Introduction
The activation of microglia, the primary immune
cells of the central nervous system (CNS), occurs in almost all
neurological disorders (1) and is
often associated with the increased production of various
pro-inflammatory mediators, including nitric oxide (NO), inducible
nitric oxide synthase (iNOS), tumor necrosis factor (TNF)-α,
interleukin (IL)-1β, nuclear factor-κB (NF-κB), caspase-3 and heat
shock protein (HSP)60 (2–6), which all contribute to
neurodegeneration (7,8). The importance of microglial
activation in neurodegeneration has prompted speculation that the
inhibition of microglial activation, in particular the control of
neurotoxic factor production, may be an effective therapeutic
option for neurodegenerative diseases. Numerous microglia-targeted
pharmacotherapies, including protein kinase C inhibitors, microglia
inhibiting factor, and various Chinese medicinal herb extracts,
have been proposed to inhibit the activation of microglia and to
promote neuronal survival in vivo (9–11).
However, the inability of these drugs to penetrate the blood-brain
barrier, in addition to side-effects that they may produce, limit
their long-term use in the clinical setting.
Naloxone is a structural analog of morphine and an
effective antagonist of the classic opioid receptors that are
widely expressed on cells of the central and peripheral nervous
systems (12). Administration of
naloxone has been demonstrated to be beneficial in the treatment of
experimental models of stroke, myocardial and brain ischemia, brain
trauma, spinal cord injuries and septic shock (13–15).
Naloxone has been demonstrated to attenuate the degeneration of
dopaminergic neurons by inhibition of β-amyloid
peptide(1–42)-induced microglial activation and degeneration of
cortical and mesencephalic neurons, suggesting that naloxone may
have potential therapeutic efficacy for the treatment of
Parkinson’s and Alzheimer’s disease (16,17).
These studies raise the possibility that naloxone binds to sites
other than opioid receptors and exerts activity that does not
involve the opioid receptor system. Compared with other microglial
inhibitors, naloxone has several advantages, including its ability
to penetrate the CNS and that it produces fewer side-effects,
presenting a potential novel therapeutic option for
neuroprotection.
Although naloxone has been demonstrated to inhibit
lipopolysaccharide (LPS)-induced microglial activation in the CNS,
the underlying mechanism is not well understood. HSP60 was
demonstrated to be released extracellularly in cardiac myocytes
during heart failure, and induces apoptosis by binding to Toll-like
receptor (TLR)-4 (18,19). The aim of the present study was to
examine whether HSP60 is involved in the neuroprotective effects of
naloxone in LPS-induced inflammatory injury in BV2 microglia.
Materials and methods
Chemicals
Naloxone and LPS were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Antibodies against β-actin, NF-κB and TLR-4
(Abcam, Cambridge, MA, USA), anti-HSP60 and anti-heat shock factor
(HSF)1 antibodies (Stressgen, San Diego, CA, USA) and an
anti-caspase-3 antibody (Cell Signaling Technology, Inc., Beverly,
MA, USA) were acquired. Proteinase inhibitor cocktails were
purchased from Merck Chemicals (Whitehouse Station, NJ, USA). IL-6,
IL-1β and TNF-α ELISA kits were from eBioscience (eBioscience, CA,
USA). Bicinchoninic acid (BCA) and enhanced chemiluminescence (ECL)
kits were acquired from Pierce (Rockford, IL, USA). Dulbecco’s
modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) came
from Gibco (Grand Island, NY, USA). Griess reagent (for identifying
NO) and nitric oxide synthase (iNOS) kits were from Jiancheng
Bioengineering Institute (Nanjing, China). The Cell Counting Kit-8
(CCK-8) was obtained from Beyotime (Nanjing, China).
Microglial cell culture
BV2 mouse microglia (Cell Bank, Shanghai, China)
were cultured in DMEM supplemented with 10% FBS, penicillin (100
U/ml) and streptomycin (100 g/ml). Cultures were maintained at 37°C
in a humidified incubator gassed with 95% O2 and 5%
CO2. Naloxone was dissolved in phosphate-buffered saline
(PBS). Cells were treated with the indicated concentrations of
naloxone for 24 h following the administration of LPS (200 ng/ml)
for the indicated time period.
