Introduction
Chronic myelogenous leukemia (CML) is a
myeloproliferative disorder of the bone marrow stem cells
characterized by genetic translocation between chromosomes 9 and
22. This translocation results in the fusion of Bcr on chromosome
22 with Abl on chromosome 9, leading to the expression of the
Bcr/Abl fusion oncoprotein. This fusion protein exhibits
constitutively active kinase activity (1), transducing signals to a variety of
downstream survival pathways, including the mitogen-activated
protein kinase kinase/extracellular signal-regulated kinase
cascade, Akt, and the signal transducers and activators of
transcription (STATs) and nuclear factor κB (NF-κB) pathways
(2–4). A number of anti-apoptotic proteins,
such as B-cell lymphoma extra large, are upregulated as a result of
the activation of these pathways. Collectively, these events
provide Bcr/Abl+ cells with a survival advantage over
normal cells, due to their reduced capacity to undergo apoptosis.
Furthermore, Bcr/Abl+ cells have exhibited varying
degrees of resistance towards conventional cytotoxic drugs
(5–7), until the recent clinical application
of imatinib mesylate (Gleevec®, STI-571). Imatinib has
been widely demonstrated to be effective in the treatment of CML
through inhibition of the tyrosine kinase activity of Bcr/Abl.
However, acquired resistance to imatinib can occur through various
mechanisms, thereby leading to continued disease progression. Thus,
the development of alternative approaches to the treatment of CML
is required.
Bortezomib (BTZ), also known as Velcade®
or PS-341, has been shown to act as an inducer of apoptosis in
multiple myeloma cells. BTZ exerts its effects through a number of
signaling cascades, predominantly the NF-κB pathway, ultimately
inducing apoptosis and thereby reversing drug resistance and
improving prognosis (8,9). The administration of BTZ as a
combination treatment with other agents has been widely
investigated; the cyclin-dependent kinase inhibitor flavopiridol
and the histone deacetylase inhibitor suberoylanilide hydroxamic
acid have both been shown to act synergistically with BTZ to induce
apoptosis in Bcr/Abl+ cells (10,11).
Arsenic trioxide (As2O3) is a
chemotherapeutic agent that acts by inducing apoptosis and
differentiation and can uniquely induce complete remission in the
majority of patients with acute promyelocytic leukemia (12–14).
Furthermore, studies have shown that As2O3
alone, and in combination with other compounds, induces apoptosis
and/or growth arrest in Bcr/Abl+ cells (15–17).
However, high concentrations of As2O3 were
used in these studies, and, due to the toxic nature of arsenic, its
use has been limited in clinical practice. In 2007, Yan et
al (18) showed that
clinically achievable concentrations of As2O3
could act synergistically with BTZ to successfully induce apoptosis
in imatinib-sensitive Bcr/Abl+ (K562) cells, in which
protein kinase Cδ (PKCδ) activation played a critical role.
This study aimed to investigate the effects of the
combination of As2O3 and BTZ on apoptosis in
imatinib-resistant Bcr/Abl+ cells and to determine
whether this combinatory approach warranted further investigation
as a potential therapeutic strategy for the treatment of CML,
particularly for cases exhibiting imatinib resistance.
Materials and methods
Reagents
Imatinib (STI571) was purchased from Selleck
Chemicals (Houston, TX, USA), prepared as a 1 mM stock solution in
dimethyl sulfoxide and stored at −20°C. BTZ (Millennium
Pharmaceuticals Inc., Cambridge, MA, USA) was dissolved in
phosphate-buffered saline (PBS) and stored at −20°C until use.
As2O3 (Sigma, St. Louis, MO, USA) was
dissolved in 1.0 N NaOH and then diluted to 1 mM with PBS.
Cell culture and viability assay
K562 and K562r cells (provided by Professor J.V.
Melo, Department of Haematology, Imperial College of London,
London, UK) were cultured in RPMI-1640 media supplemented with 10%
heat-inactivated fetal bovine serum (Gibco-BRL, Gaithersburg, MD,
USA) in a humidified atmosphere with 5% CO2 at 37°C.
K562r cells were cultured in 1 μM imatinib to maintain
drug-resistance. Cell viability and inhibition of cell growth were
estimated using a Cell Counting kit 8 (Dojindo Laboratories,
Kumamoto, Japan).
Apoptosis assessment
Apoptosis was measured using a fluorescein
isothiocyanate (FITC) Annexin V Apoptosis Detection kit (BD
Biosciences, Franklin Lakes, NJ, USA) in accordance with the
manufacturer’s instructions. Following treatment with
As2O3 and/or BTZ, 1×105 K562r
cells were collected, washed with cold PBS and resuspended with
binding buffer. A total of 5 μl annexin V-FITC was added and cells
were incubated for 5 min in the dark. Following incubation, 10
μl propidium iodide was added and the samples were incubated
for an additional 3 min. Binding buffer (400 μl) was then
added and cells were analyzed by flow cytometry.
