Introduction
The ability to evade apoptosis is one of the
hallmarks of cancer and is a crucial property of cancer cells that
confers them resistance to chemotherapeutic agents (1,2).
Understanding apoptotic resistance may assist in the development of
strategies with which to restore the sensitivity of cancer cells to
apoptosis and, ultimately, may improve the efficacy of cancer
therapy. In lung cancer, various epidermal growth factor receptor
(EGFR)-tyrosine kinase inhibitor (TKI) resistance mechanisms have
been identified, such as the second-site EGFR mutation, T790M, the
activation of the bypass path-ways, MET and AXL, and histological
transformation, and several efforts have been made to overcome
these resistance mechanisms (3,4). A
common apoptosis-associated EGFR-TKI mechanism in lung cancer is an
intrinsic deletion polymorphism in the gene encoding BIM, although
the findings regarding this are contradictory (5-7). BIM
is a pro-apoptotic member of the Bcl-2 family and plays an
essential role in the induction of cell apoptosis and tumor
metastasis (2). The upregulation
of BIM is required for apoptosis induced by EGFR and EGFR-TKIs in
tumors harboring EGFR mutations (8). Consequently, BIM has become the focus
of attention as a potential target for cancer chemotherapy.
Furthermore, the overexpression of anti-apoptotic Bcl-2 family
proteins, including Mcl-1 and Bcl-2, has been investigated and has
been identified to be associated with chemoresistance and the
prognosis of various types of cancer, including lung cancer
(9-12); however, there are a limited number
of studies on EGFR-TKI-resistant lung cancer.
The herb Coptis chinensis (known as
goldthread; CC) is widely used in Traditional Chinese medicine;
moreover, its alkaloid component, berberine, has been studied for
its multiple pharmacological activities, including anti-infectious,
anti-inflammatory and anticancer effects (13). In addition, efforts have been made
to examine the potential therapeutic and biological functions of
CC, not as a single compound, but as multi-compounds, for cancer
treatment. CC has been shown to exert an anticancer effect through
the downregulation of signal transducer and activator of
transcription (STAT)2 phosphorylation by reducing the level of
histone deacetylase 2 (HDAC2) in glioma cells and inhibiting
hepatocellular carcinoma cell growth through non-steroidal
anti-inflammatory drug (NSAID) activated gene (NAG-1) activation
(14,15). In non-small cell lung cancer
(NSCLC) cells, CC has been shown to inhibit growth and metastasis,
and to induce cell apoptosis (16). However, neither its effects on
EGFR-TKI resistant lung cancer nor its efficacy in combination with
gefitinib have been elucidated to date, at least to the best of our
knowledge.
Therefore, the present study examined the expression
of Mcl-1 and Bcl-2 in order to determine the effects of the extract
of CC (ECC) on apoptosis. The anticancer effects of ECC, as well as
combination treatment with ECC and gefitinib on gefitinib-resistant
(GR) NSCLC cells (PC9GR, A549GR and HCC827GR) were also
investigated.
Materials and methods
Cell cultures and reagents
BEAS-2B, and the GR human lung cancer cell lines,
PC9GR and A549GR, were gifts from Dr J.K. Rho, Ulsan University,
Asan Hospital. The HCC827GRKU cell line was established from HCC827
cells treated with 2 µM gefitinib for >6 months (data not
shown). All cell lines were grown in RPMI (Welgene, Inc.)
supplemented with 10% fetal bovine serum (Welgene, Inc.) and 1%
penicillin/streptomycin at 37°C in a humidified atmosphere
containing 5% CO2 for all the experiments. Gefitinib was
purchased from Selleck Chemicals and berberine was purchased from
Sigma-Aldrich; Merck KGaA. Airdried roots of CC were purchased from
Dongguk University, Ilsan Korean Medicine Hospital. CC (10 g) was
extracted in 100 ml distilled water at room temperature. After 24
h, the solution was heated to 90°C for 4 h. The extract was then
filtered, evaporated and lyophilized (yield, 12.6%). The
lyophilized extract of CC (ECC) was stored at −20°C until use. The
identification of chemical components in ECC was performed by
ultra-performance liquid chromatography (UPLC)-quadrupole
time-of-flight (QTOF) (Data S1, Fig.
S1 and Table SI). ECC was re-dissolved in RPMI to a
concentration of 1,000 µg/ml for the in vitro experiments
and in DMSO (Daejunga) to a concentration of 6 mg/ml for the in
vivo experiments.
Cell viability assays
Cell viability was measured by MTT assay. Briefly,
1×103 cells per well were seeded in 96-well culture
plates overnight and, subsequently incubated with or without the
relevant treatments of ECC, or berberine. After 72 h, 50 µl MTT
solution (0.5 mg/ml, Sigma-Aldrich; Merck KGaA) were added to each
well. Following incubation at 37°C for a further 4 h, the MTT
solution was discarded and DMSO was added. The absorbance at 750 nm
was measured using a microplate reader (SpectraMax Plus 384,
Molecular Devices, LLC). The fraction affected (Fa) and combination
index (CI) values were calculated using CompuSyn (www.combosyn.com). CI values of <1, 1, and >1
indicated synergism, additive effects and antagonism, respectively.
Cell viability assay for the co-treatment was performed with
selected concentrations of of gefitinib (PC9GR and HCC827GRKU
cells, 1 µM; A549GR cells, 2 µM) and ECC (PC9GR and HCC827GRKU
cells, 10 µg/ml; A549GR cells, 5 µg/ml) for 72 h based on the CI
values. The results were representative of a minimum of 3
independent experiments, and the error bars represent the standard
deviation (SD).
Transwell invasion assays
The invasiveness of the tumor cells was assessed via
an invasion assay in Transwell chambers comprising a Transwell
membrane (8 µm pore size, 6.5 mm in diameter, Corning Life Science,
Inc.) coated with Matrigel (100 µg/ml, 10 µl/well). The cells
(1×105) were seeded in the upper chambers in the
presence of the indicated concentrations (PC9GR cells: 0, 30 and 50
µg/ml; HCC827GRKU cells: 0 and 30 µg/ml) of ECC. The lower chambers
of the Transwell plate were filled with RPMI with 10% FBS The cells
were fixed with 70% ethanol for 10 min, stained with hematoxylin
and eosin for 5 min at room temperature, and counted under a light
microscope (Olympus-IX71, Olympus Corp.) following incubation for
24 h.
Cell migration assay
Cell migration was assessed using a wound-healing
assay. The cells (5×105) were seeded in 6-well plates
and incubated at 37°C for 24 h. After the cell monolayer was
scraped with a sterile micropipette tip, the wells were washed
several times with phosphate-buffered saline (PBS) and cultured
with the designated concentrations (PC9GR cells: 0, 30 and 50
µg/ml; A549GR cells: 0, 10 and 20 µg/ml; HCC827GRKU cells: 0 and 30
µg/ml) of ECC. The first image of each scratch from 4 independent
areas was acquired at time zero. The image of each scratch at the
same location was captured under a light microscope (Olympus-IX71,
Olympus Corp) after the indicated incubation times (0, 24 and 48
h). The healed area was measured from the captured images using
Image J software (Ver. 1.52n, NIH).
Western blot analysis
Cell were lysed with ice-cold TNN buffer (1 M
Tris-Cl pH 7.4, 0.5% NP40, 5 M NaCl., 0.5 M EDTA pH 8.0) at 4°C for
overnight. Cell lysates were centri-fuged at 16,100 × g for 15 min
and the supernatants were used as total cellular protein extracts.
