into wells of
a 96-well plate. LDH activities were determined by adding 50
µl reaction reagent from the kit, followed by incubation at
room temperature for 30 min. The reaction was stopped by the
addition of 100 µl stopping solution provided with the kit
at room temperature, and the absorbance at 560 nm was measured with
a microplate reader (EMax® Plus; Molecular Devices, LLC,
Sunnyvale, CA, USA). Cell damage was calculated as a percentage of
LDH leakage from damaged cells using the following formula:
(S-NC)/(PC-NCT) × 100.
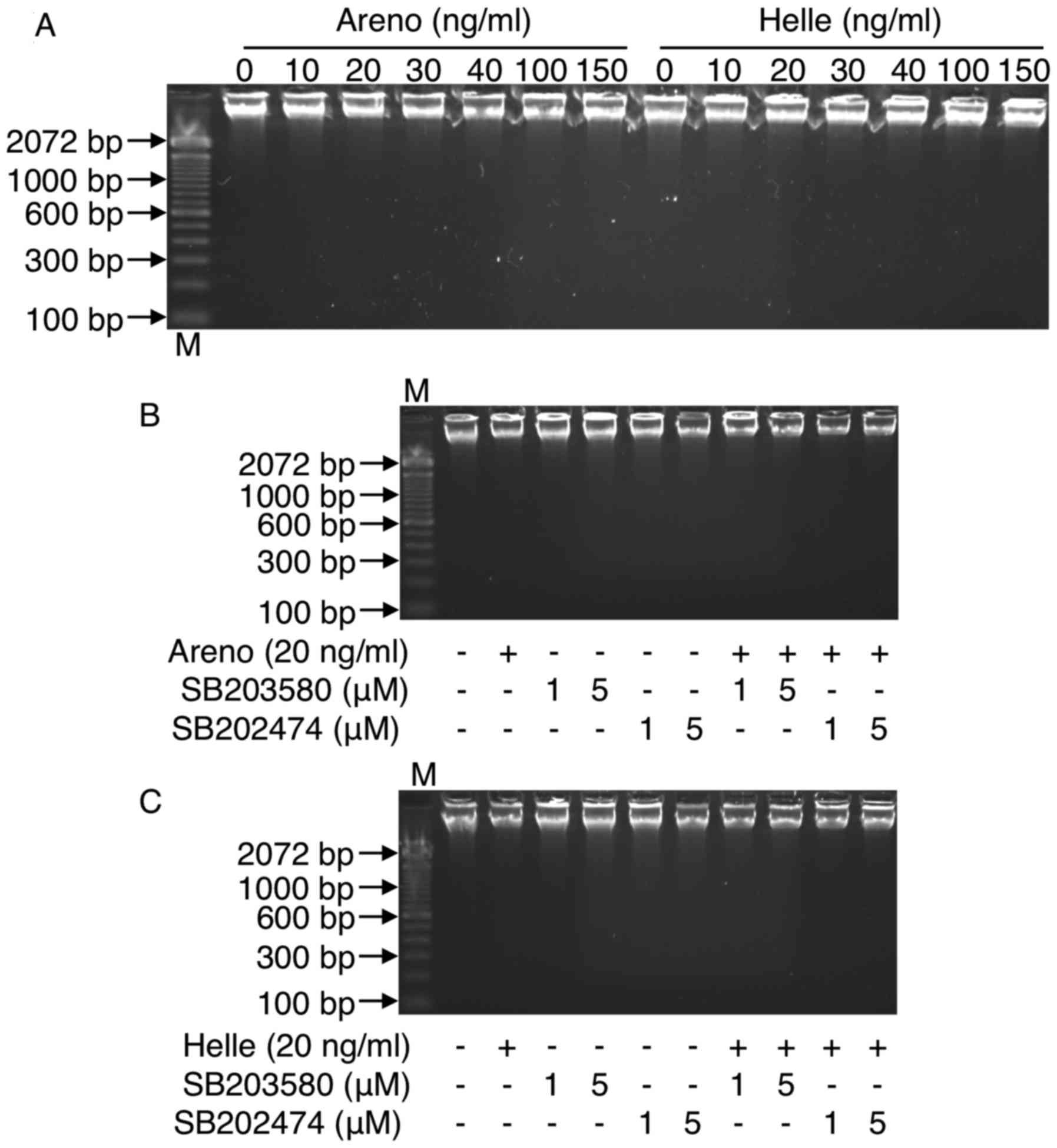
DNA fragmentation analysis
DNA fragmentation analysis was performed according
to a method described previously (32). Briefly, between 5×105
and 1×106 cells were used for the DNA preparation. The
cells were suspended in 500 µl lysis buffer (50 mM Tris-HCl,
pH 7.8, 10 mM EDTA-2Na, 0.5% sodium-N-lauroyl sarcosinate). The
suspension was incubated in an ice bath for 30 min following the
addition of 50 µl of 10% SDS with gentle agitation every 5
min. The lysates were incubated successively at 50°C for 90 min
with proteinase K (1 mg/ml), at 50°C for 30 min with RNase A (1
mg/ml) and at 60°C for 15 min following the addition of the same
volume of NaI solution [6.0 M NaI in 26 mM Tris-HCl, pH 8.0, 13 mM
EDTA-2Na, 10 mg/ml of bovine glycogen (Nacalai Tesque, Inc., Kyoto,
Japan)]. DNA was precipitated by addition of an equal volume of
100% isopropanol. DNA pellets were successively washed with 50%
isopropanol, 100% isopropanol and 70% ethanol. Extracted DNA was
dissolved in TE buffer (10 mM Tris-HCl, pH 7.8, 1 mM EDTA), and the
DNA concentrations were determined by staining with Hoechst 33258
as described (33). The DNA
samples (~20 µg DNA in 20 µl TE buffer) and 100 bp
DNA ladder (Invitrogen; Thermo Fisher Scientific, Inc.) as a DNA
size marker were electrophoresed on a 2% agarose X gel using TBE
buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA). Gels were
visualized using 0.5 µg/ml ethidium bromide staining (at
room temperature, 30 min), followed by viewing under a Luminograph
II chemiluminescent imaging system (ATTO Corporation, Tokyo,
Japan).
