Introduction
c-Jun N-terminal protein kinase (JNK) is a subfamily
of the mitogen-activated protein kinase (MAPK) superfamily
(1). JNK has two ubiquitously
expressed isoforms, JNK1 and JNK2, and a tissue-specific isoform,
JNK3; all of which have two different splicing forms (p54 and p46)
(2). JNK is activated by
sequential protein phosphorylation through a MAP kinase module in
response to a variety of extracellular stimuli, including tumor
necrosis factor-α (TNF-α) (3) and
UV light (4). Activation of JNK is
regulated by scaffold proteins, including JNK-interacting proteins,
β-arrestin, and nuclear factor of κ light polypeptide gene enhancer
in B-cells (NF-κB) (5). JNK
regulates multiple cellular activities, ranging from gene
expression to apoptosis, and there is evidence that JNK also
contributes to cell survival. Genetic evidence from a previous
study reveals that JNK1 and JNK2 are involved in the survival of
neuronal cells in mouse fore and hindbrain regions during
development (5).
Activation of tumor necrosis factor receptor 1
(TNFR1) by TNF-α leads to the recruitment of the
TNFRSF1A-associated via death domain (TRADD) protein, which in turn
recruits Fas (TNFRSF1A)-associated via death domain, leading to
caspase-8 activation and apoptosis via the extrinsic pathway
(6). However, activation of TNF-α
is not always cytotoxic to cells, as recruitment of TNF-α receptor
associated factor 2 and receptor TNFRSF-interacting
serine-threonine kinase by TRADD can activate NF-κB and JNK, which
have a pivotal role in cell proliferation and survival (7). The involvement of JNK in
TNF-α-mediated apoptosis is the subject of much debate (8). Inhibition of NF-κB results in
sustained activation of JNK, which may directly promote
TNF-α-mediated apoptosis (9).
Despite the fact that sustained activation of JNK promotes cell
death, the molecular mechanisms by which JNK contributes to
TNF-α-mediated apoptosis remain to be addressed.
The intrinsic apoptotic pathway is the result of
activation of mitochondria-mediated cell death events, including
changes in mitochondrial membrane permeability and the subsequent
release of proapoptotic factors (10). Although the apoptotic pathway
through death receptors and the pathway through mitochondria are
capable of operating independently, previous studies suggest that a
crosstalk exists between the two pathways (11).
The second mitochondria-derived activator of
caspases (Smac/DIABLO) is a 29-kDa mitochondrial protein which,
following an apoptotic trigger, is processed to a 23-kDa mature
protein and translocates to the cytosol (12). In addition to its interaction with
X-linked inhibitor of apoptosis protein (XIAP), Smac/DIABLO has
been shown to bind other inhibitor of apoptosis (IAP) proteins
including c-IAP1, c-IAP2, survivin and baculoviral Op-IAP (13,14).
Mature Smac/DIABLO binds the BIR3 domain of XIAP with an N-terminus
recognition motif similar to that which binds caspase-9. This same
amino terminal sequence of Smac/DIABLO also forms a stable complex
with the BIR2 domain of XIAP, meaning that Smac/DIABLO can act as a
competitor in the XIAP-dependent inhibition of caspase-3 and
caspase-7 (12).
Previous studies have shown that JNK mediates the
release of cytochrome c (15) and Smac (16). Furthermore, JNK has been linked to
apoptosis; specifically, two serine residues (Ser63 and 73) within
c-Jun are direct targets for JNK (17). However, the mechanism by which Smac
is released from the mitochondria has yet to be elucidated. The aim
of the present study was to investigate whether JNK1 is activated
during apoptosis induced by tumor necrosis factor-α (TNF-α) and
whether the activation of JNK1 is functionally associated with Smac
release from the mitochondria in HeLa cells.
Materials and methods
Reagents
Cycloheximide was obtained from Sigma-Aldrich (St.
Louis, MO, USA) and dissolved in phosphate-buffered saline (PBS) at
a concentration of 5 mg/ml. TNF-α was obtained from Biosource
International Inc. (Camarillo, CA, USA). Caspase-3 substrate,
Ac-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC), was
purchased from BD Biosciences Pharmingen (San Diego, CA, USA).