Western blot analysis
Equal amounts of protein were loaded on 8–14%
SDS-polyacrylamide gels, subjected to SDS-PAGE and transferred to
nitrocellulose membranes (Amersham Pharmacia Biotech, Amersham,
UK). The blots were stained with 0.2% Ponceau S red to ensure equal
protein loading. Following blocking with 5% nonfat milk in PBS, the
membranes were probed with primary antibodies targeting c-Abl,
PKCδ, poly (ADP-ribose) polymerase (PARP) (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and cleaved caspase-3
(Cell Signaling Technology, Inc., Beverly, MA, USA). Membranes were
subsequently incubated with horseradish peroxidase (HRP)-linked
secondary antibodies (Cell Signaling Technology, Inc.) and the
signal was detected using a chemiluminescence
Phototope®-HRP kit (Cell Signaling Technology, Inc.).
Blots were stripped and reprobed with a mouse anti-β-actin
monoclonal antibody (Oncogene, San Diego, CA, USA) as a loading
control.
Quantitative polymerase chain reaction
(qPCR) analysis of Bcr/Abl mRNA
Total RNA was isolated by TRIzol™ reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) and RNA was
treated with DNase (Promega Corp., Madison, WI, USA). Complementary
DNA was synthesized according to the manufacturer’s instructions.
The analysis of Bcr/Abl and β-actin was performed by qPCR using
SYBR Green PCR Master Mix reagents (Applied Biosystems, Foster
City, CA, USA) using an ABI PRISM 7900 system (Applied Biosystems).
The specific primers used for detecting p210 Bcr/Abl were as
follows: Forward primer (5′-CTGGCCCAACGATGGCGA-3′) and reverse
primer (5′-CACTCAGACCCTGAGGCTCAA-3′). Primers were synthesized by
Sangon Biotechnology (Shanghai, China).
Measurement of reactive oxygen species
(ROS)
Cells were incubated with either
As2O3, BTZ or the two in combination for the
indicated times. Following incubation, cells were washed twice with
PBS and treated with 10 μM 2′,7′-dichlorodihydrofluorescein
diacetate (Molecular Probes®/Invitrogen Life
Technologies) for 30 min at 37°C, in the dark. Cells were then
washed with PBS once. Red fluorescence was detected by
fluorescence-activated cell sorting at an excitation wavelength of
485 nm and an emission wavelength of 535 nm.
Statistical analysis
All experiments were repeated in triplicate. The
SPSS 11.0 (SPSS Inc., Chicago, IL, USA) software package was used
for statistical analysis. P<0.05 was considered to indicate a
statistically significant difference. Results are presented as the
mean ± standard deviation.
Results
K562r cells are more resistant than K562
cells to imatinib treatment
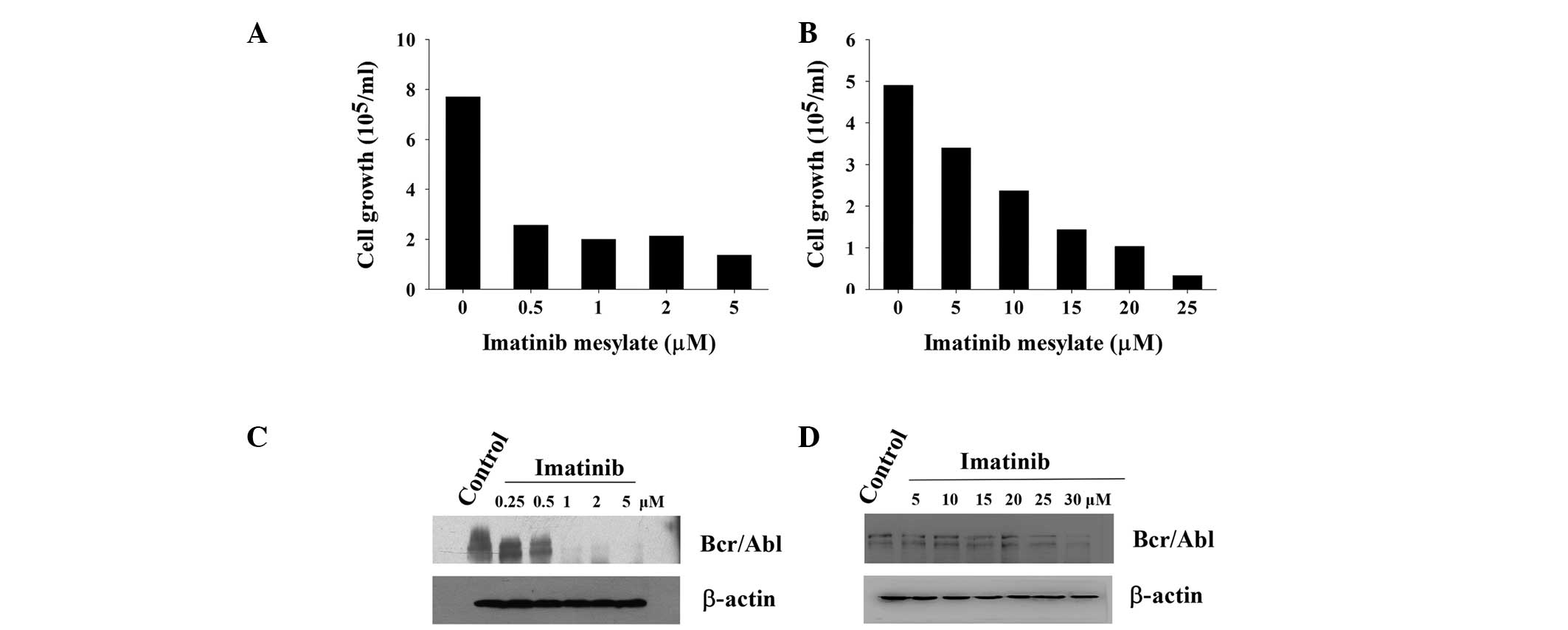
In order to determine the resistance characteristics
of K562r cells, K562 (imatinib-sensitive) or K562r
(imatinib-resistant) cells were incubated with varying
concentrations of imatinib. Of note, 0.5 μM imatinib induced
50% inhibition of K562 cell growth, whereas up to 10 μM
imatinib was required to inhibit 50% cell growth in K562r cells
(Fig. 1A and B). Western blot
analysis showed that 1 μM imatinib caused the downregulation
of Bcr/Abl protein expression in K562 cells, whereas 25 μM
imatinib was required to observe measurable downregulation in K562r
cells (Fig. 1C and D).