Cell viability assay
Cell viability was measured using the CCK-8. Cells
(7.5×103 cells in 100 μl culture medium/well) were
seeded in 96-well plates. CCK-8 solution (10 μl) was added to each
well and the cultures were incubated at 37°C for 90 min. Absorbance
at 450 nm was measured using an immunoreader (Bio-Rad, Beijing,
China). The results were plotted as the mean ± standard deviation
of three separate experiments having four determinations per
experiment for each experimental condition. The cell survival ratio
was calculated by normalization to control.
Enzyme-linked immunosorbent assay
(ELISA)
The levels of IL-6, IL-1β and TNF-α in culture
medium were quantified according to the manufacturer’s
instructions. Absorbance was determined at 450 nm using a
microplate reader (Bio-Rad).
Western blotting
Following treatment, BV2 cells were washed with PBS
three times and lysed with radioimmunoprecipitation assay buffer
(Sigma-Aldrich). The protein concentration was determined with the
BCA kit according to the manufacturer’s instructions. Equal
quantities of protein were loaded and run on SDS/polyacrylamide
gels and then transferred to a polyvinylidine fluoride membrane.
Membranes were blocked with 5% dried milk and incubated with
primary antibodies in Tris-buffered saline with Tween 20 overnight
at 4°C. After being rinsed in milk-TBST, blots were incubated in
the horseradish peroxidase-conjugated secondary antibodies. The
target proteins were detected using the ECL kit and X-ray films
(Kodak, Shanghai, China).
Statistical analysis
Statistical significance was determined using
one-way analysis of variance. P<0.05 was considered to indicate
a statistically significant difference. All data are presented as
the mean ± standard error of the mean.
Results
Naloxone promotes the viability of BV2
microglia
To determine whether naloxone has an effect on the
apoptosis of LPS-stimulated BV2 cells, CCK-8 assay was performed.
The results indicated that treatment of microglia with 0.1–2.0 μM
naloxone for up to 24 h significantly increased the viability of
LPS-stimulated BV2 cells, compared with that of the LPS group
(Fig. 1). Naloxone at a
concentration of 1.0 μM exhibited the maximal protection, so this
concentration was selected for the subsequent experiments. The
results indicated that naloxone has a positive effect on the
viability of LPS-stimulated BV2 microglia.
Naloxone inhibits HSP60 protein
expression and release in LPS-stimulated BV2 microglia
Levels of HSP60 expression and release were measured
in activated BV2 cells. Western blot results indicated that LPS
induced an increase in the expression levels of HSP60 compared with
levels in controls, and that naloxone significantly inhibited this
increase (Fig. 2A). The HSP60
promoter has been reported to have a heat shock element that is the
binding site for HSF-1, which regulates HSP60 gene expression
(20). Therefore, HSF-1 levels
were investigated, and demonstrated to exhibit the same expression
pattern as that of HSP60, indicating that HSP60 expression is
driven by HSF-1 (Fig. 2B). HSP60
has been reported to translocate extracellularly upon stress in
order to exert injury effects (11). ELISA results demonstrated that
HSP60 was released upon LPS-induced activation of BV2 cells, and
this increased extracellular HSP60 may be suppressed by naloxone
(Fig. 2C).
Naloxone inhibits TLR-4, NF-κB and
caspase-3 expression
TLR-4 mediates inflammatory responses in activated
microglia. In the present study, it was demonstrated that TLR-4
expression levels in LPS-stimulated BV2 cells were reduced by
naloxone (Fig. 3A). The expression
levels of the p65 subunit of NF-κB, which is a pivotal factor in
the TLR-4 pathway, increased following LPS stimulation, but this
effect was markedly inhibited by the addition of naloxone (Fig. 3B). Caspase-3 is upstream of NF-κB
in this signaling pathway, and inhibition of caspase-3 has been
demonstrated to prevent neuronal loss in brain diseases involving
activated microglia. Thus, in the current study, the effects of
naloxone on caspase-3 expression were examined (Fig. 3C). Caspase-3 expression was
suppressed in LPS-stimulated BV2 cells following naloxone
exposure.