Statistical analysis
Experiments were independently repeated three times,
and the results are presented as the means ± standard deviation of
three assays. Statistical analysis was performed using GraphPad
Prism® 6 software. The Student's t-test was used to
compare sample means between two groups, and one-way analysis of
variance followed by Dunnett's post hoc test was used to compare
sample means among three or more groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
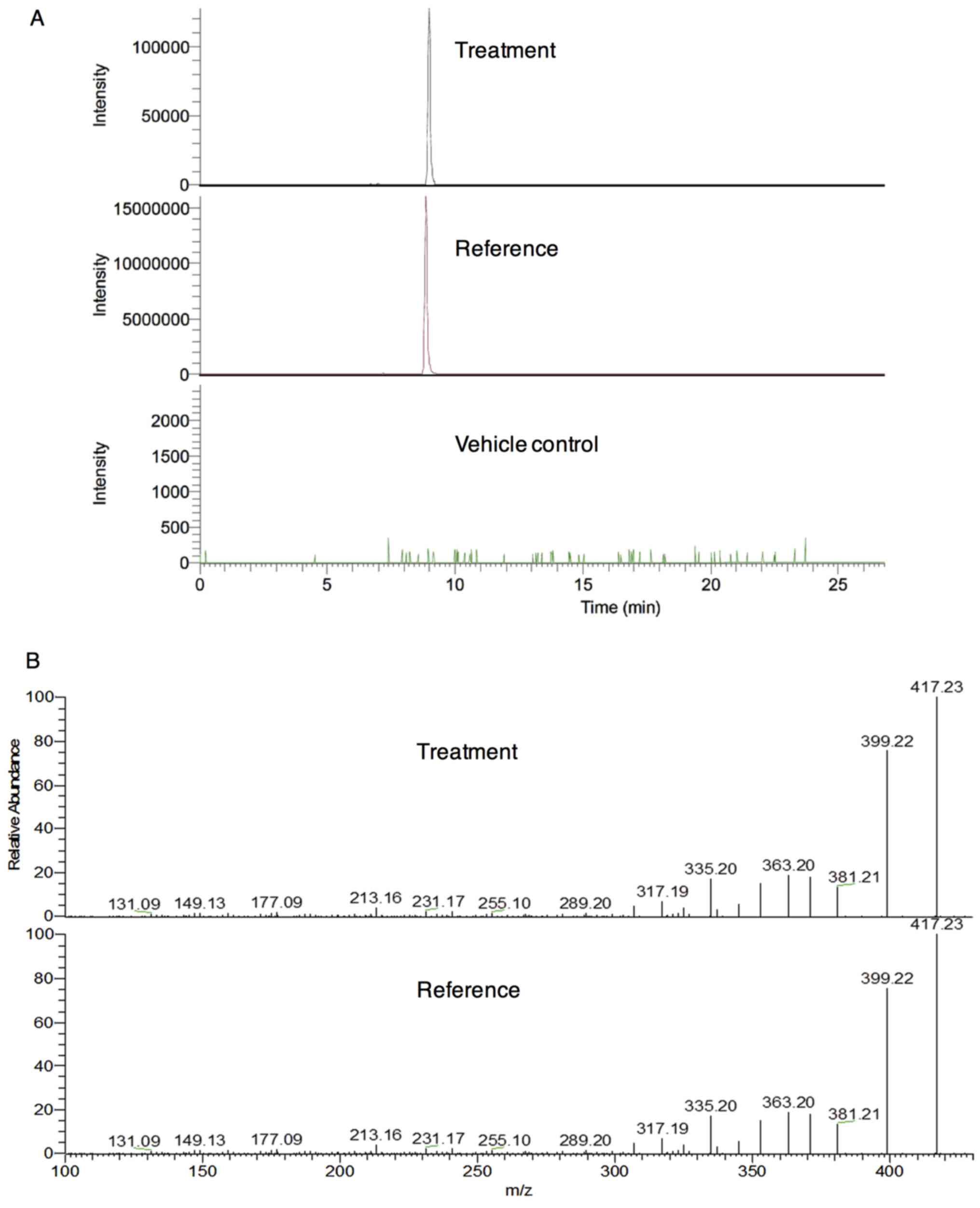
Presence of arenobufagin, but not
hellebrigenin, in cerebrospinal fluid of rats
First, whether the two active bufadienolides were
able to cross the BBB was investigated. The [M+H]+ ion
of arenobufagin at m/z 417.2264
(C24H33O6, Cal.417.2272, error
−1.88 ppm) with a retention time of 8.97 min was only detected in
the cerebrospinal fluid of rats who received a single oral dose of
arenobufagin, instead of saline (vehicle control) (Fig. 1A). However, the [M+H]+
ion of hellebrigenin at m/z 417.2277
(C24H33O6, Cal.417.2272, error
1.26 ppm) with a retention time of 8.91 min was hardly detected in
the cerebrospinal fluid of rats who received a single oral dose of
hellebrigenin due to its very low signal intensity. The further
identification of arenobufagin in cerebrospinal fluids was
performed using MS/MS experiments (Fig. 1B). These results indicated that
arenobufagin, but not hellebrigenin, were able to cross the
BBB.
Cytotoxic effects of arenobufagin and
hellebrigenin in human glioblastoma cell line U-87
A significant decrease in cell viability was
observed in a dose-dependent manner in U-87 cells following
treatment with various concentrations of arenobufagin and
hellebrigenin for 48 h. The IC50 values of arenobufagin
and hellebrigenin were 24.9±2.8 ng/ml and 23.5±2.4 ng/ml,
respectively (Fig. 2A and B). In
comparison, under the same treatment conditions, no cytocidal
effect was observed in mouse primary astrocytes when treated with
arenobufagin (Fig. 2C), and a
slight increase in viability was observed following treatment with
hellebrigenin at concentrations >10 ng/ml (Fig. 2D). Morphological alterations of
U-87 cells were also observed using an inverted microscope. As
shown in Fig. 2E and F, treatment
with arenobufagin and hellebrigenin at concentrations >100 ng/ml
for 48 h, caused a large number of U-87 cells to detach from the
plate. In contrast, concentrations <40 ng/ml of each drug
exhibited little effect on cell morphology, although an evident
decrease in the number of cells/field was observed, indicating an