The protein concentrations were determined by Bradford assay
(Microplate reader, model-680, Bio-rad). Protein denaturation (20
µg/lane) was carried out by sodium dodecyl sulfate (SDS) and
mercaptoethanol loading and electrophoresed on a 12% acrylamide gel
(this excluded caspase-3 which was electrophoresed on a 15%
acrylamide gel). This was followed by transfer onto nitrocellulose
membranes (GE Healthcare Life Science, Inc.). The membranes were
blocked with 5% non-fat dry milk (SK1400.500, BioShop) in TBST (247
mM Tris, 1.37 M NaCl, 27 mM KCl, 1% Tween-20, pH 7.6) at room
temperature for 1 h. These membranes were, subsequently, probed
with the indicated primary antibodies at 4°C for overnight and
incubated with the appropriate goat anti-mouse IgG (1:5,000,
sc-2005, Santa Cruz Biotechnology, Inc.) or goat anti-rabbit IgG
(1:5,000, sc-2004, Santa Cruz Biotechnology, Inc.) at room
temperature for 1 h. Secondary antibodies were conjugated with
horseradish peroxidase prior to signal detection using the enhanced
chemiluminescence system (Translab) in accordance with the
manufacturer's instructions. The primary antibodies (dilution, cat.
no.) against EGFR (1:1,000, #2232), AKT (1:1,000, #4691), p-AKT
(1:1,000, #4691), caspase-3 (1:1,000, #9662) and poly(ADP-ribose)
polymerase (PARP) (1:1,000, #9542) were purchased from Cell
Signaling Technology, Inc. The antibodies against p-EGFR (1:1,000,
sc-101668), MET (1:1,000, sc-161), Bcl-2 (1:1,000, sc-492), Mcl-1
(1:1,000, sc-819), Bcl-xL (1:1,000, sc-7195) and β-actin (1:20,000,
sc-47778) were purchased from Santa Cruz Biotechnology, Inc.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was
synthesized from the total RNA using a reverse transcription kit
(LaboPass, Cosmo Genetech) in accordance with the manufacturer's
instructions. qPCR was conducted using gene-specific primers with
SYBR-Green Q Master (LaboPass) on an ABI 7500 Real Time PCR System
(Applied Biosystems). The following PCR primers were used:
Bcl-2 sense, 5′-AAG GGG GAA ACA CCA GAA TC-3′ and antisense,
5′-ATC CTT CCC AGA GGA AAA GC-3′; Mcl-1 sense, 5′-TGC TGG
AGT AGG AGC TGG TT-3′ and antisense, 5′-CCT CTT GCC ACT TGC TTT
TC-3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
sense, 5′-GCC ATC GTC ACC AAC TGG GAC-3′ and antisense, 5′-CGA TTT
CCC GCT CGG CCG TGG-3′. The PCR thermocycling conditions consisted
of 95°C for 5 min, followed by 40 cycles of 95°C for 10 sec and
62°C for 30 sec. The Ct values of the target genes were normalized
to those of an endogenous reference gene (GAPDH) using the
ΔΔCq method (17). Each gene was
analyzed in triplicate in 2 independent experiments.
Cell cycle analysis
GR cells were harvested following treatment with ECC
(PC9GR cells, 50 µg/ml; A549GR cells, 30 µg/ml) for the indicated
time periods (0, 24 and 48 h) and dissociated into single cells.
The cells were fixed with 95% ethanol, incubated at −20°C for at
least 1 h, and washed with PBS. The cells were then resuspended in
PBS with 0.1 mg/ml RNase A, 50 mg/ml propidium iodide (PI), and
0.05% Triton X-100 for 15 min at room temperature in the dark and
washed with PBS. The stained samples were analyzed using a FACS
Canto 2 (BD Biosciences) within 1 h of staining. All experiments
were performed in triplicate.
Terminal deoxynucleotidyl transferase
dUTP nick-end labelling (TUNEL) assay
Cells were seeded on a coverslip with complete
medium and incubated with or without the indicated concentrations
(PC9GR cells, 50 µg/ml; A549GR cells, 30 µg/ml) of ECC. Following
incubation at 37°C for 24 and 48 h, the cells were fixed with 4%
paraformaldehyde for 25 min at 4°C and washed twice with PBS at
room temperature. The cells were then permeabilized with 0.2%
Triton X-100 in PBS at room temperature for 5 min and washed twice
with PBS. TUNEL assay of the nuclei was performed, and the labeled
cells were viewed under a fluorescent microscope (Olympus-IX71,
Olympus Corp.), as described in the manufacturer's protocol
(DeadEnd™ Fluorometric TUNEL System; Promega).
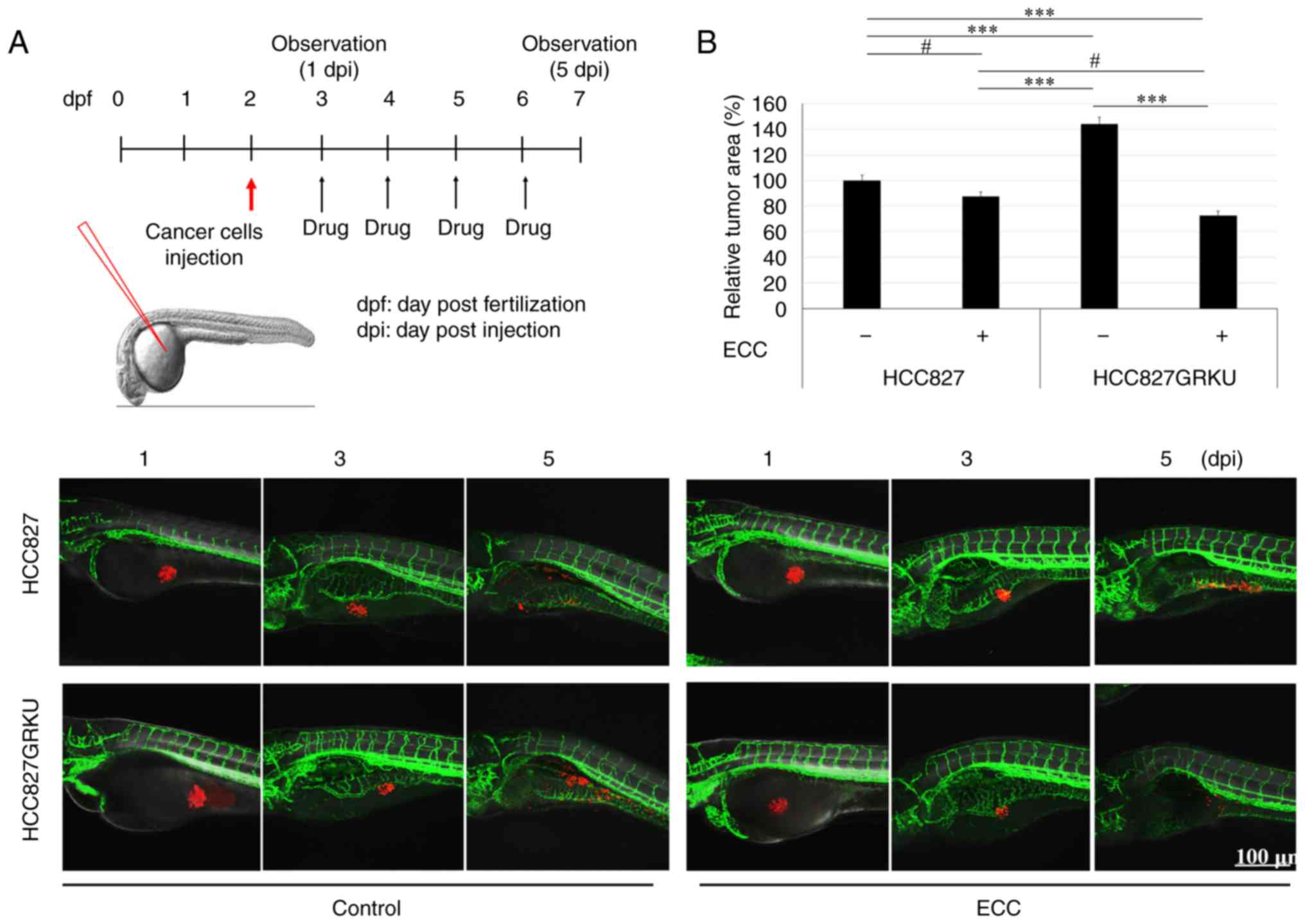
In vivo zebrafish tumor model
Zebrafish (Danio rerio) and embryos were bred
and maintained according to standard procedures. All animal
experimental protocols were approved by the Committee for Ethics of
Animal Experimentation of the Sookmyung Women's University and
performed as previously described (18). Approximately 50 fluorescent cell
tracker CM-Dil-labeled HCC827 or HCC827GRKU cells were injected
into the yolk sac of each zebrafish embryo (100 embryos for each
treatment), after which the embryos were maintained at 33°C and
drug treatments were administered every 24 h for 4 days.