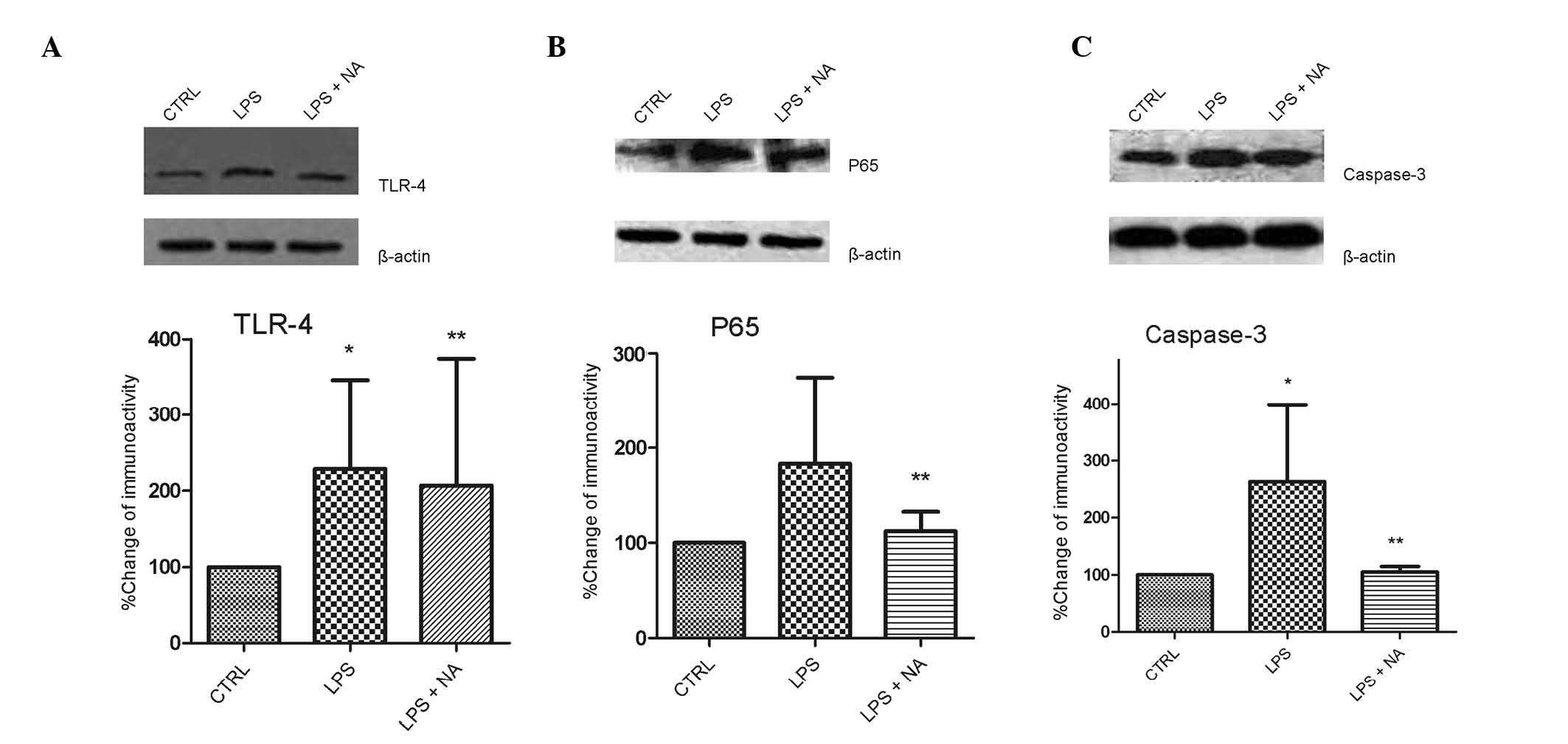 | Figure 3NA inhibited the increased expression
of TLR-4, NF-κB and caspase-3 in LPS-stimulated BV2 microglia.