inhibition of cell viability. In comparison, almost no
morphological alterations were observed in the primary astrocytes
following treatment with the two bufadienolides at concentrations
≤150 ng/ml (Fig. 2G).
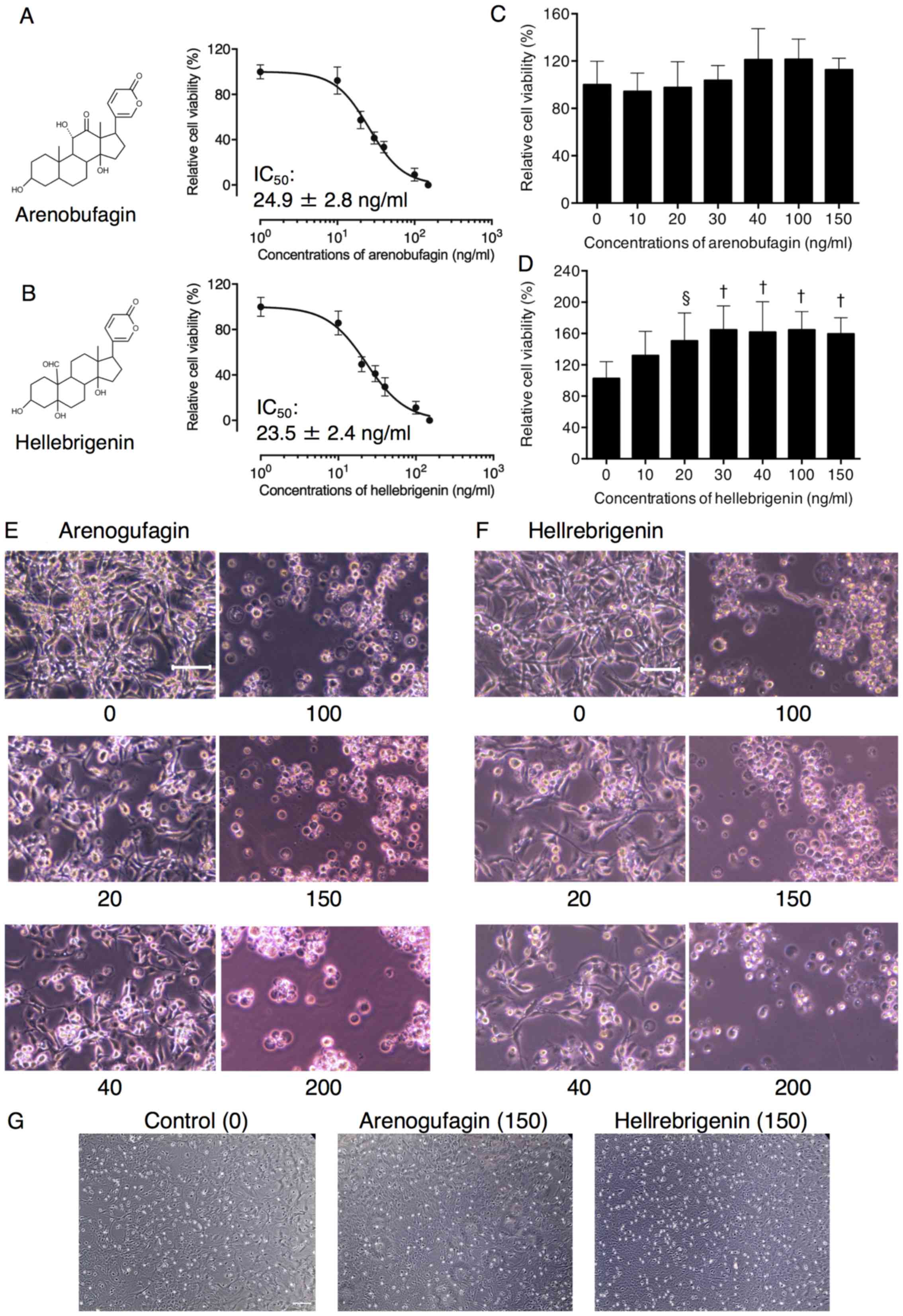 | Figure 2Cytotoxic effects of Areno and Helle
in human glioblastoma cell line, U-87. Cell viability of U-87 [(A)
Areno treatment; (B) Helle treatment and primary mouse astrocytes;
(C) Areno treatment; (D) Helle treatment] was determined using an
XTT assay following treatment with various concentrations (10, 20,
30, 40, 100 and 150 ng/ml) of each single drug for 48 h. Relative
cell viability was calculated as the ratio of the absorbance at 450
nm of each treatment group against those of the corresponding
untreated control group. Data are presented as the mean ± standard
deviation (n≥3). §P<0.001 and †P<0.0001
vs. control. Following treatment with various concentrations (20,
40, 100, 150 and 200 ng/ml) of (E) Areno and (F) Helle for 48 h,
the morphological alterations of U-87 were evaluated as described
in the materials and methods. (G) The morphological alterations of
primary mouse astrocytes were also evaluated following treatment
with as high as 150 ng/ml of Areno and Helle. Representative images
of the morphological alterations are shown from three independent
experiments. Scale bar, 100 µm. Areno, arenobufagin; Helle,
hellebrigenin. |
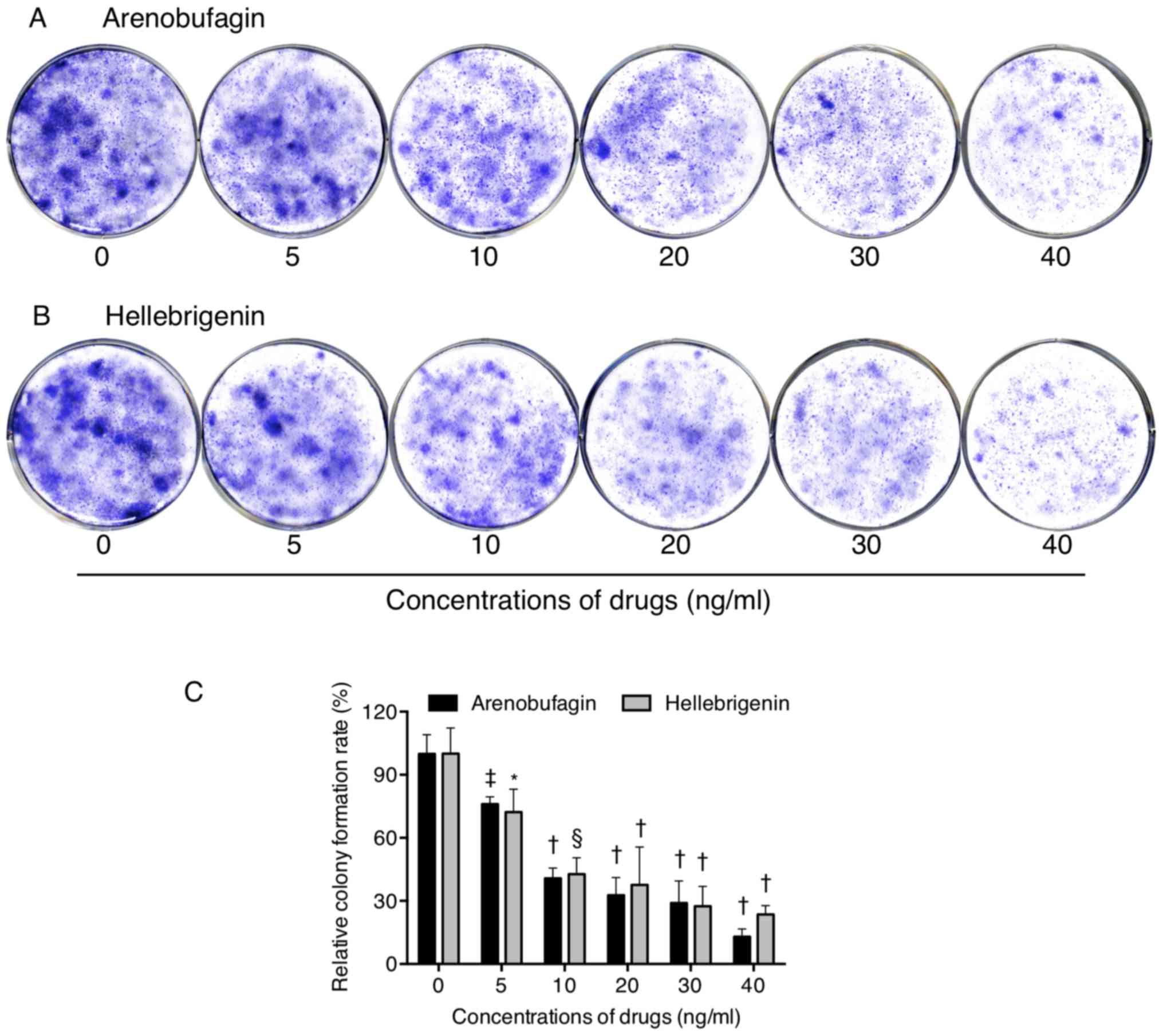
Inhibition of colony formation of U-87
cells by arenobufagin and hellebrigenin
To explore whether exposure to arenobufagin and
hellebrigenin suppressed the surviving fraction of U-87 cells, a
colony formation assay was performed. As shown in Fig. 3, significant suppression of the
colony numbers of U-87 was observed following long-term treatment
with arenobufagin or hellebrigenin, at concentrations ≥5 ng/ml,
indicating their potency against the survival of the cells.
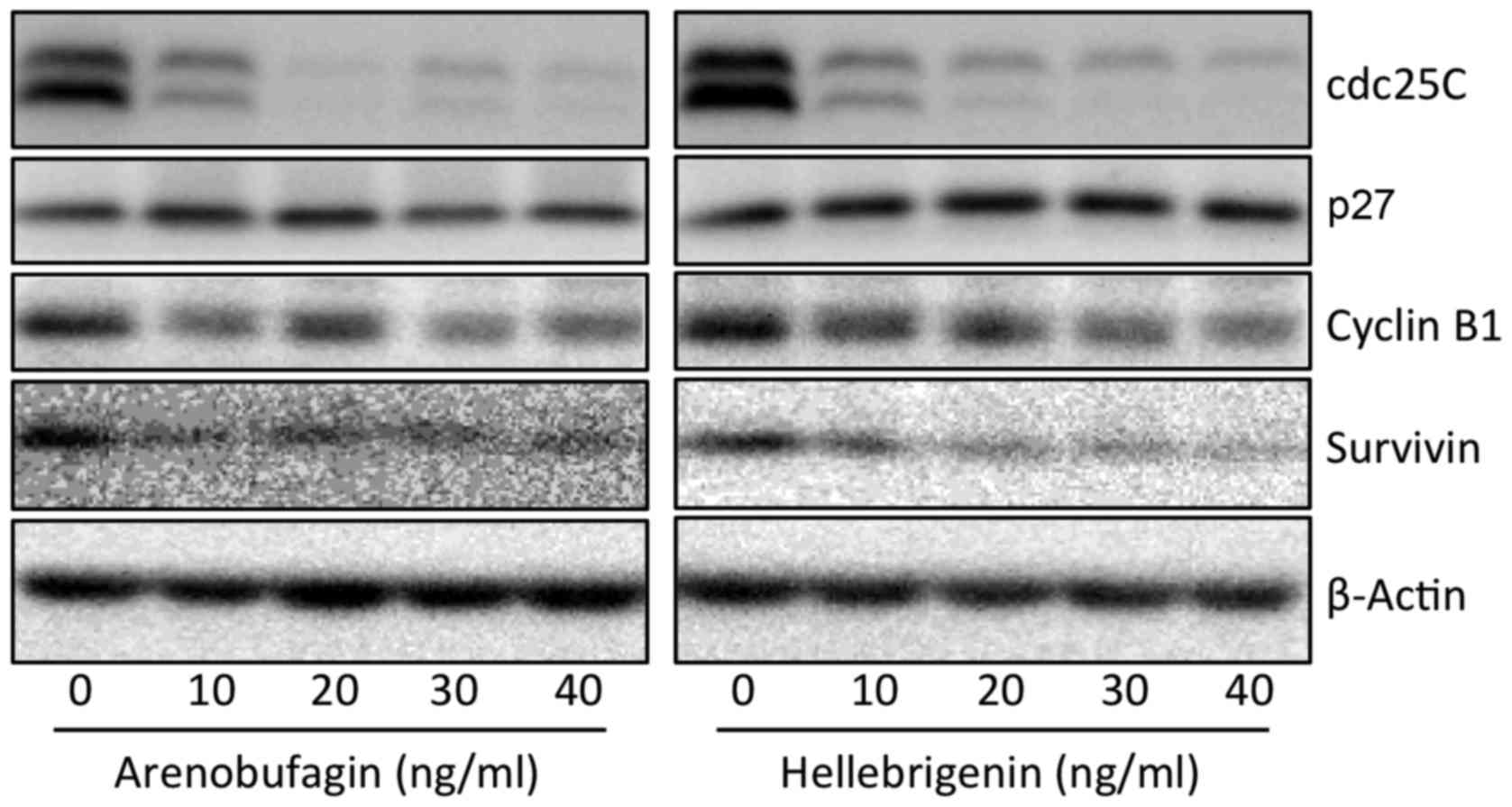
Effect of arenobufagin and hellebrigenin
on the cell cycle, and the expression level of cell
cycle-associated proteins in U-87 cells
To explore whether cell cycle arrest is involved in
the cytotoxic effects of arenobufagin and hellebrigenin, cell cycle
analyses were performed using flow cytometry (Figs. 4 and 5). In comparison with control group
(untreated cells), a significant increase in the number of cells in
the G2/M phase was observed (Figs. 4A and C, and 5A and C) following treatment with various
concentrations of the two drugs for 48 h. Concomitantly, a
significant decrease in the number of cells in the