Fluorescence image acquisition was performed using a Zeiss LSM700
confocal microscope (Carl Zeiss AG). The area penetrated by the
CM-Dil-labeled cancer cells was quantified using ImageJ software
(ver. 1.52n, NIH) and normalized to the cancer cells (100%) in
non-treated zebrafish embryos for each group.
Dose selection of ECC
For the in vitro assay, the IC50
value of 3 days for the PC9GR (32.73 µg/ml) and A549GR (20 µg/ml)
cells and an approximately 1.5-fold higher concentration than the
IC50 value of 3 days were selected. For the HCC827GRKU,
one concentration was selected, which was approximately 1.5-fold
higher than the IC50 value of 3 days. For the in
vivo assay, the toxicity of ECC was tested on zebrafish embryo
development at a series of concentrations of ECC (0.1, 1 and 5
µl/ml); the embryos were observed until 7 days post-fertilization
(dpf) and the concentration which did not affect the survival of
the zebrafish was selected. The zebrafish embryos died from overall
necrosis, a terminated heart beat and no movement from mechanical
stimulation at 4 dpf following treatment with 5 µl/ml of ECC, but
survived and developed normally following treatment with 0.1 and 1
µl/ml of ECC. Subsequently, further tests were performed to select
the ECC concentration which was most effective against cancer cells
from a series of ECC concentrations (0.1, 0.2 and 1.0 µl/ml), which
was 0.2 µl/ml.
Statistical analysis
The experiments were repeated at least twice. The
values are expressed as the means ± standard deviation and were
compared using a two-tailed Student's t-test or ANOVA. If the
P-value obtained by one-way ANOVA was <0.05, P-values between
the groups were compared with a post hoc test, such as the
Bonferroni and Tukey's HSD. A value of P≤0.05 was considered to
indicate a statistically significant difference.
Results
ECC inhibits the viability and mobility
of the GR NSCLC cell lines, PC9GR, A549GR and HCC827GR