Cells were pretreated with LPS for 0.5 h, followed by an incubation
with 1.0 μM naloxone for 24 h. The lysates were probed by
immunoblotting with antibodies against TLR-4, p65, caspase-3 and
β-actin. The graphs display ratios of the signal intensities of (A)
TLR-4/β-actin, (B) p65/β-actin and (C) caspase-3/β-actin. Each
experiment was derived from at least 6 independent cultures.
*P<0.05 vs. CTRL, **P<0.05 vs. LPS
group. CTRL, control; LPS, lipopolysaccharide; NA, naloxone; TLR,
Toll-like receptor; NF-κB, nuclear factor-κB. |
Naloxone inhibits the production of
proinflammatory factors
Using ELISA assay, naloxone suppression of the
release of proinflammatory factors, including NO, iNOS, TNF-α,
IL-1β and IL-6, was investigated in LPS-stimulated BV2 cells. As
shown in Fig. 4, 24-h naloxone
treatment of LPS-stimulated BV2 cells resulted in significant
reductions in the levels of the aforementioned factors in culture
media compared those following LPS stimulation alone. These results
implied that naloxone effectively suppresses the production of
neurotoxic factors in activated microglia.
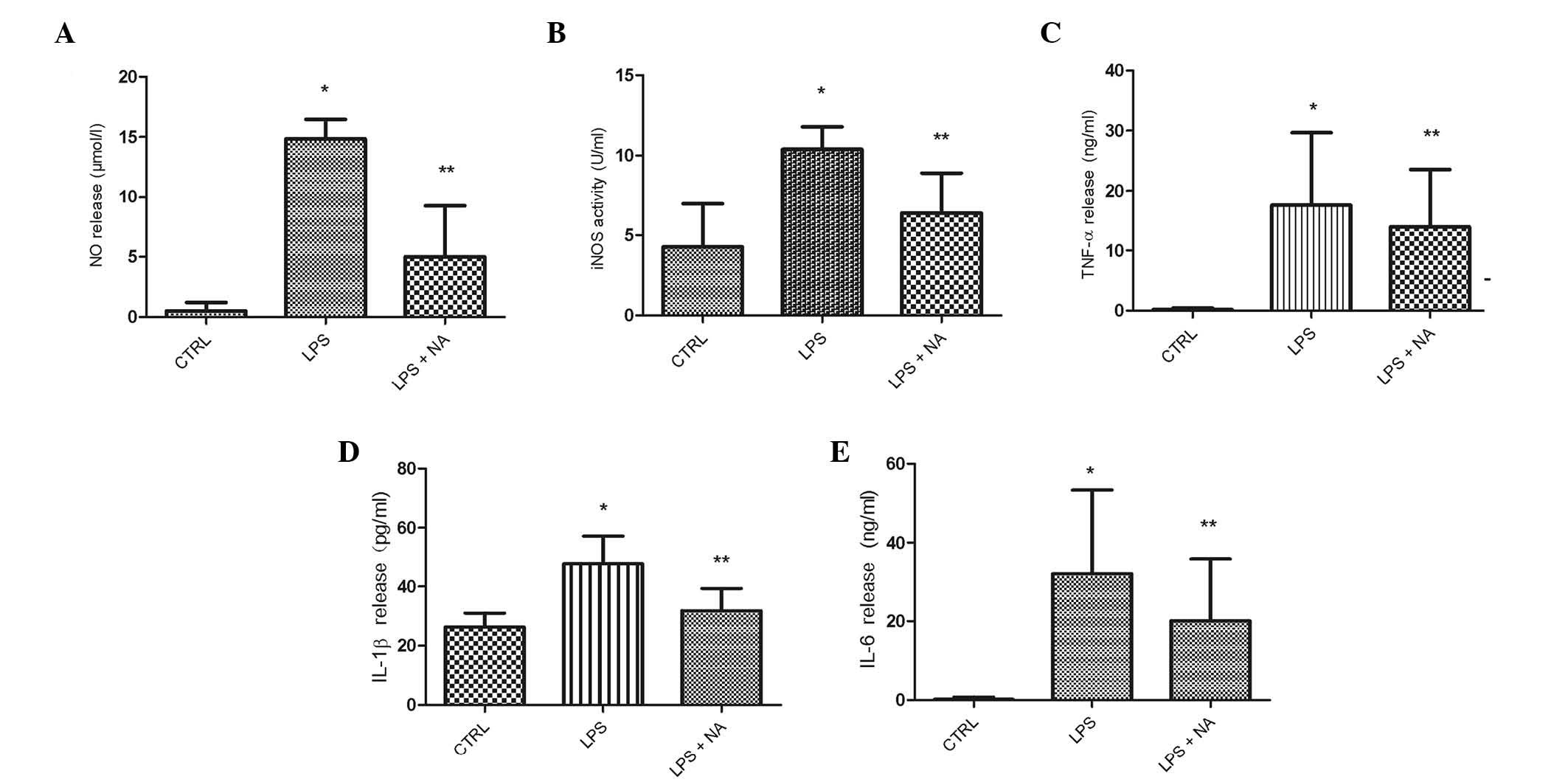 | Figure 4NA reduced the release of NO, iNOS,
TNF-α, IL-6 and IL-1β in LPS-stimulated BV2 microglia. Cells were
pretreated with LPS for 0.5 h, followed by an incubation with 1.0
μM NA for 24 h. Extracellular levels of (A) NO and (B) iNOS were
assayed with Griess reagent and iNOS kits, respectively, and levels
of (C) TNF-α, (D) IL-1β and (E) IL-6 were measured using ELISA
assay. The results are presented as the mean ± standard error of
three separate experiments performed in triplicate.
*P<0.05 vs. CTRL. **P<0.05 vs. LPS
group. NO, nitric oxide; CTRL, control; LPS, lipopolysaccharide;
NA, naloxone; iNOS, inducible nitric oxide synthase; TNF, tumor