G0/G1 and S phase was also observed (Figs. 4A and C, and 5A and C). Furthermore, following
treatment with arenobufagin or hellebrigenin at 20 ng/ml, which was
almost equal to their IC50 values, for 24, 48 and 72 h,
G2/M cell cycle arrest-inducing activity of each drug
was observed at 24-h post-treatment, which reached the maximum at
48-h post-treatment, and was sustained for up to 72 h (Figs. 4B and D, and 5B and D). A significant decrease in the
number of cells in the G0/G1 and S phase was
also concomitantly observed (Figs. 4B
and D, and 5B and D). As shown
in Fig. 6, compared with the
control group, the expression of Cdc25C and Cyclin B1 was
remark-ably downregulated following treatment with arenobufagin or
hellebrigenin in a dose-dependent manner, although almost no
evident alteration was observed in the expression levels of p27.
Similarly, a dose-dependent downregulation in the expression of
survivin was also observed.
 | Figure 4Effect of Areno on the U-87 cell
cycle profile. Following treatment with (A) various concentrations
of Areno (10, 20, 30 and 40 ng/ml) for 48 h, or (B) 20 ng/ml Areno
for 24, 48 and 72 h, cell cycle analysis was performed using a
FACSCanto™ flow cytometer as described in the materials and
methods. ModFit LT™ v3.0 was then used to calculate the number of
cells in each G0/G1, S and G2/M
phase fraction following treatment (C) with the indicated
concentrations of Areno for 48 h, or (D) with 20 ng/ml Areno for
the indicated time periods. A representative FACS histogram from
three independent experiments is shown. Data are presented as the
means ± standard deviation from three independent experiments.
*P<0.05, ‡P<0.01,
§P<0.001 and †P<0.0001 vs. the
respective control. Areno, arenobufagin. |
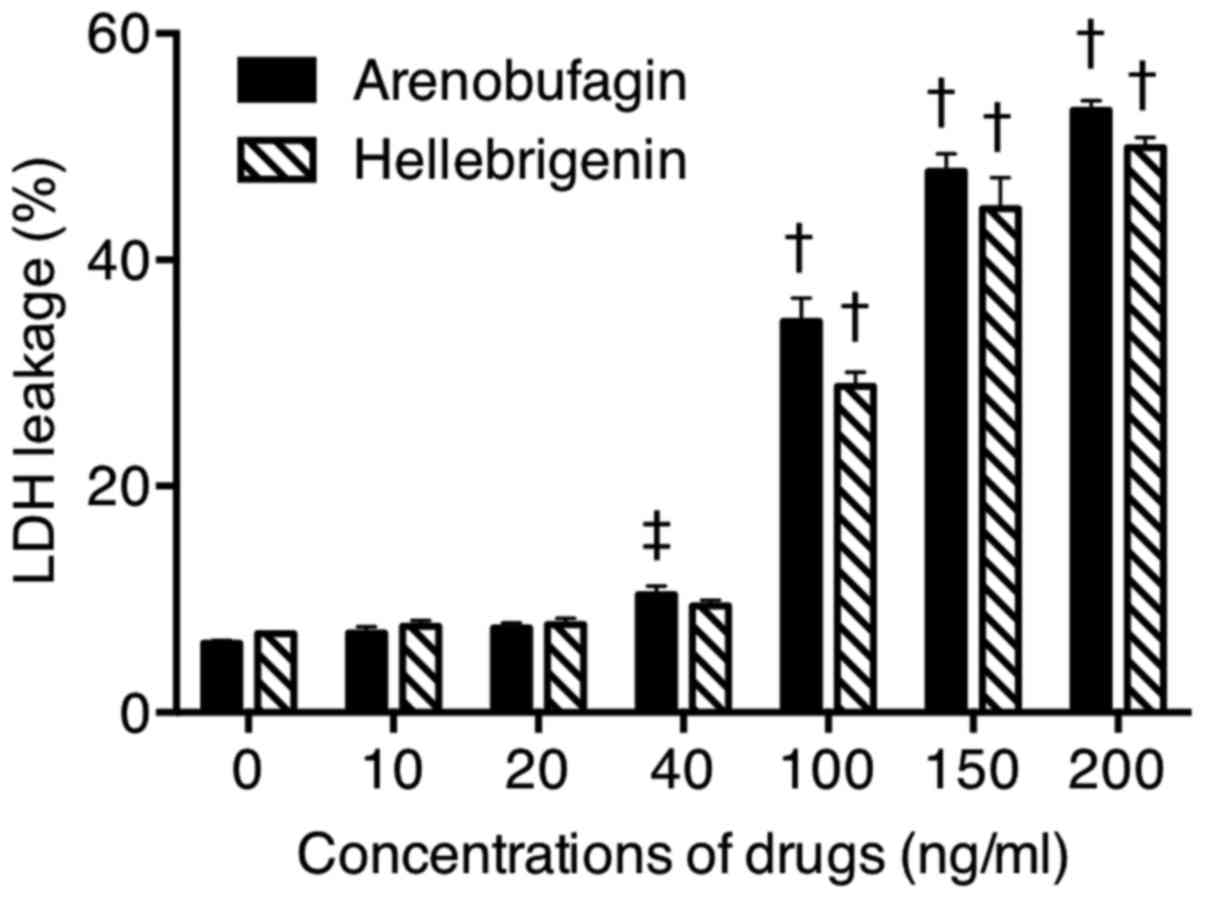
Effect of arenobufagin or hellebrigenin
on LDH release from U-87 cells
The release of LDH provides an accurate measure of
the cell membrane integrity and cell viability (13,14,31).
Following treatment with various concentrations of arenobufagin and
hellebrigenin for 48 h, LDH leakage analysis was performed to
examine whether the treatments affected cell membrane integrity. A
dose-dependent increased in LDH leakage was observed in U-87 cells
treated with arenobufagin or hellebrigenin (Fig. 7). Notably, no significant
difference was observed between cells treated with each drug at 20
ng/ml and their respective control group (0 ng/ml), indicating that
cell membrane damage was more likely due to relatively high
concentrations, but not relatively low concentrations, of each
drug.