BTZ synergistically interacts with
As2O3 to induce apoptosis in K562r cells
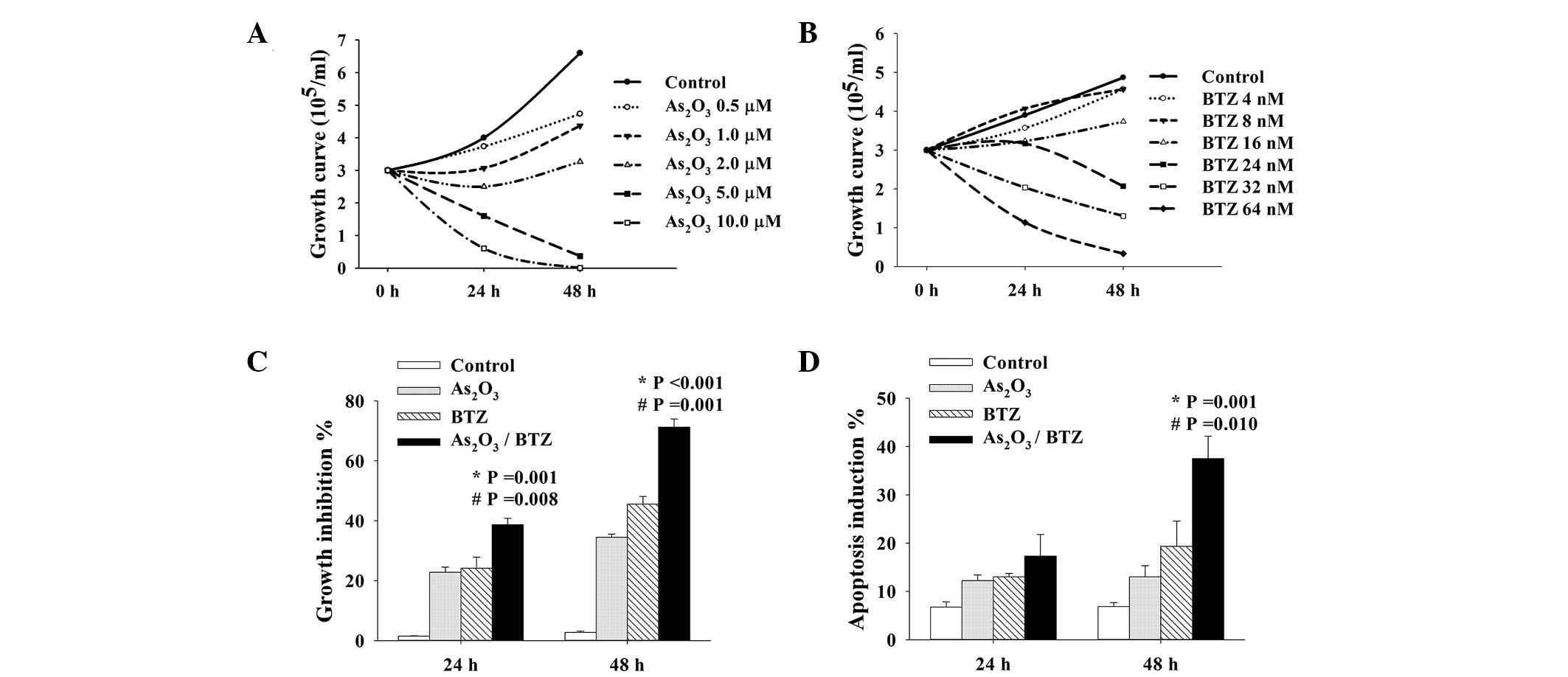
Treatment with either BTZ or
As2O3 alone inhibited cell growth in K562r
cells in a dose- and time-dependent manner (Fig. 2A and B). To evaluate the potential
synergistic effects of these two agents, BTZ and
As2O3 were used at concentrations of 24 nM
and 2 μM, respectively, the individual half maximal concentration
(IC50) of these agents in the inhibition of K562r cell
growth. Neither of these concentrations elicited significant
apoptosis-inducing effects. After 24–48 h of combined incubation
with BTZ and As2O3, growth inhibition and
apoptosis induction were significantly enhanced in K562r cells as
compared with those administered a single treatment (P<0.05)
(Fig. 2C and D).