necrosis factor; IL, interleukin; ELISA, enzyme-linked
immunosorbent assay. |
Discussion
HSP60 is primarily considered to be a mitochondrial
protein, but a number of studies have established that HSP60 is
also involved in apoptosis. When HSP60 is in the cytosol or
mitochondria, it is antiapoptotic and protective. However, when
HSP60 is in the plasma membrane or extracellular space, it is
associated with apoptosis. Previous studies detected HSP60
expression on the exofacial surface of myocytes, where it was a
potential antibody target or innate immune system ligand of TLR-4
(18,19). TLR-4 has been shown to be present
in microglia (21), and HSP60 is a
ligand for TLR-4 in the immune system (22).
A number of studies have identified neuroprotective
effects of naloxone, and it was demonstrated to inhibit LPS-induced
microglial activation in the CNS (16,17).
However, the mechanisms involved remain unclear. In the present
study, it was hypothesized that LPS triggered HSP60 release from
microglia, and extracellular HSP60 binds to TLR-4 on the surface of
microglia, inducing apoptosis by activating the NF-κB pathway. The
results supported these hypotheses. A pretreatment of 1 μM naloxone
effectively suppressed the activation of microglia. While LPS
stimulation increased the expression and release of HSP60, naloxone
significantly suppressed these effects. HSP expression is regulated
by HSFs. In the current study, HSF-1 expression levels increased as
did those of HSP60 when triggered by LPS and then were inhibited by
naloxone. Thus, the inhibitory effect of naloxone on HSP60 may
occur through inhibition of its transcription factor HSF-1. The
overexpressed HSP60 released extracellularly may act as an innate
immune signal to further activate microglia.