Effect of arenobufagin or hellebrigenin
on DNA fragmentation in U-87 cells
To explore whether apoptosis is involved in the
cytotoxic effects of arenobufagin and hellebrigenin, DNA
fragmentation analysis was preformed using DNA gel electrophoresis.
As shown in Fig. 8, no DNA
fragmentation was observed in U-87 cells when treated with
arenobufagin or hellebrigenin, even at the concentrations >100
ng/ml, which significantly induced a large number of U-87 cells to
lose their adhesive properties (Fig.
2E and F) and LDH release (Fig.
7). These results indicated that apoptosis induction was less
likely to contribute to the cytotoxicity caused by arenobufagin or
hellebrigenin regardless of the drug concentration. These results
were also supported by the cell cycle analysis, demonstrating
almost no accumulation of cells in the sub-G1 phase, which
typically represents apoptotic cells (Figs. 4 and 5).
Effect of arenobufagin or hellebrigenin
on the activation of the PI3K/Akt and MAPK signaling pathway in
U-87 cells
To explore whether the PI3K/Akt and/or MAPK
signaling pathway are involved in the cytocidal effects of
arenobufagin and hellebrigenin, the activation of Akt, p38, Erk and
JNK was determined in U-87 cells following treatment with various
concentrations of arenobufagin and hellebrigenin for 48 h. As shown
in Fig. 9, a substantial increase
in the expression levels of phospho-p38 MAPK over the endogenous
levels was detected in the cells when treated with arenobufagin or
hellebrigenin, although almost no alterations in the expression
levels of total p38 MAPK expression was observed. Furthermore,
similar phenomena were observed in the expression levels of
phospho-MAPKAPK2 (MK2), a direct downstream target of p38 MAPK. In
contrast, the expression level of phospho-Akt was slightly
increased along with a modest decrease in the total Akt expression.
Similarly, a modest increase in the expression level of phospho-Erk
and total Erk was also observed in the cells, although the highest
concentrations of hellebrigenin (40 ng/ml) slightly downregulated
the expression of phospho-Erk. These results indicated that the
activation of p38 MAPK was involved in the cytocidal effects of
arenobufagin and hellebrigenin, although the partial participation
of Akt and Erk may not be completely excluded. In addition, the
expression level of phospho-JNK was not detected regardless of the
presence or absence of each drug, indicating that there was no
association between JNK and the cytocidal effects of the two
drugs.
 | Figure 9Effect of Areno or Helle on the
activation of the PI3K/Akt and MAPK signaling pathways in U-87
cells. Following treatment with various concentrations (10, 20, 30
and 40 ng/ml) of Areno and Helle for 48 h, the expression profiles
of both phosphorylated and/or total forms of Akt, p38, MK2 and Erk
were analyzed using western blotting. Representative images of the
expression profile of each protein are shown from three independent
experiments. p-Akt, p-p38, p-MK2 and p-Erk represent phospho-Akt,
phospho-p38, phospho-MK2 and phospho-Erk, their phosphorylated
active form, respectively. Areno, arenobufagin; Helle,
hellebrigenin. |
Enhancement of cytotoxicity of
arenobufagin and hellebrigenin by a specific inhibitor of p38 MAPK
in U-87 cells
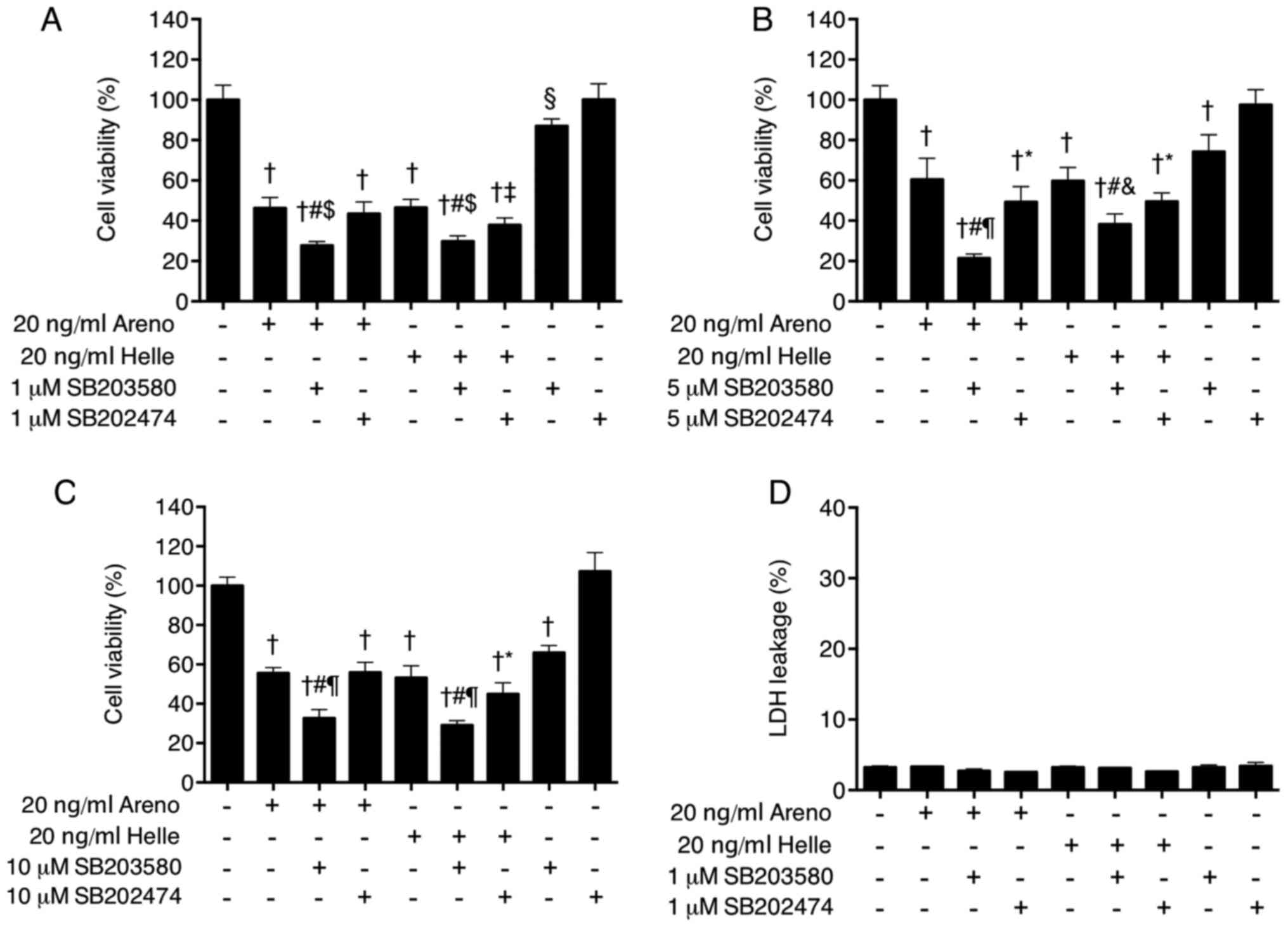
To provide further confirm the direct involvement of
p38 MAPK in the cytocidal effects of arenobufagin and
hellebrigenin, alterations to cell viability were determined in
U-87 cells following treatment for 48 h with 20 ng/ml arenobufagin
or hellebri-genin in the presence or absence of various
concentrations of SB203580, a specific inhibitor of p38 MAPK.
Consistent with the IC50 values of each drug (Fig. 2), treatment with 20 ng/ml of
arenobufagin or hellebrigenin alone significantly reduced cell
viability by ~50% (Fig. 10A–C).
Compared with the groups treated with arenobufagin or hellebrigenin
alone, combining various concentrations of SB203580 (1, 5 and 10
µM) with each drug significantly enhanced the cytotoxicities
of the two drugs (Fig. 10A–C).
Although significant reductions in the viability following
treatment with each drug in combination with SB202474, a negative
control for SB203580, were also observed compared with single drug
treatment, the efficacy of each drug was increased more by the
addition of SB203580 in comparison with SB202474, indicating the
critical role of p38 MAPK. Notably, a dose-dependent decease in the
cell viability was observed in cells treated with various
concentrations of SB202580 (1, 5 and 10 µM) alone, but not
SB202474 alone. In order to evaluate whether there was an
association between the activation of the p38 MAPK pathway and
other cellular responses, alterations to cell cycle arrest, LDH
leakage and DNA fragmentation were further determined in cells. No
alteration in LDH leakage (Fig.