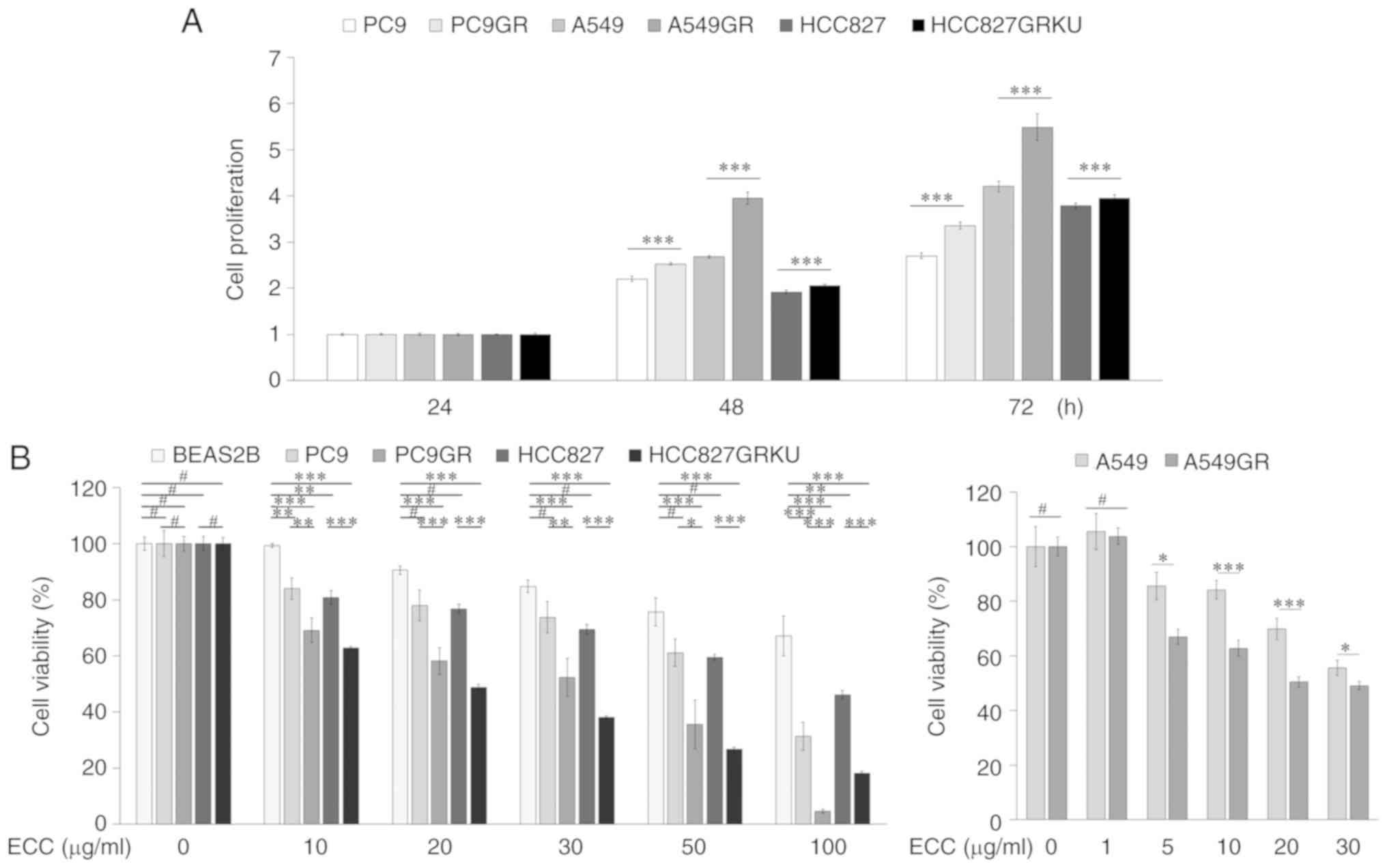
Given that, as previously demonstrated, the GR cell
lines, PC9GR, A549GR and HCC827GR, exhibit an enhanced viability
upon gefitinib treatment (18-20)
and in this study, were found to proliferate more rapidly than
their parental cells (Fig. 1A), an
MTT assay was performed on these GR NSCLC cells, as well as on
their parental cells PC9, A549 and HCC827 and the normal bronchus
cell line, BEAS-2B, to examine the effects of ECC on cell
viability. The effects of berberine (Data S1), a known alkaloid
extracted from CC, on cell viability and cytotoxicity was examined
using an MTT assay in order to compare its effects to those of ECC,
which is a multi-compound formulation. ECC suppressed the viability
of the GR cells more effectively than that of the parental cells
and exerted minimal cytotoxic effects on the BEAS-2B cells
(Fig. 1B and Table I). However, berberine was less
toxic to the PC9GR cells than the PC9 cells, and exerted
significant cytotoxic effects on the BEAS-2B cells; moreover, the
BEAS-2B cells were even more sensitive to berberine than the HCC827
lung cancer cells (Fig. S2). The
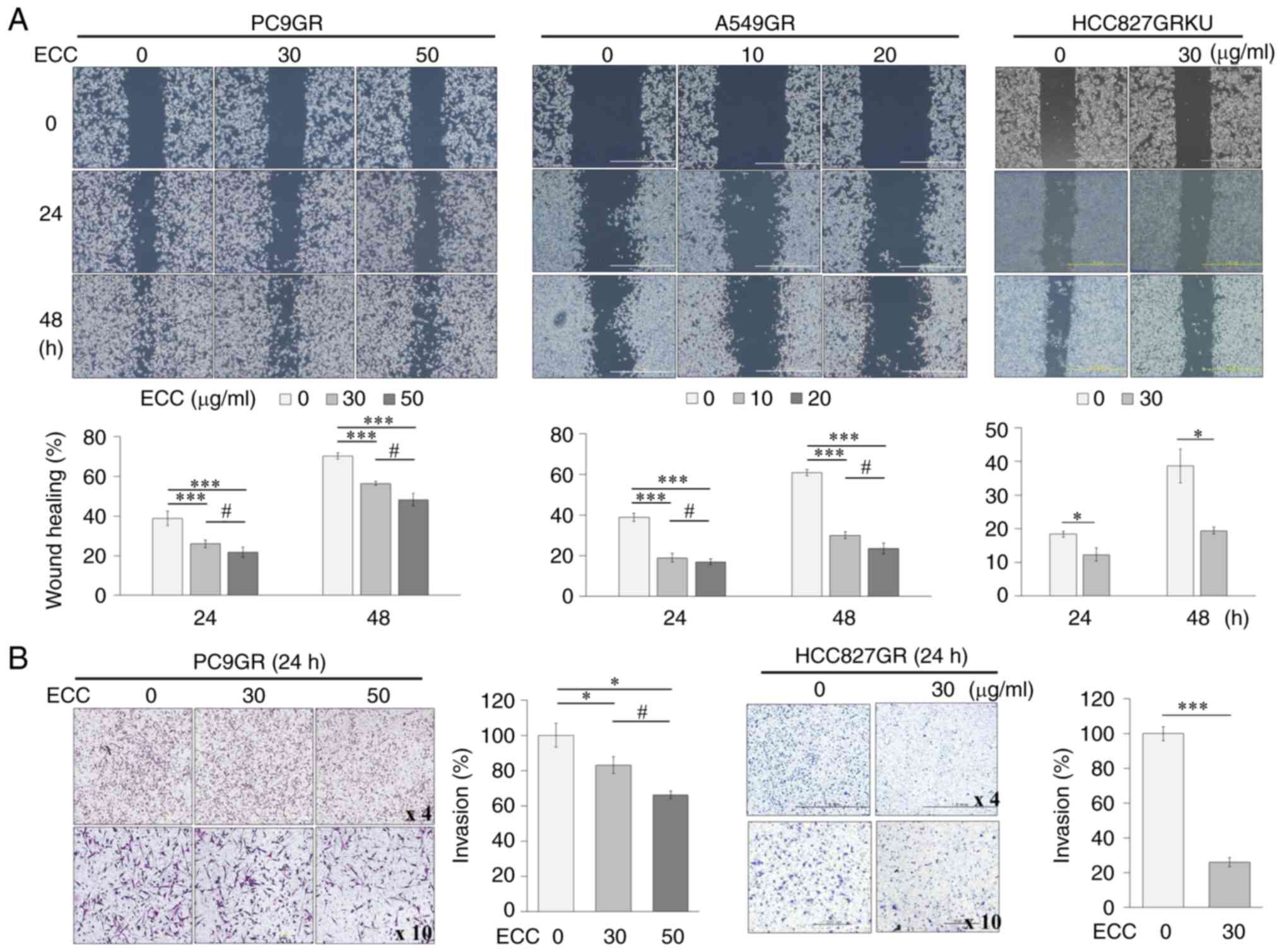
effects of ECC on the migratory and the invasive potential of the
PC9GR, A549GR and HCC827GR cells were examined using in
vitro migration and invasion assays. Treatment with ECC
inhibited the migration and invasion of the GR cells in a dose- and
time-dependent manner (Fig. 2);
however, a limitation of the present study should be stated here in
that 10% FBS may have affected cell proliferation. The invasion
assay could not be performed for the A549GR cells, as the cells do
not attach effectively on Matrigel. Collectively, these results
revealed that ECC exerted anticancer effects on the GR cells.
 | Table IIC50 values of ECC,
gefitinib and berberine in GR and parental cells. |
Table I
IC50 values of ECC,
gefitinib and berberine in GR and parental cells.