Combined treatment with BTZ and
As2O3 results in enhanced caspase-3
activation and PARP and PKCδ cleavage
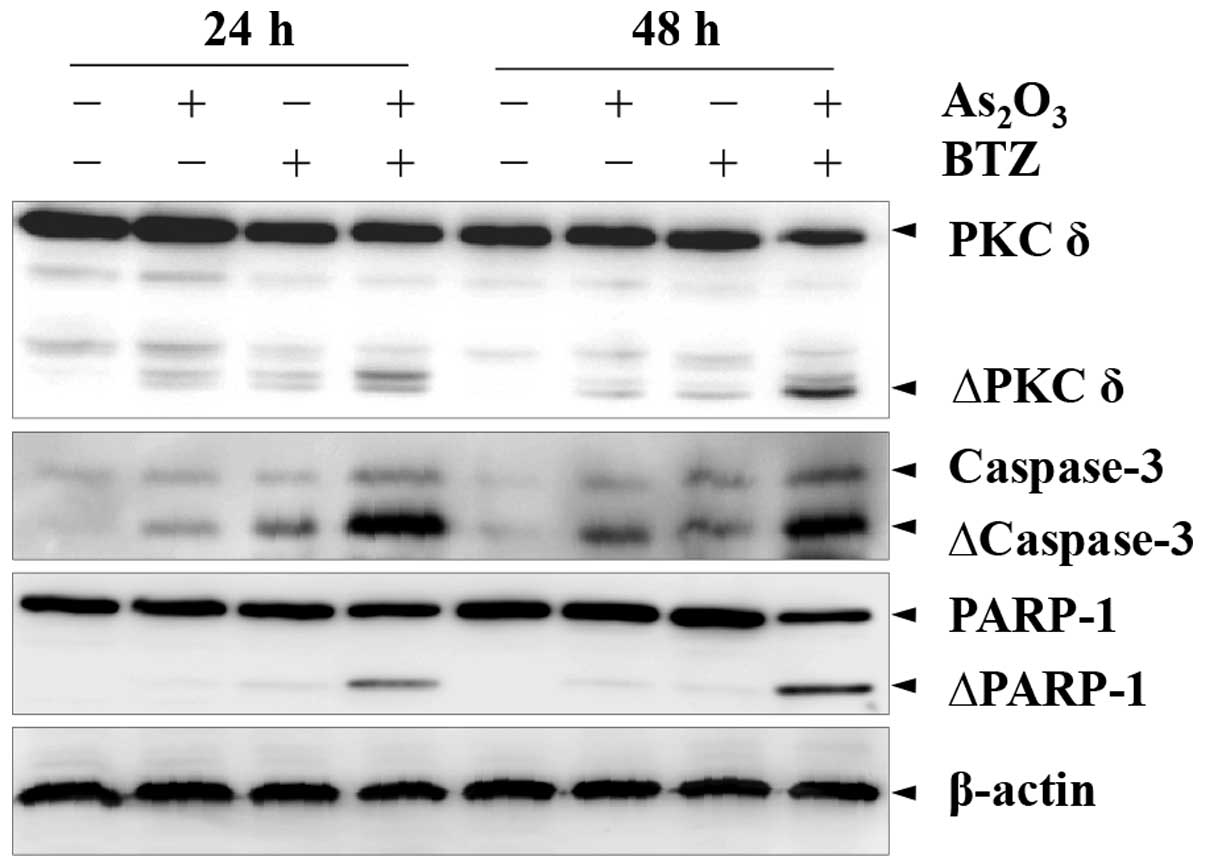
Western blot analysis was performed to assess
apoptosis-associated events during combined treatment with BTZ and
As2O3 in K562r cells. Treatment with 24 nM
BTZ or 2 μM As2O3 alone resulted in
only minimal effects, whereas combined treatment with BTZ and
As2O3 for up to 48 h resulted in a
significant increase in caspases-3 activation, as well as enhanced
PARP and PKCδ cleavage (Fig.
3).