TLR-4-mediated microglial activation induced cell
death, which is a mechanism by which activated immune cells are
eliminated. Kim et al (18)
demonstrated that extracellular HSP60 activated TLR-4 by binding to
it at a different site to that which LPS binds, leading to cytokine
production and cardiac myocytes apoptosis. Release of TNF-α may
then lead to further myocyte apoptosis and increased HSP60
expression through activation of NF-κB (23). In the present study, it was
demonstrated that TLR-4 may be activated on microglia not only by
LPS but also by extracellular HSP60, and that activated TLR-4 was
effectively inhibited by naloxone. LPS and HSP60 binding to TLR-4
may trigger a cascade of signaling events resulting in the
activation of downstream effector molecules such as NF-κB and
caspase 3 and culminating in the production of proimflammatory
immune mediators, including TNF-α, IL-1β, IL-6 and IL-8. Release of
NO and chemokines by these cells has also been reported (24). NF-κB is the major transcription
factor in the induction of the transcription of pro-inflammatory
genes. The activation of NF-κB has been demonstrated to lead to
ischemia-induced neuronal injury (25). Caspase-3 is crucial for apoptosis
and CNS inflammation (26) and
when caspase-3/7 is blocked, activated microglia are non-toxic to
neighboring neurons (27).
Activation of the NF-κB-p65 cascade induces HSP60 production and
release following oxidative stress (19) possibly by the binding of NF-κB to
the HSP60 gene promoter (23).
TNF-α is also a mediator of NF-κB signaling and drives the
increased expression of HSP60, which can be reversed by p65
inhibition (23). Levels of HSP60,
NF-κB and TNF-α are simultaneously decreased by naloxone. By
demonstrating the marked inhibition of the expression of caspase-3
and the NF-κB downstream mediator p65, and production of NO, iNOS,
TNF-α, IL-1β and IL-6 by naloxone treatment, the findings of the
present study suggest that the neuroprotective and
anti-inflammatory effects of naloxone may be due to inhibition of
the HSP60-TLR-4-NF-κB pathway.
In summary, to the best of our knowledge, the
results of the present study indicated for the first time that
naloxone may exert its neuroprotective action through
HSP60-TLR-4-NF-κB inhibition to prevent the overactivation of
microglia.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 31060140, 31260243,
81060034, 81060126 and 81060112); The Project Sponsored by the SRF
for ROCS, State Education Ministry (SEM); The Program for New
Century Excellent Talents in University, SEM for Professor Yin
Wang; and the Grant of 2012 from Ningxia Medical University for Mr
Yunhong Li.
References
|
1
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanisch UK and Kettenmann H: Microglia:
active sensor and versatile effector cells in the normal and
pathologic brain. Nat Neurosci. 10:1387–1394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gehrmann J, Matsumoto Y and Kreutzberg GW:
Microglia: intrinsic immuneffector cell of the brain. Brain Res
Rev. 20:269–287. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Innamorato NG, Lastres-Becker I and
Cuadrado A: Role of microglial redox balance in modulation of
neuroinflammation. Curr Opin Neurol. 22:308–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lynch MA: The multifaceted profile of
activated microglia. Mol Neurobiol. 40:139–156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li YH, Teng P, Wang Y, Zhang YM, Ma CJ, Pu
J, et al: Expression and regulation of HSP60 in activated microglia
cells. J Ningxia Med Coll. 8:712–714. 2011.
|
|
7
|
Zhang D, Sun L, Zhu H, Wang L, Wu W, Xie J
and Gu J: Microglial LOX-1 reacts with extracellular HSP60 to
bridge neuroinflammation and neurotoxicity. Neurochem Int.
61:1021–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lehnardt S, Schott E, Trimbuch T, Laubisch
D, Krueger C, Wulczyn G, Nitsch R and Werber JR: A vicious cycle
involving release of heat shock protein 60 from injured cells and
activation of toll-like receptor 4 mediates neurodegeneration in
the CNS. J Neurosci. 28:2320–2331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thanos S, Mey J and Wild M: Treatment of
the adult retina with microglia-suppressing factors retards
axotomy-induced neuronal degradation and enhances axonal
regeneration in vivo and in vitro. J Neurosci. 13:455–466.
1993.
|
|
10
|
Thanos S: The Relationship of Microglial
Cells to Dying Neurons During Natural Neuronal Cell Death and
Axotomy-induced Degeneration of the Rat Retina. Eur J Neurosci.