10D), G2/M cell cycle arrest (Fig. 11) or DNA fragmentation (Fig. 8B and C) was observed in the cells
treated with a combination of each drug and SB203580 when compared
with treatment with each drug alone, indicating no association
between the activation of p38 MAPK signaling pathway and these
cellular responses.
Discussion
Due to the restrictive nature of the BBB, the
delivery of anticancer drugs to the central nervous system remains
a major challenge to the treatment of glioblastoma. Much effort has
been directed towards developing a novel system such as
nanoparticulate formulation for brain delivery of drugs (34,35).
In the present study, the qualitative assessment demonstrated for
the first time the existence of arenobufagin, but not
hellebrigenin, in the cerebrospinal fluid of rats who received a
single oral dose of the drug, directly indicating the capacity of
arenobufagin to cross the BBB. In fact, previous biodistribution
studies have demonstrated that other active bufadienolide compounds
with similar structures to arenobufagin, including bufotalin,
bufalin, resibufogenin and cinobufagin, were primarily distributed
in the brain, lung and spleen (35,36),
and the longest retention was observed in the brain (35), as a result of the intravenous
delivery of these compounds to mice. Compared with the chemical
structure of arenobufagin, hellebrigenin contains a formyl group
instead of a methyl group at position 19 of the molecule. Thus, it
was suggested that the difference in their chemical structure may
be associated with the differing ability to cross the BBB, although
further investigation is required to confirm this. Collectively,
these results indicate the possibility of the clinical application
of arenobufagin for patients with glioblastoma. However,
quantitative evaluation of arenobufagin levels in cerebrospinal
fluid is required to clarify whether its exact concentration is
adequate to achieve the inhibition of brain cancer cell
viability.
It was further demonstrated that arenobufagin and
hellebrigenin exhibited similar dose-dependent cytotoxic effects
against U-87 cells as evidenced by the XTT assay, as well as
morphological alterations and colony formation inhibitory
properties. In comparison, the same treatments did not cause any
detectable toxic effects in mouse primary astrocytes, and the
addition of hellebrigenin even resulted in enhanced viability of
the cells, although we are unable to provide a plausible
explanation for the phenomena at present. Similarly, it was
previously demonstrated that arenobufagin possesses selective
cytotoxic activity against human tumor cells, including U-87 and a
pancreatic cancer cell line SW1990 rather than PBMCs (9). These results provide further evidence
for the efficacy of arenobufagin and hellebrigenin against
cancerous glial cells with high potency, selectivity and
tolerability, although elucidation of their physiological
mechanisms underlying differential responses between tumor and
normal cells is required. In addition, Meng et al (8) demonstrated that cinobufacini
possesses effective anticancer activity in patients with
hepatocellular carcinoma and pancreatic cancer with low toxicity
and few side effects. Collectively, these findings provide a
rationale for the clinical application of arenobufagin and/or
hellebrigenin for patients with glioblastoma.
Active bufadienolide compounds, including bufalin,
arenobufagin and hellebrigenin, have been demonstrated to induce
G2/M cell cycle arrest in human hepatocellular carcinoma
cells (16-18). In line with these previous
findings, treatment with arenobufagin or hellebrigenin caused
dose-and time-dependent accumulation of cells in the
G2/M phase, along with a significant decrease in the
number in G0/G1 and S phase cells.
Concomitantly, a remarkable downregulation of the expression levels
of Cdc25C and Cyclin B1, key regulators involved in the
G2/M cell cycle transition (37,38),
was further observed in the treated cells compared with the control
(untreated cells). A number of previous studies have demonstrated
that in glioblastoma cell lines, downregulation of the expression
levels of Cdc25C and Cyclin B1 has been implicated in the
G2/M phase arrest induced by various antitumor agents,
including arsenic trioxide (39),
tivozanib, a pan-inhibitor of vascular endothelial growth factor
(40), and knockdown of β-arrestin
1, a mediator of tachykinin receptor neurokinin-1 signaling pathway
responsible for cell proliferation (41). Survivin, an important
cancer-associated protein, is highly expressed in the majority of
human tumor cells, including primary human glioblastoma cells
(19), and its inhibition has been
considered as a compelling strategy for cancer therapy (42). It has been demonstrated that
nuclear survivin expression may be a useful biomarker for
predicting the prognosis in patients with glioblastoma (43). Furthermore, suppression of survivin
by small interfering RNA results in reduced proliferation and
G2/M cell cycle arrest in human cancer cells, including
HeLa, a human cervical cancer cell line, and MCF-7, a human breast
cancer cell line (44). In the
present study, similar to Cdc25C and Cyclin B1, a dose-dependent
downregulation of survivin expression was observed, suggesting
survivin serves an important role in the G2/M phase
arrest induced by arenobufagin and hellebrigenin.
It was further demonstrated that necrotic cell death
was observed in U-87 cells when treated with a relatively high
concentration (>100 ng/ml), but not a relatively low
concentration (<40 ng/ml) of each drug as evidence by the LDH
leakage assay. However, apoptotic DNA fragmentation was not
observed in the cells regardless of drug concentrations used,
although arenobufagin and hellebrigenin have been reported to
induce apoptosis in human hepatocellular carcinoma cells (16,45),
suggesting whether induction of apoptosis by arenobufagin or
hellebrigenin may be cell-specific. Collectively, rather than
apoptosis induction, cell cycle arrest and necrotic cell death are
more likely to contribute the cytotoxicity induced by a relatively
low and high concentration of each drug.
Increasing evidence has indicated an important role
for the PI3K/Akt signaling pathway in the regulation of cell
survival and death in response to diverse stimuli, including the
majority of chemotherapeutic agents (46,47).
In addition, members of the MAPK family, consisting of p38, Erk and
JNK, are also involved in a range of cellular functions, including
proliferation, cell cycle arrest and cell death (26,27,48,49).
In the present study, evident activation of p38 MAPK pathway was
observed in U-87 cells when treated with arenobufagin or
hellebrigenin as evidenced by a marked increase in the expression
levels of phospho-p38 and its direct downstream target, phospho-MK2
(25). In comparison to the
expression pattern of phospho-p38, only a modest increase in the
expression level of phospho-Akt and phospho-Erk was observed in the
treated cells, and no expression of phospho-JNK was detected
regardless of treatment with the two drugs. These results thus
suggest that the cytotoxicities of arenobufagin and hellebrigenin
are primarily associated with p38 MAPK, rather than Akt, Erk and
JNK, and led to the exploration of the possible functional
interactions between the activation of the p38 MAPK pathway and the
cytotoxicities of the two drugs. It was surprising that a specific
inhibitor of p38 MAPK per se caused a significant decrease in the
cell viability, and enhanced the cytotoxicity of arenobufagin or
hellebrigenin as evidenced by the XTT assays, suggesting that p38
MAPK serves an important pro-survival role in the cells. In
agreement with these findings, previous studies have demonstrated a
pro-survival role for p38 MAPK in different types of cells,
including human glioblastoma cells (48,50).
Sooman et al (27) reported
that p38 MAPK phosphorylation may be a prognostic marker for
patients with high-grade glioma, and vandetanib combined with a p38
MAPK inhibitor may be a useful combination chemotherapy for
patients with glioma. Considering the results of previous studies
and the observations in the present study, we hypothesized that
combining a p38 MAPK inhibitor with arenobufagin or hellebrigenin
may improve the efficacy of both drugs, and may provide more
therapeutic benefits to patients with glioblastoma, although the
precise contribution of the p38 MAPK pathway to the cytotoxicity of
the two drugs warrants further investigation in a glioblastoma
xenograft model. Although the activation of p38 MAPK has been
associated with cell cycle arrest, apoptotic and necrotic cell
death in other cancer cells, including HL-60, a human promyelocytic
cell line and MCF-7 (21,26), no alteration in G2/M
cell cycle arrest, LDH leakage or DNA fragmentation was observed in
U-87 cells treated with the two drugs combined with a specific
inhibitor of p38 MAPK when compared with treatment with each drug
alone. This suggests that these cellular responses are independent
of the activation of the p38 MAPK pathway. These findings are
supported by a previous study, which revealed that p38 MAPK
promotes cell survival in response to DNA damage, but is not
required for the G2 DNA damage checkpoint in human
cancer cells, including HeLa and A549, a human lung cancer cell
line (25).