| Treatment | Cells lines and
IC50 values
|
|---|
| PC9 | PC9GR | A549 | A549GR | HCC827 | HCC827GRKU | BEAS-2B |
|---|
| Gefitinib (µM) | 0.008 | 8.79 | 15.34 | 18.65 | 0.01 | 10 | N/A |
| ECC (µg/ml) | 69.80 | 32.73 | 30 | 20 | 85.33 | 19.07 | 178.08 |
| Berberine (µM) | 2.81 | 7.73 | 13.99 | <1 | 48.1 | 13.3 | 33.01 |
ECC induces the apoptosis of GR NSCLC
cells (PC9GR, and A549GR cells)
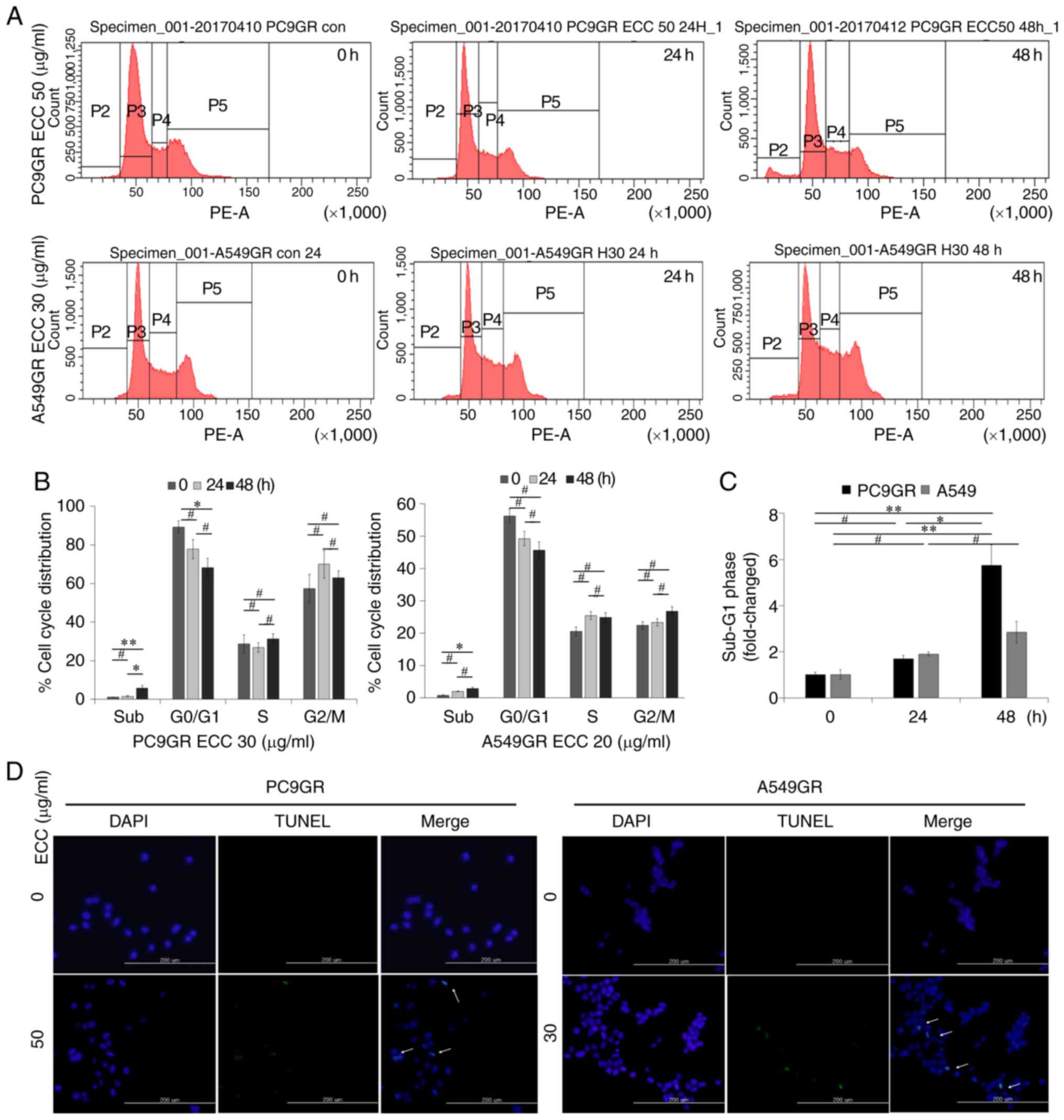
To elucidate the mechanisms through which ECC
affects GR cell viability, cell cycle and apoptosis analyses were
performed using PI-stained cells through FACS analysis and TUNEL
assay. The distribution of GR cells in the cell cycle phase was
analyzed following treatment with the indicated concentrations of
ECC for 24 and 48 h. ECC treatment increased the percentage of GR
cells in the sub-G1 phase (i.e., dead cells) in a time-dependent
manner (Fig. 3A-C). Apoptosis
induced by ECC was confirmed by TUNEL assay, which revealed an
increase in the number of TUNEL-positive cells upon ECC treatment
(Fig. 3D). Thus, these data
indicated that the anticancer effects of ECC on GR cells resulted
from the induction of cell cycle arrest and cell death.
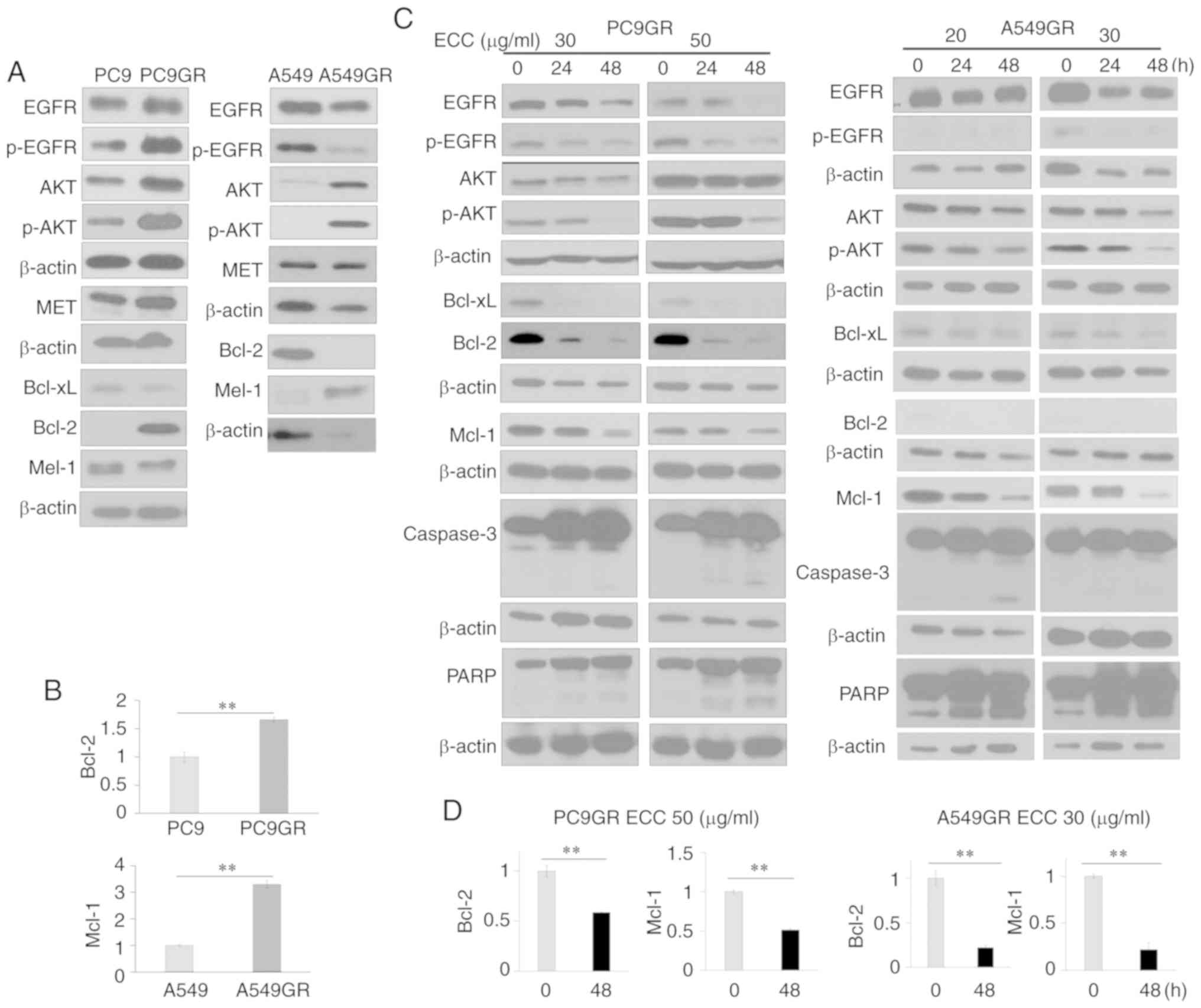
ECC suppresses the EGFR-AKT pathway and
the expression of the anti-apoptotic proteins, Mcl-1 and Bcl-2
Increased cell survival owing to the impairment of
an essential pathway for EGFR-TKI-mediated apoptosis has been
suggested as a mechanism responsible for resistance to EGFR-TKIs.