Combined treatment with BTZ and
As2O3 downregulates Bcr/Abl mRNA and protein
expression
The Bcr/Abl kinase can inhibit apoptosis through
multiple mechanisms, leading Bcr/Abl+ cells to develop
resistance to apoptosis induced by conventional agents (5–7). In
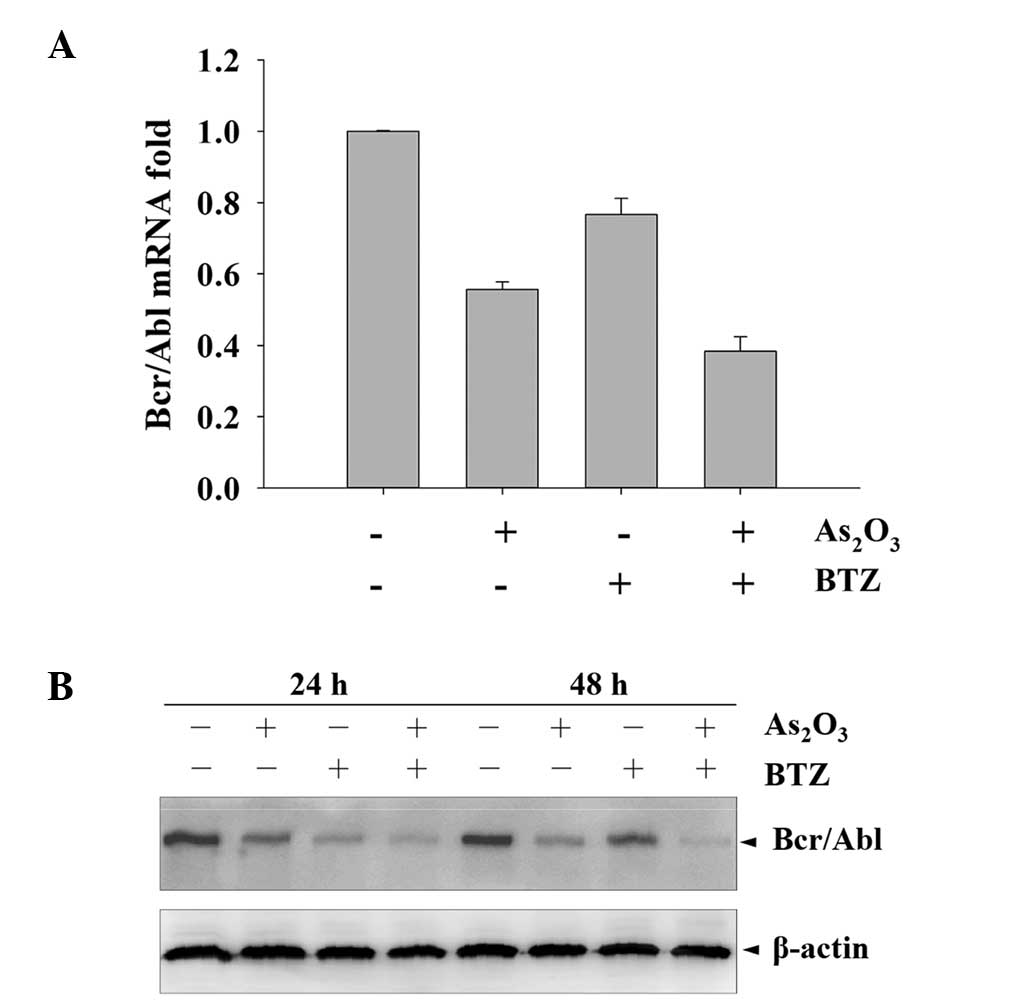
the present study, the effects of 24 nM BTZ and/or 2 μM
As2O3 on Bcr/Abl mRNA and protein expression
were examined. The results showed that single treatment with either
BTZ or As2O3 alone could decrease Bcr/Abl
mRNA and protein expression to a moderate extent; however, combined
treatment with the two agents resulted in a significant decrease in
Bcr/Abl expression in K562r cells (P<0.01), most markedly after
48 h of incubation (Fig. 4A and
B). This effect was further confirmed by the Bcr/Abl protein
downregulation analyzed by western blotting.
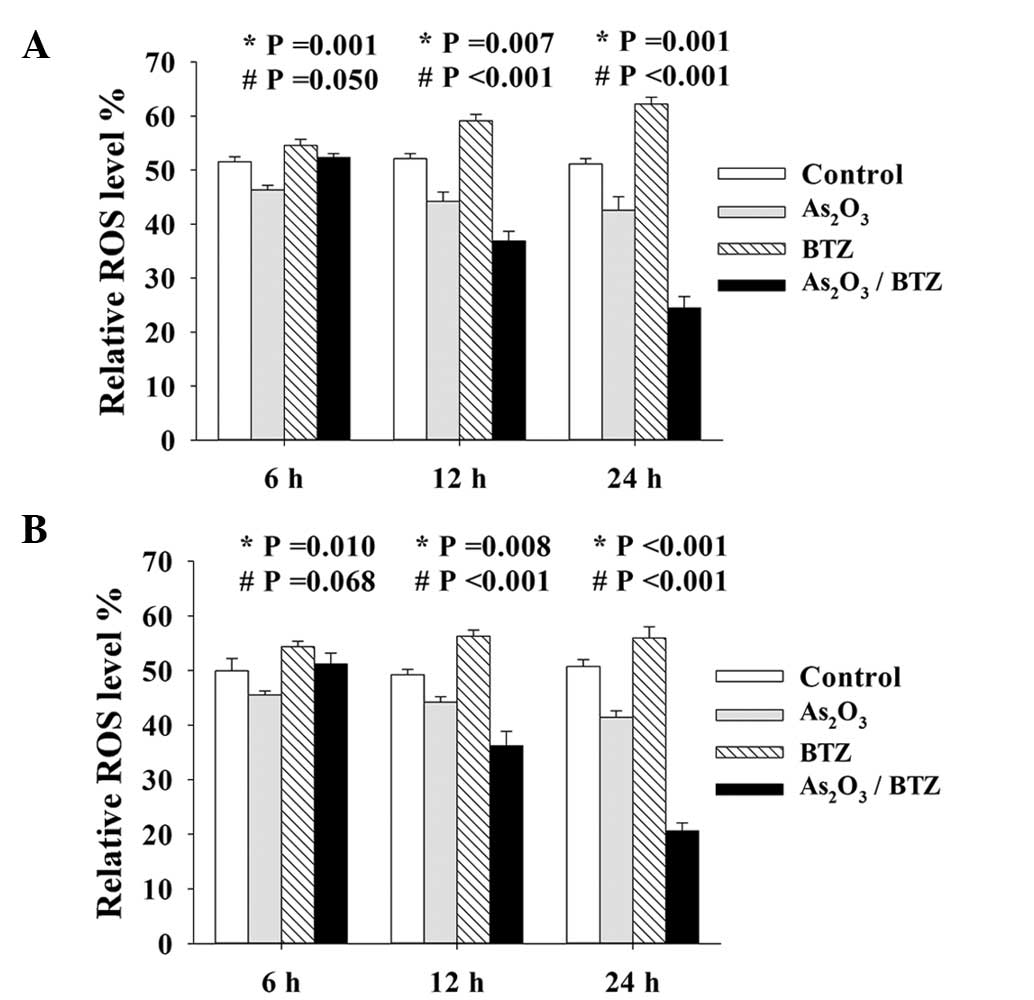
BTZ enhances
As2O3-induced downregulation of ROS in K562r
cells
As2O3 has been reported to
exert pro- or anti-apoptotic effects via manipulating cellular
oxidative stress, whilst BTZ is considered to be an inducer of ROS
(19–21). The effects of
As2O3 and/or BTZ on ROS production in K562r
cells were next investigated. The results showed that treatment
with As2O3 alone exhibited a marginal
decrease in ROS production, whilst that with BTZ elicited an
increase in ROS levels. Of note, although
As2O3 and BTZ exerted opposing effects,
combined treatment with both agents significantly downregulated ROS
production. Parallel experiments were performed in K562 cells and
comparable results were produced (Fig.