A report revealed that U87 cells are not the
original glioblastoma cell line that was established in 1968 at the
University of Uppsala (51). The
authors of the report demonstrated that the cell line is of central
nervous system origin and is likely to be a bona fide human
glioblastoma cell line, with an unknown patient origin. Marampon
et al (52) demonstrated
that histone deacetylase (HDAC)4 and 6 expression is significantly
associated with negative prognostic factors in patients with
glioblastoma multiforme, treated with temozolomide and
radiotherapy. They hypothesized that HDAC4 and/or HDAC6 knockdown
may lead to chromatin decondensation, favoring DNA damage. By using
U87 cells stably transfected with HDAC4 or HDAC6 short hairpin RNA,
it was demonstrated that these two HDACs differentially regulated
the molecular mechanisms responsible for DNA double-strand break
repair, by protecting glioblastoma multiforme cells from cell death
mediated by radiotherapy-induced senescence, apoptosis or autophagy
(52). These results demonstrate
that scientific findings observed in the clinical samples of
patients with glioblastoma multiforme may be successfully verified
in the U87 cells line despite the aforementioned misidentification
issue, suggesting the outcomes of the current study are appropriate
for the evaluation of the effects of arenobufagin and
hellebrigenin.
The present study demonstrated that arenobufagin and
hellebrigenin exhibited distinct cytotoxicity against cancerous
glial cells with high potency, selectivity and tolerability, and
further demonstrated that the inhibition of cell proliferation and
colony formation, G2/M cell cycle arrest as well as
necrotic cell death were attributed to their toxicities.
Considering the important role of survivin in diverse cellular
responses in various types of cancer cells, including glioblastoma
(19,42,43),
the cytotoxicity of both drugs against cancer cells may be
primarily attributed to the downregulation of survivin expression.
The efforts to evaluate the mechanisms underlying functional
interactions between survivin and cellular responses, including
cell cycle arrest and necrotic cell death, in the treated U-87
cells in our laboratory are ongoing. Given that p38 MAPK serves an
essential role in promoting glioblastoma cell survival (27,50),
and as an optimal chemotherapeutic regimen for glioblastoma has not
been defined at present (53,54),
developing a combination regimen of arenobufagin/hellebrigenin plus
a p38 MAPK inhibitor with the aim of preventing the activation of
the p38 MAPK pathway may provide novel insight into approaches
designed to treat glioblastoma. To the best of our knowledge, the
results of the present study demonstrated, for the first time, the
existence of arenobufagin in cerebrospinal fluid of rats who
received a single oral dose of the drug, supporting the clinical
application of arenobufagin and/or hellebrigenin for patients with
glioblastoma.
Acknowledgments
Not applicable.
Funding
This study was supported in part by grants from the
National Standardization Project of Chinese Medicine (grant no.
ZYBZH-C-AH-01).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LH and BY contributed equally to this study; LH, RS
and BY performed experiments; BY and LH analyzed results and
presented; HH, NS, HZ, BB and NT assisted interpretation of the
result with BY; BY designed the research and wrote the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental procedures complied with the Animal
Ethics Committee Guidelines of Beijing Animals Science Biology
Technology Co., Ltd. (Beijing, China; registration no. 170703002)
and was approved by the Committee of Animal Care and Welfare of
Tokyo University of Pharmacy and Life Sciences (Tokyo, Japan;
registration no. P17-60).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Tonn JC and Brada M: High-grade
malignant glioma: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 21(Suppl 5): v190–v193. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sathornsumetee S, Reardon DA, Desjardins
A, Quinn JA, Vredenburgh JJ and Rich JN: Molecularly targeted
therapy for malignant glioma. Cancer. 110:13–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Z, Zhai XF, Su YH, Wan XY, Li J, Xie
JM and Gao B: Clinical observation of cinobufacini injection used
to treat moderate and advanced primary liver cancer. Zhong Xi Yi
Jie He Xue Bao. 1:184–186. 2003.In Chinese. View Article : Google Scholar
|
|
7
|
Qin TJ, Zhao XH, Yun J, Zhang LX, Ruan ZP
and Pan BR: Efficacy and safety of gemcitabine-oxaliplatin combined
with huachansu in patients with advanced gallbladder carcinoma.
World J Gastroenterol. 14:5210–5216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge
Y, Newman RA, Cohen L, Liu L, Thornton B, et al: Pilot study of
huachansu in patients with hepatocellular carcinoma, nonsmall-cell
lung cancer, or pancreatic cancer. Cancer. 115:5309–5318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan B, He J, Kisoh K, Hayashi H, Tanaka
S, Si N, Zhao HY, Hirano T, Bian B and Takagi N: Effects of active
bufadienolide compounds on human cancer cells and
CD4+CD25+Foxp3+ regulatory T cells
in mitogen-activated human peripheral blood mononuclear cells.
Oncol Rep. 36:1377–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Facciabene A, Motz GT and Coukos G:
T-regulatory cells: Key players in tumor immune escape and
angiogenesis. Cancer Res. 72:2162–2171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maruyama T, Kono K, Mizukami Y, Kawaguchi
Y, Mimura K, Watanabe M, Izawa S and Fujii H: Distribution of Th17
cells and FoxP3(+) regulatory T cells in tumor-infiltrating
lymphocytes, tumor-draining lymph nodes and peripheral blood
lymphocytes in patients with gastric cancer. Cancer Sci.
101:1947–1954. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakaguchi S: Naturally arising
Foxp3-expressing CD25+CD4+ regulatory T cells
in immunological tolerance to self and non-self. Nat Immunol.
6:345–352. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kon A, Yuan B, Hanazawa T, Kikuchi H, Sato
M, Furutani R, Takagi N and Toyoda H: Contribution of membrane
progesterone receptor α to the induction of progesterone-mediated
apoptosis associated with mitochondrial membrane disruption and
caspase cascade activation in Jurkat cell lines. Oncol Rep.
30:1965–1970. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao M, Yuan B, Wang X, Sato A, Sakuma K,
Kaneko K, Komuro H, Okazaki A, Hayashi H, Toyoda H, et al:
Synergistic cytotoxic effects of arsenite and tetrandrine in human
breast cancer cell line MCF-7. Int J Oncol. 51:587–598. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan B, Okusumi S, Yoshino Y, Moriyama C,
Tanaka S, Hirano T, Takagi N and Toyoda H: Delphinidin induces
cyto-toxicity and potentiates cytocidal effect in combination with
arsenite in an acute promyelocytic leukemia NB4 cell line. Oncol
Rep. 34:431–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng LJ, Hu LP, Peng QL, Yang XL, Bai LL,
Yiu A, Li Y, Tian HY, Ye WC and Zhang DM: Hellebrigenin induces
cell cycle arrest and apoptosis in human hepatocellular carcinoma
HepG2 cells through inhibition of Akt. Chem Biol Interact.
219:184–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng LJ, Peng QL, Wang LH, Xu J, Liu JS,
Li YJ, Zhuo ZJ, Bai LL, Hu LP, Chen WM, et al: Arenobufagin
intercalates with DNA leading to G2 cell cycle arrest via ATM/ATR
pathway. Oncotarget. 6:34258–34275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu CM, Tsai Y, Wan L and Tsai FJ: Bufalin
induces G2/M phase arrest and triggers autophagy via the TNF, JNK,
BECN-1 and ATG8 pathway in human hepatoma cells. Int J Oncol.
43:338–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chakravarti A, Zhai GG, Zhang M, Malhotra
R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q and Loeffler J:
Survivin enhances radiation resistance in primary human
glioblastoma cells via caspase-independent mechanisms. Oncogene.