To investigate this pathway in GR cells, the expression of the EGFR
pathway and anti-apoptotic proteins was examined in GR cells and
compared with that in their parental cells. The expression of
AKT/p-AKT and the anti-apoptotic proteins, Mcl-1 and/or Bcl-2, was
increased in the GR cells (Figs. 4A
and B, and S3A). As ECC
exerted anti-survival and pro-apoptotic effects, the effects of ECC
on the expression of AKT/p-AKT, Mcl-1 and Bcl-2 were then examined.
ECC treatment resulted in the suppression of the expression of
these molecules (Figs. 4C and
S3B). The effects of ECC on the
expression of caspase-3 and PARP, which act as Mcl-1/Bcl-2
downstream effectors in the apoptotic pathway in GR cells were then
further examined. The expression of Mcl-1 and Bcl-2 was decreased,
and the cleaved forms of caspase-3 and PARP were increased in a
time- and dose-dependent manner (Fig.
4C). The suppression of the expression of Mcl-1 and Bcl-2 by
ECC was confirmed by RT-qPCR (Figs.
4D and S3C).
ECC synergistically enhances the activity
of gefitinib in GR NSCLC cells in vitro
The ability of ECC to enhance the effects of
gefitinib on GR cells was evaluated by MTT assay using cells
treated with a combination of ECC and gefitinib. Combination
treatment reduced GR cell viability in comparison to treatment with
gefitinib or ECC alone (Fig. 5A).
To elucidate the mechanisms through which ECC restores the
antitumor activities of EGFR-TKIs, the activities of EGFR and its
downstream molecule, AKT, as well as the anti-apoptotic proteins
Bcl-2 and Mcl-1, were examined in GR cells. As expected,
combination treatment resulted in the most effective inhibitory
effects (Fig. 5B). It should be
noted that in the PC9GR cells, combination treatment only decreased
Bcl-2 expression at 48 h, not at 24 h (Fig. 5B). The reasons for this are not
clear. In addition, whether the combination treatment was able to
enhance the inhibitory effects gefitinib on cell viability through
a synergistic effect was examined. This was determined by the Fa-CI
plot (Chu-Talalay Plot; www.combosyn.com) median effect analysis, which
revealed that the combination index (CI) was smaller than 1
(Fig. 5C) (21), indicating synergistic growth
inhibition of the GR cells by this treatment combination.
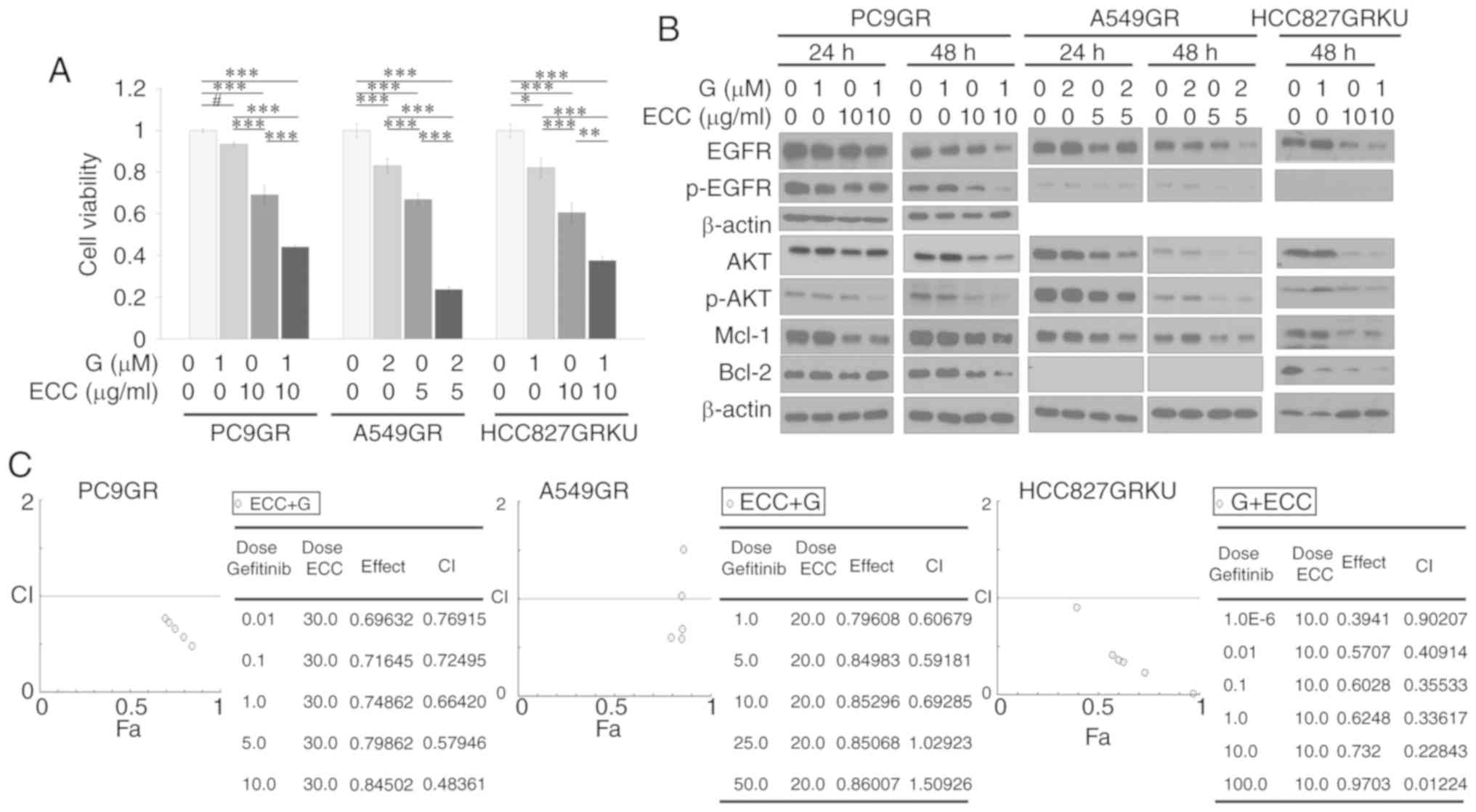 | Figure 5ECC synergistically enhances the
activity of gefitinib in GR cells. (A) Cell viability was assessed
by MTT assay following treatment of the PC9GR, A549GR and
HCC827GRKU cells with the indicated concentrations of of gefitinib
alone, ECC alone, or co-treatment with gefitinib and ECC for 72 h.
(B) Western blot analysis was performed with the indicated
antibodies following treatment with the indicated concentrations of
gefitinib, ECC and the combination of gefitinib and ECC for 24
and/or 48 h. (C) The CI of gefitinib and ECC were calculated and
the Fa-CI plots generated by the Chou-Talalay method using data
obtained from MTT assay following co-treatment with the indicated
concentrations of each drug for 72 h. CI values of <1, 1 and
>1 indicate synergism, additive effects and antagonism,
respectively. The results are shown as the means ± SD of triplicate
experiments. *P<0.05, **P<0.01 and
***P<0.001; the hash symbol (#) indicates that there
were no significant differences. ECC, extract of Coptis
chinensis; GR, gefitinib-resistant. |
Cancer cell-specific/sensitive toxicity
of ECC in vivo
The suppression of tumorigenicity in vivo by
ECC treatment in lung cancer cells was examined in xenograft
zebrafish models. CM-Dil-labeled HCC827 or HCC827GRKU cells (red)
were grafted into Tg(flk1:EGFP) zebrafish embryos and either
DMSO, 0.5 µM gefitinib, 0.2 µl/ml ECC, or the combination of
gefitinib and ECC were added to the embryo culture water, and
refreshed every 24 h for 5 days (Fig.