5A and B).
Discussion
Despite the success of imatinib, the emergence of
drug resistance and disease progression in CML remains a continuing
problem (22). In the present
in vitro study, it was found that the IC50 of
imatinib in K562r cells was up to 20-fold greater than that in K562
cells and clinically unachievable concentrations of up to 20
μM were required to downregulate Bcr/Abl protein. Thus, the
identification of alternative drugs or targets is of great
importance. It has been suggested that, instead of inhibiting the
tyrosine kinase activity, targeting the signaling pathways
downstream of Bcr/Abl or downregulating Bcr/Abl expression could be
an effective strategy to overcome imatinib resistance (23).
Following the success of BTZ in the treatment of
myeloma, proteasome inhibitors have gained interest as novel
treatment strategies for cancer. As summarized by a number of
studies (10,11,18,24),
proteasome inhibitors, including BTZ, may exhibit anti-leukemic
activities when used alone and may increase the sensitivity of
cancer cells to various anti-cancer agents, including flavopiridol,
histone deacetylase inhibitors and As2O3. The
combination treatment of As2O3 and BTZ has
been widely reported to induce apoptosis in numerous leukemia cell
lines (18,25–27).
Among these studies, a report by Yan et al (18) showed that
As2O3 and BTZ interacted synergistically to
enhance apoptosis in imatinib-sensitive K562 cells, predominantly
through PKCδ activation and decreased NF-κB activity. In the
present study, the data showed that clinically achievable
concentrations of BTZ and As2O3 could
synergistically inhibit cell growth and induce apoptosis in K562r
cells that were resistant to imatinib treatment. Thus, these data
support the potential use of this combinatory therapy in the
treatment of imatinib-resistant leukemia.
The caspase cascade is a protease system that
directly facilitates apoptosis. Activation of caspase-3, the
endpoint of this cascade, is critical in cleaving numerous
substrates, including the repair enzyme PARP, leading to DNA strand
breaks and eventually apoptosis. Caspase-3-mediated cleavage of
PARP has been shown to induce apoptosis in various malignant cell
lines, including Caco-2 colon cancer cells (28). Consistent with previous reports,
the present study also identified that the serial activity of
caspases was involved in inducing apoptosis in combined treatment
with BTZ and As2O3 in CML cells, further
supporting the use of this treatment in chemotherapies for CML
(18,26–27).
The Bcr/Abl kinase plays a key role in the
pathogenesis of Bcr/Abl+ malignancies by blocking
apoptosis through multiple pathways, including NF-κB, Akt and STAT
signaling cascades (29). As
reported both in vitro and in vivo, the
overexpression of Bcr/Abl kinase represents a major mechanism of
resistance to imatinib (22,30).
Wei et al (31) reported
that alantolactone could significantly induce apoptosis in K562r
cells and that this mechanism involved the knockdown of Bcr/Abl
protein expression, resulting in increased apoptosis. Thus, 24 nM
BTZ and 2 μM As2O3 were tested in
combination in K562r cells and the expression levels of Bcr/Abl
were analyzed. Of note, while each agent alone minimally affected
the expression of Bcr/Abl, combined treatment led to a marked
downregulation of Bcr/Abl mRNA and protein, thus providing
important insights into the mechanisms through which these drugs
exert their effects.
ROS act as important regulators of apoptosis through
numerous signaling pathways (19).
A previous study reported that As2O3 and BTZ
alone or in combination with other agents could induce ROS
production and subsequent apoptosis in several cell lines,
including NB4 cells (20).
However, certain studies have shown contradictory results, such as
a study by Stepnik et al (21), which showed that 5 μM
As2O3 induced >20-fold increase in the
generation of ROS in HL-60 cells after 6 h of exposure, whereas a
35% decrease was noted in K562 cells under the same conditions.
This indicated that the ROS response in cells was strongly
dependent on the cell origin, concentration of drugs, time of
exposure and other parameters (21). Additionally, the Bcr/Abl kinase is
known to enhance ROS production in certain Bcr/Abl-expressing
hematopoietic cells and imatinib could effectively reduce ROS
production during apoptosis (32–34).