3:1189–1207. 1991. View Article : Google Scholar
|
|
11
|
Teng P, Li YH, Cheng W, Zhou L, Shen Y and
Wang Y: Neuroprotective effects of Lycium barbarum polysaccharides
in lipopolysaccharide-induced BV2 microglial cells. Mol Med Rep.
7:1977–1981. 2013.PubMed/NCBI
|
|
12
|
Smith AP and Lee NM: Pharmacology of
dynorphin. Annu Rev Pharmacol Toxicol. 28:123–140. 1988. View Article : Google Scholar
|
|
13
|
Faden AI and Salzman S: Pharmacological
strategies in CNS trauma. Trends Pharmacol Sci. 13:29–35. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fallis RJ, Fisher M and Lobo RA: A double
blind trial of naloxone in the treatment of stroke. Stroke.
15:627–629. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kan MN, Chen YT and Lee AY: Naloxone
reversal of ischemic arrhythmia is stereospecific and suggests role
of endogenous opioid peptides in ischemic heart disease. Proc Soc
Exp Biol Med. 200:518–521. 1992. View Article : Google Scholar
|
|
16
|
Liu B, Du L and Hong JS: Naloxone protects
rat dopaminergic neurons against inflammatory damage through
inhibition of microglia activation and superoxide generation. J
Pharmacol Exp Ther. 293:607–617. 2000.
|
|
17
|
Liu Y, Qin L, Wilson BC, An L, Hong JS and
Liu B: Inhibition by naloxone stereoisomers of β-amyloid peptide
(1–42)-induced superoxide production in microglia and degeneration
of cortical and mesencephalic neurons. J Pharmacol Exp Ther.
302:1212–1219. 2002.
|
|
18
|
Kim SC, Stice JP, Chen L, Jung JS, Gupta
S, Wang Y, Baumgarten G, Trial J and Knowlton AA: Extracellular
heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res.
105:1186–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin L, Kim SC, Wang Y, Gupta S, et al:
HSP60 in heart failure: abnormal distribution and role in cardiac
myocyte apoptosis. Am J Physiol Heart Circ Physiol.
293:H2238–H2247. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hansen JJ, Bross P, Westergaard M, Nielsen
MN, Eiberg H, et al: Genomic structure of human mitochondrial
chaperonin genes: HSP60 and HSP10 are localised head to head on
chromosome 2 separated by a bidirectional promoter. Hum Genet.
112:71–77. 2003. View Article : Google Scholar
|
|
21
|
Aloisi F: Immune function of microglia.
Glia. 36:165–179. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kol A, Lichtman AH, Finberg RW, Libby P
and Kurt-Jones EA: Cutting edge: heat shock protein (HSP) 60
activates the innate immune response: CD14 is an essential receptor
for HSP60 activation of mononuclear cells. J Immunol. 164:13–17.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Chen L, Hagiwara N and Knowlton
AA: Regulation of heat shock protein 60 and 72 expression in the
failing heart. J Mol Cell Cardiol. 48:360–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohashi K, Burkart V, Flohé S and Kolb H:
Cutting edge: heat shock protein 60 is a putative endogenous ligand
of the toll-like receptor-4 complex. J Immunol. 164:558–561. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J, Zhou Y, Mueller-Steiner S, Chen
LF, Kwon H, Yi S, Mucke L and Gan L: SIRT1 protects against
microglia-dependent amyloid-β toxicity through inhibiting NF-κB
signaling. J Biol Chem. 280:40364–40374. 2005.PubMed/NCBI
|
|
26
|
Soria JA, Arroyo DS, Gaviglio EA,
Rodriguez-Galan MC, Wang JM and Iribarren P: Interleukin 4 induces
the apoptosis of mouse microglial cells by a caspase-dependent
mechanism. Neurobiol Dis. 43:616–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burguillos MA, Deierborg T, Kavanagh E,
Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P,
Englund E, Venero JL and Joseph B: Caspase signalling controls
microglia activation and neurotoxicity. Nature. 472:319–324. 2011.
View Article : Google Scholar : PubMed/NCBI
|
