6A). The anticancer effects of ECC were confirmed in both the
HCC827 and HCC827GRKU cells. This result was consistent with the
in vitro results, in which the HCC827GRKU cells were more
sensitive than the HCC827 cells to ECC (Fig. 6B). In addition, the gefitinib-,
ECC-, and the gefitinib and ECC combination-treated embryos were
compared and found to have significantly fewer cancer cells than
the control group; however, no significant differences were
observed between the treatment groups (Fig. 6C). Unexpectedly, the treatment of
zebrafish with ECC alone or in combination with gefitinib resulted
in the significantly increased survival of the zebrafish compared
to treatment with gefitinib alone (Fig. 6D). This result indicated that ECC
had a more specific toxicity against cancer cells than normal
cells, consistent with the in vitro results.
Discussion
EGFR-TKIs are some of the most effective therapeutic
drugs against NSCLCs with EGFR mutations. However, the various
adaptive and acquired resistance mechanisms reported have
significantly limited the efficacy of EGFR-TKIs and, consequently,
the current chemotherapeutic strategies for NSCLCs. Therefore,
there is a need to overcome GR resistance to EGFR-TKIs, as GR
resistance in lung cancer results in more aggressive cells with an
increased viability, proliferation and metastatic ability.
Apoptosis is the natural process through which the
elimination of unwanted or damaged cells that present a threat to
the health of an organism occurs. This process is highly
controlled, and the Bcl family of proteins, which comprises
anti-apoptotic proteins and pro-apoptotic proteins, serves as the
main regulator of this process (22,23).
The anti-apoptotic proteins, Mcl-1 and Bcl-2, play important roles
in the maintenance of cell viability and survival, but not
proliferation, through the interaction of several other regulators
of apoptosis. The overexpression of Mcl-1 and/or Bcl-2 has been
shown to facilitate chemoresistance in various types of cancer
(24-26), and has been suggested as a
therapeutic target. Therefore, efforts have been made to develop
drugs targeting Mcl-1 and Bcl-2 to induce chemosensitization and
overcome chemoresistance (27-29).
Mcl-1 has been suggested to be a critical survival factor in lung
cancer as it promotes cancer cell migration ability and
epithelial-mesenchymal transition (11,30-32).
A previous study demonstrated that the overexpression of Mcl-1
increased the viability of cancer cells following exposure to
cytotoxic chemotherapeutic agents and EGFR-TKIs (30).
Conversely, the inhibition of Bcl-2 by various
methods (gene suppression and inhibitors) has been shown to
increase the sensitivity of lung cancer cells to EGFR-TKIs
(26,30,33).
This highlights the role of Mcl-1 and Bcl-2 in EGFR-TKI resistance,
and indicates that combination treatment comprising EGFR-TKI and
Mcl-1 and/or Bcl-2 may exert synergistic effects.
Therefore, the present study evaluated the
expression of the anti-apoptotic molecules, Mcl-1, Bcl-2 and
Bcl-xL, as well as EGFR signaling molecules, in established GR lung
cancer cell and compared it with the expression in their parental
cell PC9, A549 and HCC827. The PC9GR cells exhibited a higher
expression of EGFR signaling molecules and Bcl-2, but not of Mcl-1.
The A549GR cells exhibited a higher expression of AKT/p-AKT and
Mcl-1, but not of EGFR/p-EGFR and Bcl-2. The HCC827GRKU cells
exhibited a higher expression of AKT/p-AKT, Bcl-2 and Mcl-1. These
data suggested that the cellular response to EGFR-TKIs is dependent
on the EGFR mutation status, as described in other studies
(34,35), which results in a variety of
resistance mechanisms to EGFR-TKIs. In the present study, cell
viability, migration and invasion assays were thus performed to
investigate the physiological anticancer effects of ECC in GR
cells. ECC effectively inhibited GR cell viability, migration and
invasion. Simultaneously, ECC suppressed EGFR signaling through
AKT, regardless of the EGFR mutation status, resulting in the
inhibition of GR cell survival. However, ECC exerted minimal
toxicity on the normal bronchial cell line, BEAS-2B, unlike the
alkaloid extract, berberine; this was confirmed in the in
vivo model. The dose of ECC in the in vivo model was
selected, such that it did not pathophysiologically affect survival
or induce damage in the zebrafish themselves, but only affected the
cancer cells. Therefore, even if the anticancer effect of ECC was
not greater than that of gefitinib alone or the combination
treatment in vivo, the increased survival of zebrafish
indicated the specific toxicity of ECC against cancer cells, but
not against normal cells, which is crucial for the development of
novel anticancer drugs. Cell cycle and TUNEL assays revealed that
ECC induced the apoptosis of GR cells through the suppression of
Mcl-1, Bcl-2 and Bcl-xl, and the promotion of the expression of
cleaved caspase-3 and PARP. As ECC suppressed anti-apoptotic and
EGFR/AKT signaling, its synergistic effects with the EGFR-TKI,
gefitinib, in GR cells to overcome EGFR-TKI resistance, were
evaluated in cells treated with both gefitinib and ECC, and the CI
was calculated. The results revealed that ECC treatment
re-sensitized the GR cells to gefitinib synergistically through the
inhibition of EGFR-AKT signaling and the anti-apoptotic proteins,
Bcl-2 and Mcl-1.
The fact that in the present study, no tumor
xenograft mouse model was used validating the anti-tumor effect of
ECC addition to the zebrafish tumor model, which can provide more
convincing evidence to the present data and the fact that ECC is a
multi-component formulation, rather than a single compound, may be
a limitation of this study. However, it should considered that the
very weak cytotoxic effects of ECC on normal cells compared to
those of berberine, a known alkaloid extracted from ECC, may arise
due to the multi-component nature of ECC.
Collectively, the present study found that ECC
exerted anti-cancer effects through the suppression of EGFR/AKT
signaling and induced apoptosis via the suppression of the
anti-apoptotic proteins, Mcl-1 and Bcl-2, which were overexpressed
in GR cells. Moreover, combination treatment with ECC
synergistically enhanced GR cell sensitivity to gefitinib,
regardless of the EGFR mutation status in vitro and
increased the viability of normal cells and survival of zebrafish
in vivo. These results indicated the potential role of ECC
in the treatment of EGFR-TKI-resistant NSCLCs, particularly in
combination with EGFR-TKI therapy, with minimal side-effects.
Supplementary Data
Acknowledgements
The abstract was presented and published as a poster
(no. P-35-049) in supplement (vol. 9, issue S1): The 44th FEBS
Congress July 6-11, 2010 in Krakow, Poland.
Funding
The present study was supported by the Bio-Synergy
Research Project (NRF-2014M3A9C4066487) of the Ministry of Science,
ICT and Future Planning through the National Research Foundation
and by the Basic Science Research Program Grants
(NRF-2017R1A2B4003233 and NRF-2019R1A2C1083909) from the National
Research Foundation of Korea, which is funded by the Ministry of
Education, Science and Technology, Republic of Korea.
Availability of data and materials
All data generated or analyzed during the study are
included in this published article or are available from the
corresponding author upon reasonable request.