In the present study, the effects of the
BTZ/As2O3 combination on ROS production at 6,
12 and 24 h of incubation were investigated. Of note, 2 μM
As2O3 moderately decreased ROS levels, while
20 nM BTZ had only a minor promoting effect on ROS levels; however,
the combined treatment markedly downregulated ROS production in a
time-dependent manner. These data showed that
BTZ/As2O3 treatment could directly reduce ROS
production in K562r cells and that Bcr/Abl downregulation further
enhanced this effect.
Collectively, this study showed that BTZ and
As2O3 synergistically induced apoptosis in
Bcr/Abl+ K562r cells via caspase-3 activation and
downregulation of Bcr/Abl kinase; reduced ROS production appeared
to be involved in the underlying mechanism. These findings indicate
that combined treatment of BTZ and As2O3 may
be a potential therapeutic regimen for the treatment of
imatinib-resistant CML.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation (grant nos. 81170509 and 30700334). The
authors would like to thank Professor J.V. Melo for kindly
providing the K562 and K562r cells.
References
|
1
|
Schiffer CA, Hehlmann R and Larson R:
Perspectives on the treatment of chronic phase and advanced phase
CML and Philadelphia chromosome positive ALL(1). Leukemia.
17:691–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Voss J, Posern G, Hannemann JR, Wiedemann
LM, Turhan AG, Poirel H, Bernard OA, Adermann K, Kardinal C and
Feller SM: The leukaemic oncoproteins Bcr-Abl and Tel-Abl
(ETV6/Abl) have altered substrate preferences and activate similar
intracellular signalling pathways. Oncogene. 19:1684–1690. 2000.
View Article : Google Scholar
|
|
3
|
Danial NN and Rothman P: JAK-STAT
signaling activated by Abl oncogenes. Oncogene. 19:2523–2531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kirchner D, Duyster J, Ottmann O, Schmid
RM, Bergmann L and Munzert G: Mechanisms of Bcr-Abl-mediated
NF-kappaB/Rel activation. Exp Hematol. 31:504–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bedi A, Barber JP, Bedi GC, el-Deiry WS,
Sidransky D, Vala MS, Akhtar AJ, Hilton J and Jones RJ:
BCR-ABL-mediated inhibition of apoptosis with delay of G2/M
transition after DNA damage: a mechanism of resistance to multiple
anticancer agents. Blood. 86:1148–1158. 1995.PubMed/NCBI
|
|
6
|
Bueno-da-Silva AE, Brumatti G, Russo FO,
Green DR and Amarante-Mendes GP: Bcr-Abl-mediated resistance to
apoptosis is independent of constant tyrosine-kinase activity. Cell
Death Differ. 10:592–598. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amarante-Mendes GP, McGahon AJ, Nishioka
WK, Afar DE, Witte ON and Green DR: Bcl-2-independent
Bcr-Abl-mediated resistance to apoptosis: protection is correlated
with up regulation of Bcl-xL. Oncogene. 16:1383–1390. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cavo M: Proteasome inhibitor bortezomib
for the treatment of multiple myeloma. Leukemia. 20:1341–1352.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caravita T, de Fabritiis P, Palumbo A,
Amadori S and Boccadoro M: Bortezomib: efficacy comparisons in
solid tumors and hematologic malignancies. Nat Clinl Pract Oncol.
3:374–387. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai Y, Rahmani M, Pei XY, Dent P and Grant
S: Bortezomib and flavopiridol interact synergistically to induce
apoptosis in chronic myeloid leukemia cells resistant to imatinib
mesylate through both Bcr/Abl-dependent and -independent
mechanisms. Blood. 104:509–518. 2004. View Article : Google Scholar
|
|
11
|
Yu C, Rahmani M, Conrad D, Subler M, Dent
P and Grant S: The proteasome inhibitor bortezomib interacts
synergistically with histone deacetylase inhibitors to induce
apoptosis in Bcr/Abl+ cells sensitive and resistant to
STI571. Blood. 102:3765–3774. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW,
Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z and Wang ZY: Use
of arsenic trioxide (As2O3) in the treatment
of acute promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360.
1997.PubMed/NCBI
|
|
13
|
Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ,
Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang
P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY and Chen Z: In vitro
studies on cellular and molecular mechanisms of arsenic trioxide
(As2O3) in the treatment of acute
promyelocytic leukemia: As2O3 induces NB4
cell apoptosis with downregulation of Bcl-2 expression and
modulation of PML-RAR alpha/PML proteins. Blood. 88:1052–1061.
1996.PubMed/NCBI
|
|
14
|
Mathews V, George B, Lakshmi KM,
Viswabandya A, Bajel A, Balasubramanian P, Shaji RV, Srivastava VM,
Srivastava A and Chandy M: Single-agent arsenic trioxide in the
treatment of newly diagnosed acute promyelocytic leukemia: durable
remissions with minimal toxicity. Blood. 107:2627–2632. 2006.
View Article : Google Scholar
|
|
15
|
Perkins C, Kim CN, Fang G and Bhalla KN:
Arsenic induces apoptosis of multidrug-resistant human myeloid
leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2,
or Bcl-x(L). Blood. 95:1014–1022. 2000.
|
|
16
|
Porosnicu M, Nimmanapalli R, Nguyen D,
Worthington E, Perkins C and Bhalla KN: Co-treatment with
As2O3 enhances selective cytotoxic effects of
STI-571 against Brc-Abl-positive acute leukemia cells. Leukemia.