23:7494–7506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kikuchi H, Yuan B, Yuhara E, Takagi N and
Toyoda H: Involvement of histone H3 phosphorylation through p38
MAPK pathway activation in casticin-induced cytocidal effects
against the human promyelocytic cell line HL-60. Int J Oncol.
43:2046–2056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao J, Qian CJ, Ye B, Zhang X and Liang Y:
ERK inhibition enhances TSA-induced gastric cancer cell apoptosis
via NF-κB-dependent and Notch-independent mechanism. Life Sci.
91:186–193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kikuchi H, Yuan B, Yuhara E, Imai M,
Furutani R, Fukushima S, Hazama S, Hirobe C, Ohyama K, Takagi N, et
al: Involvement of histone H3 phosphorylation via the activation of
p38 MAPK pathway and intracellular redox status in cytotoxicity of
HL-60 cells induced by Vitex agnus-castus fruit extract. Int J
Oncol. 45:843–852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zanotto-Filho A, Braganhol E, Battastini
AM and Moreira JC: Proteasome inhibitor MG132 induces selective
apoptosis in glioblastoma cells through inhibition of PI3K/Akt and
NFkappaB pathways, mitochondrial dysfunction, and activation of
p38-JNK1/2 signaling. Invest New Drugs. 30:2252–2262. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Phong MS, Van Horn RD, Li S,
Tucker-Kellogg G, Surana U and Ye XS: p38 mitogen-activated protein
kinase promotes cell survival in response to DNA damage but is not
required for the G(2) DNA damage checkpoint in human cancer cells.
Mol Cell Biol. 30:3816–3826. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pereira L, Igea A, Canovas B, Dolado I and
Nebreda AR: Inhibition of p38 MAPK sensitizes tumour cells to
cisplatin-induced apoptosis mediated by reactive oxygen species and
JNK. EMBO Mol Med. 5:1759–1774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sooman L, Lennartsson J, Gullbo J,
Bergqvist M, Tsakonas G, Johansson F, Edqvist PH, Pontén F, Jaiswal
A, Navani S, et al: Vandetanib combined with a p38 MAPK inhibitor
synergistically reduces glioblastoma cell survival. Med Oncol.
30:6382013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Heerden FR and Vleggaar R: A revised
13C NMR spectral assignment of hellebrigenin. Magn Reson Chem.
26:464–467. 1988. View Article : Google Scholar
|
|
29
|
Hayashi H, Campenot RB, Vance DE and Vance
JE: Glial lipo-proteins stimulate axon growth of central nervous
system neurons in compartmented cultures. J Biol Chem.
279:14009–14015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cao YD, Zhang LZ, Wang MS, Tang TY and Su
WH: Two methods to collect cerebrospinal fluid in SD rat. Acta Acad
Med XuZhou. 25:317–319. 2005.
|
|
31
|
Yoshino Y, Yuan B, Kaise T, Takeichi M,
Tanaka S, Hirano T, Kroetz DL and Toyoda H: Contribution of
aquaporin 9 and multidrug resistance-associated protein 2 to
differential sensitivity to arsenite between primary cultured
chorion and amnion cells prepared from human fetal membranes.
Toxicol Appl Pharmacol. 257:198–208. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yuan B, Ohyama K, Bessho T, Uchide N and
Toyoda H: Imbalance between ROS production and elimination results
in apoptosis induction in primary smooth chorion trophoblast cells
prepared from human fetal membrane tissues. Life Sci. 82:623–630.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cesarone CF, Bolognesi C and Santi L:
Improved microfluorometric DNA determination in biological material
using 33258 Hoechst. Anal Biochem. 100:188–197. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kreuter J: Nanoparticulate systems for
brain delivery of drugs. Adv Drug Deliv Rev. 47:65–81. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li F, Weng Y, Wang L, He H, Yang J and
Tang X: The efficacy and safety of bufadienolides-loaded
nanostructured lipid carriers. Int J Pharm. 393:203–211. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu CL and Hou HM: Plasma pharmacokinetics
and tissue distribution of bufotalin in mice following single-bolus
injection and constant-rate infusion of bufotalin solution. Eur J
Drug Metab Pharmacokinet. 35:115–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gavet O and Pines J: Progressive
activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell.
18:533–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Perdiguero E and Nebreda AR: Regulation of
Cdc25C activity during the meiotic G2/M transition. Cell Cycle.
3:733–737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao S, Tsuchida T, Kawakami K, Shi C and
Kawamoto K: Effect of As2O3 on cell cycle progression and cyclins
D1 and B1 expression in two glioblastoma cell lines differing in
p53 status. Int J Oncol. 21:49–55. 2002.PubMed/NCBI
|
|
40
|
Momeny M, Moghaddaskho F, Gortany NK,
Yousefi H, Sabourinejad Z, Zarrinrad G, Mirshahvaladi S, Eyvani H,
Barghi F, Ahmadinia L, et al: Blockade of vascular endothelial
growth factor receptors by tivozanib has potential anti-tumour
effects on human glioblastoma cells. Sci Rep. 7:440752017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang YX, Li XF, Yuan GQ, Hu H, Song XY,
Li JY, Miao XK, Zhou TX, Yang WL, Zhang XW, et al: β-Arrestin 1 has
an essential role in neurokinin-1 receptor-mediated glioblastoma
cell proliferation and G2/M phase transition. J Biol Chem.
292:8933–8947. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen X, Duan N, Zhang C and Zhang W:
Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic
Strategies. J Cancer. 7:314–323. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shirai K, Suzuki Y, Oka K, Noda SE, Katoh
H, Suzuki Y, Itoh J, Itoh H, Ishiuchi S, Sakurai H, et al: Nuclear
survivin expression predicts poorer prognosis in glioblastoma. J
Neurooncol. 91:353–358. 2009. View Article : Google Scholar
|
|
44
|
Li Y, Liu D, Zhou Y, Li Y, Xie J, Lee RJ,
Cai Y and Teng L: Silencing of survivin expression leads to reduced
proliferation and cell cycle arrest in cancer cells. J Cancer.
6:1187–1194. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang DM, Liu JS, Deng LJ, Chen MF, Yiu A,
Cao HH, Tian HY, Fung KP, Kurihara H, Pan JX, et al: Arenobufagin,
a natural bufadienolide from toad venom, induces apoptosis and
autophagy in human hepatocellular carcinoma cells through
inhibition of PI3K/Akt/mTOR pathway. Carcinogenesis. 34:1331–1342.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gutiérrez-Uzquiza Á, Arechederra M,
Bragado P, Aguirre-Ghiso JA and Porras A: p38α mediates cell
survival in response to oxidative stress via induction of
antioxidant genes: Effect on the p70S6K pathway. J Biol Chem.
287:2632–2642. 2012. View Article : Google Scholar
|
|
49
|
Chen Y, Yang W, Zhang X, Yang S, Peng G,
Wu T, Zhou Y, Huang C, Reinach PS, Li W, et al: MK2 inhibitor
reduces alkali burn-induced inflammation in rat cornea. Sci Rep.
6:281452016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yoshino Y, Aoyagi M, Tamaki M, Duan L,
Morimoto T and Ohno K: Activation of p38 MAPK and/or JNK
contributes to increased levels of VEGF secretion in human
malignant glioma cells. Int J Oncol. 29:981–987. 2006.PubMed/NCBI
|
|
51
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Marampon F, Megiorni F, Camero S,
Crescioli C, McDowell HP, Sferra R, Vetuschi A, Pompili S, Ventura
L, De Felice F, et al: HDAC4 and HDAC6 sustain DNA double strand
break repair and stem-like phenotype by promoting radioresistance
in glioblastoma cells. Cancer Lett. 397:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fine HA, Dear KB, Loeffler JS, Black PM
and Canellos GP: Meta-analysis of radiation therapy with and
without adjuvant chemotherapy for malignant gliomas in adults.
Cancer. 71:2585–2597. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stewart LA: Chemotherapy in adult
high-grade glioma: A systematic review and meta-analysis of
individual patient data from 12 randomised trials. Lancet.
359:1011–1018. 2002. View Article : Google Scholar : PubMed/NCBI
|