Authors' contributions
JYL conceived and designed the experiments; JHK,
ESK, DK, SHP, EJK, JR, WMY, IJH, HS and IJH conducted the
experiments; JHK, ESK, DK, SP, EJK, JR, MJK, WMY, IJH, MJP, WMY and
JYL analyzed and interpreted the results. All authors reviewed the
manuscript and all authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experimental protocols were approved by
the Committee for Ethics of Animal Experimentation of Sookmyung
Women's University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernald K and Kurokawa M: Evading
apoptosis in cancer. Trends Cell Biol. 23:620–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.PubMed/NCBI
|
|
4
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko
TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, et al: A
common BIM deletion polymorphism mediates intrinsic resistance and
inferior responses to tyrosine kinase inhibitors in cancer. Nat
Med. 18:521–528. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao M, Zhang Y, Cai W, Li J, Zhou F,
Cheng N, Ren R, Zhao C, Li X, Ren S, et al: The Bim deletion
polymorphism clinical profile and its relation with tyrosine kinase
inhibitor resistance in Chinese patients with non-small cell lung
cancer. Cancer. 120:2299–2307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu SG, Liu YN, Yu CJ, Yang PC and Shih JY:
Association of BIM deletion polymorphism with intrinsic resistance
to EGFR tyrosine kinase inhibitors in patients with lung
adenocarcinoma. JAMA Oncol. 2:826–828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Costa DB, Halmos B, Kumar A, Schumer ST,
Huberman MS, Boggon TJ, Tenen DG and Kobayashi S: BIM mediates EGFR
tyrosine kinase inhibitor-induced apoptosis in lung cancers with
oncogenic EGFR mutations. PLoS Med. 4:1669–1680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pandey MK, Prasad S, Tyagi AK, Deb L,
Huang J, Karelia DN, Amin SG and Aggarwal BB: Targeting cell
survival proteins for cancer cell death. Pharmaceuticals (Basel).
9. pii: E11. 2016, View Article : Google Scholar
|
|
10
|
Delbridge AR and Strasser A: The BCL-2
protein family, BH3-mimetics and cancer therapy. Cell Death Differ.
22:1071–1080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang TM, Barbone D, Fennell DA and
Broaddus VC: Bcl-2 family proteins contribute to apoptotic
resistance in lung cancer multicellular spheroids. Am J Respir Cell
Mol Biol. 41:14–23. 2009. View Article : Google Scholar :
|
|
12
|
Martin B, Paesmans M, Berghmans T, Branle
F, Ghisdal L, Mascaux C, Meert AP, Steels E, Vallot F, Verdebout
JM, et al: Role of Bcl-2 as a prognostic factor for survival in
lung cancer: A systematic review of the literature with
meta-analysis. Br J Cancer. 89:55–64. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang F, Mei W, Tian D and Huang D: An
evidence-based perspective of Coptis chinensis (Chinese Goldthread)
for cancer patients. Evidence-based anticancer materia medica. Cho
WCS: Springer; NY: pp. 111–130. 2011, View Article : Google Scholar
|
|
14
|
Li J, Ni L, Li B, Wang M, Ding Z, Xiong C
and Lu X: Coptis chinensis affects the function of glioma cells
through the down-regulation of phosphorylation of STAT by reducing
HDAC3. BMC Complement Altern Med. 17:5242017. View Article : Google Scholar
|
|
15
|
Auyeung K and Ko J: Coptis chinensis
inhibits hepatocellular carcinoma cell growth through nonsteroidal
anti-inflammatory drug-activated gene activation. Int J Mol Med.
24:571–577. 2009.PubMed/NCBI
|
|
16
|
Lulu N, Jiangan L, Weixing Z, Zhiyi Z, Hui
L, Jianhui T, Haizhou L and Hongli R: Coptis chinensis inhibits
growth and metastasis and induces cell apoptosis in non-small cell
lung cancer cells. Int J Clin Exp Med. 10:16037–16048. 2017.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Park SH, Kim JH, Ko E, Kim JY, Park MJ,
Kim MJ, Seo H, Li S and Lee JY: Resistance to gefitinib and
cross-resistance to irreversible EGFR-TKIs mediated by disruption
of the Keap1-Nrf2 pathway in human lung cancer cells. FASEB J.
32:5862–5873. 2018. View Article : Google Scholar
|
|
19
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar
|
|
20
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Lee SS, Kim CH, Yoo YD and Lee JC: The role of MET
activation in determining the sensitivity to epidermal growth
factor receptor tyrosine kinase inhibitors. Mol Cancer Res.
7:1736–1743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chou TC and Martin N: CompuSyn software
for drug combinations and for general dose-effect analysis, and
user's guide. ComboSyn, Inc.; Paramus, NJ: 2007
|
|
22
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
23
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei G, Margolin AA, Haery L, Brown E,
Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R and Golub TR:
Chemical genomics identifies small-molecule MCL1 repressors and
BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 21:547–562.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karnak D and Xu L: Chemosensitization of
prostate cancer by modulating Bcl-2 family proteins. Curr Drug
Targets. 11:699–707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu DW, Chen CY, Chu CL and Lee H: Paxillin
confers resistance to tyrosine kinase inhibitors in EGFR-mutant
lung cancers via modulating BIM and Mcl-1 protein stability.
Oncogene. 35:621–630. 2016. View Article : Google Scholar
|
|
27
|
Belmar J and Fesik SW: Small molecule
Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther.
145:76–84. 2015. View Article : Google Scholar :
|
|
28
|
Brumatti G and Ekert PG: Seeking a MCL-1
inhibitor. Cell Death Differ. 20:1440–1441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan R, Ruvolo VR, Wei J, Konopleva M, Reed
JC, Pellecchia M, Andreeff M and Ruvolo PP: Inhibition of Mcl-1
with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737
resistance in acute myeloid leukemia. Blood. 126:363–372. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song L, Coppola D, Livingston S, Cress D
and Haura EB: Mcl-1 regulates survival and sensitivity to diverse
apoptotic stimuli in human non-small cell lung cancer cells. Cancer
Biol Ther. 4:267–276. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toge M, Yokoyama S, Kato S, Sakurai H,
Senda K, Doki Y, Hayakawa Y, Yoshimura N and Saiki I: Critical
contribution of MCL-1 in EMT-associated chemo-resistance in A549
non-small cell lung cancer. Int J Oncol. 46:1844–1848. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang H, Guttikonda S, Roberts L, Uziel T,
Semizarov D, Elmore SW, Leverson JD and Lam LT: Mcl-1 is critical
for survival in a subgroup of non-small-cell lung cancer cell
lines. Oncogene. 30:1963–1968. 2011. View Article : Google Scholar
|
|
33
|
Zhang J, Wang S, Wang L, Wang R, Chen S,
Pan B, Sun Y and Chen H: Prognostic value of Bcl-2 expression in
patients with non-small-cell lung cancer: A meta-analysis and
systemic review. Onco Targets Ther. 8:3361–3369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lohinai Z, Hoda MA, Fabian K, Ostoros G,
Raso E, Barbai T, Timar J, Kovalszky I, Cserepes M, Rozsas A, et
al: Distinct epidemiology and clinical consequence of classic
versus rare EGFR mutations in lung adenocarcinoma. J Thorac Oncol.
10:738–746. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li K, Yang M, Liang N and Li S:
Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR
mutations in non-small cell lung cancer: Perplexity and solution
(Review). Oncol Rep. 37:1347–1358. 2017. View Article : Google Scholar : PubMed/NCBI
|