15:772–778. 2001.PubMed/NCBI
|
|
17
|
Potin S, Bertoglio J and Bréard J:
Involvement of a Rho-ROCK-JNK pathway in arsenic trioxide-induced
apoptosis in chronic myelogenous leukemia cells. FEBS Lett.
581:118–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan H, Wang YC, Li D, Wang Y, Liu W, Wu YL
and Chen GQ: Arsenic trioxide and proteasome inhibitor bortezomib
synergistically induce apoptosis in leukemic cells: the role of
protein kinase Cdelta. Leukemia. 21:1488–1495. 2007. View Article : Google Scholar
|
|
19
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou WC, Jie C, Kenedy AA, Jones RJ, Trush
MA and Dang CV: Role of NADPH oxidase in arsenic-induced reactive
oxygen species formation and cytotoxicity in myeloid leukemia
cells. Proc Natl Acad Sci USA. 101:4578–4583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stepnik M, Ferlińska M, Smok-Pieniążek A,
Gradecka-Meesters D, Arkusz J and Stańczyk M: Assessment of the
involvement of oxidative stress and Mitogen-Activated Protein
Kinase signaling pathways in the cytotoxic effects of arsenic
trioxide and its combination with sulindac or its metabolites:
sulindac sulfide and sulindac sulfone on human leukemic cell lines.
Med Oncol. 29:1161–1172. 2012.
|
|
22
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Radujkovic A, Schad M, Topaly J, Veldwijk
MR, Laufs S, Schultheis BS, Jauch A, Melo JV, Fruehauf S and Zeller
WJ: Synergistic activity of imatinib and 17-AAG in
imatinib-resistant CML cells overexpressing BCR-ABL - Inhibition of
P-glycoprotein function by 17-AAG. Leukemia. 19:1198–1206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li QF, Yan J, Zhang K, Yang YF, Xiao FJ,
Wu CT, Wang H and Wang LS: Bortezomib and sphingosine kinase
inhibitor interact synergistically to induces apoptosis in
BCR/ABl+ cells sensitive and resistant to STI571 through
down-regulation Mcl-1. Biochem Biophys Res Commun. 405:31–36. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wen J, Feng Y, Huang W, Chen H, Liao B,
Rice L, Preti HA, Kamble RT, Zu Y, Ballon DJ and Chang CC: Enhanced
antimyeloma cytotoxicity by the combination of arsenic trioxide and
bortezomib is further potentiated by p38 MAPK inhibition. Leuk Res.
34:85–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Canestraro M, Galimberti S, Savli H,
Palumbo GA, Tibullo D, Nagy B, Guerrini F, Piaggi S, Cine N,
Metelli MR and Petrini M: Synergistic antiproliferative effect of
arsenic trioxide combined with bortezomib in HL60 cell line and
primary blasts from patients affected by myeloproliferative
disorders. Cancer Gene Cytogenet. 199:110–120. 2010. View Article : Google Scholar
|
|
27
|
He Y, Yang JM, Wang JM, Zhou H, Lü SQ and
Hu XX: Synergistic effects of arsenic trioxide and proteasome
inhibitor bortezomib on apoptosis induction in Raji cell line.
Zhongguo Shi Yan Xue Ye Xue Za Zhi. 16:794–798. 2008.(In
Chinese).
|
|
28
|
Ruemmele FM, Dionne S, Qureshi I, Sarma
DS, Levy E and Seidman EG: Butyrate mediates Caco-2 cell apoptosis
via up-regulation of pro-apoptotic BAK and inducing caspase-3
mediated cleavage of poly-(ADP-ribose) polymerase (PARP). Cell
Death Differ. 6:729–735. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fernandez-Luna JL: Bcr-Abl and inhibition
of apoptosis in chronic myelogenous leukemia cells. Apoptosis.
5:315–318. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bianchini M, De Brasi C, Gargallo P,
Gonzalez M, Bengió R and Larripa I: Specific assessment of BCR-ABL
transcript overexpression and imatinib resistance in chronic
myeloid leukemia patients. Eur J Haematol. 82:292–300. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei W, Huang H, Zhao S, Liu W, Liu CX,
Chen L, Li JM, Wu YL and Yan H: Alantolactone induces apoptosis in
chronic myelogenous leukemia sensitive or resistant to imatinib
through NF-κB inhibition and Bcr/Abl protein deletion. Apoptosis.
18:1060–1070. 2013.
|
|
32
|
Sattler M, Verma S, Shrikhande G, Byrne
CH, Pride YB, Winkler T, Greenfield EA, Salgia R and Griffin JD:
The BCR/ABL tyrosine kinase induces production of reactive oxygen
species in hematopoietic cells. J Biol Chem. 275:24273–24278. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stein SJ and Baldwin AS: NF-κB suppresses
ROS levels in BCR-ABL(+) cells to prevent activation of JNK and
cell death. Oncogene. 30:4557–4566. 2011.
|
|
34
|
Naughton R, Quiney C, Turner SD and Cotter
TG: Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway.
Leukemia. 23:1432–1440. 2009. View Article : Google Scholar : PubMed/NCBI
|