Cell culture, apoptotic induction and
transfection
HeLa cells (American Type Culture Collection,
Manassas, VA, USA) were maintained at 37°C and 5% CO2 in
DMEM supplemented with 10% heated-inactivated FBS and
antibiotics-antimycotics (all Gibco-BRL, Carlsbad, CA, USA). For
apoptotic induction, cells were treated with 5 ng/ml TNF-α and 5
μg/ml cycloheximide. As TNF-α alone had no effect on the viability
of the HeLa cells, the protein synthesis inhibitor cycloheximide
was used in combination with TNF-α for the induction of apoptosis
in HeLa cells. Transient transfection was performed in 100-mm
culture plates using PolyFect (Qiagen, Valencia, CA, USA). Cells
(5×105) were seeded and transfected using 10 μg total
DNA. The transfected cells were incubated for 24 h and subsequently
treated for 0, 3 or 6 h with TNF-α (5 ng/ml) and cycloheximide (5
μg/ml) for subcellular fractionation, caspase-3 activity assay and
western blotting.
Western blot analysis
Cell pellets were washed with PBS and lysed in lysis
buffer (0.5% Triton X-100, 20 mM Tris, pH 7.5, 2 mM
MgCl2, 1 mM dithiothreitol (DTT), 1 mM EGTA, 50 mM
β-glycerophosphate, 25 mM NaF, 1 mM Na3VO4,
100 μg/ml phenylmethanesulfonyl fluoride (PMSF), 10 mM protease
inhibitor cocktail in phosphate buffer, pH 7.0) on ice for 1 h,
then centrifuged at 15,000 × g for 20 min at 4°C. Lysates were
subjected to SDS-PAGE and transferred to a polyvinylidene
difluoride membrane (Gibco-BRL). The membrane was blocked with 5%
non-fat dried milk in PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM
Na2HPO4-7H2O, 1.4 mM
KH2PO4, pH 7.4) containing 0.05% Tween 20 and
incubated in a 1:1,000 dilution of the following antibodies: Rabbit
polyclonal anti-JNK1, rabbit polyclonal anti-poly ADP-ribose
polymerase (PARP), rabbit polyclonal anti-β-actin, and rabbit
polyclonal anti-cytochrome c oxidase IV (COX IV) (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), mouse monoclonal
anti-phospho-JNK, mouse monoclonal anti-Smac/DIABLO and mouse
monoclonal anti-FLAG antibodies (Cell Signaling Technology, Inc.,
Beverly, MA, USA). Bands were visualized with horseradish
peroxidase-conjugated antibodies (Pierce, Rockford, IL, USA) and
the Enhanced Chemiluminescence system (Pierce, Rockford, IL,
USA).
Preparation of cytosolic and
mitochondrial fractions
The cell pellet was washed with PBS and was
suspended in 2 volumes of buffer A (20 mM HEPES, pH 7.5, 1.5 mM
MgCl2, 10 mM KCl, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, and
10 mM protease inhibitor cocktail in phosphate buffer, pH 7.0)
containing 250 nM sucrose. The pellet was homogenized using a
Dounce homogenizer (BBI-8530718, Thomas Scientific, Swedesboro, NJ,
USA), centrifuged at 800 × g for 10 min at 4°C and the supernatant
was collected and further centrifuged at 100,000 × g for 1 h at
4°C. The supernatant was used as cytosolic extract, and the cell
pellet was lysed and used as the mitochondrial fraction
Caspase-3 activity assay
Cells were cultured in 100-mm dishes and treated
with TNF-α and cycloheximide for the indicated time periods. Cell
lysates were prepared in the lysis buffer (50 mM Tris-HCl, pH 7.5,
150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1 mM PMSF)
supplemented with 10 mM protease inhibitor cocktail in phosphate
buffer, pH 7.0. The lysates were centrifuged at 10,000 × g for 10
min and the supernatant was collected. Cell lysates were added to
the reaction buffer (20 mM HEPES pH 7.5, 10% glycerol, 2 mM DTT)
containing 25 μM Ac-DEVD-AMC in 96-well plates. Lysates were
incubated at 37°C for 1 h. Fluorescence of the cleavage product was
measured using a SpectraFluor F129003 (Tecan, Maenndorf,
Switzerland) at excitation and emission wavelengths of 360 nm and
460 nm, respectively
Peptide filter binding assay
The kinase reaction was performed as follows:
Recombinant JNK1 was incubated with synthetic peptide in the
presence of 5X reaction buffer (40 mM MOPS, pH 7.0, 1 mM EDTA),
[32-P] ATP and distilled water. The reaction tube was placed in a
microcentrifuge and a pulse-spin was used to wash the components
into the base of the tube. The tube was incubated for 10 min at
30°C and the reactants subsequently transferred onto the center of
P81 phosphocellulose paper (Millipore, Billerica, MA, USA) and
washed 3 times with 0.75% phosphoric acid for 5 min at room
temperature. The assay squares were washed once with acetone for 5
min at room temperature and transferred to scintillation vials, in
which scintillation cocktail was added. The sample was read in a
scintillation counter. Scintillation vials, cocktail and counter
were purchased from PerkinElmer (Waltham, MA, USA).
Results
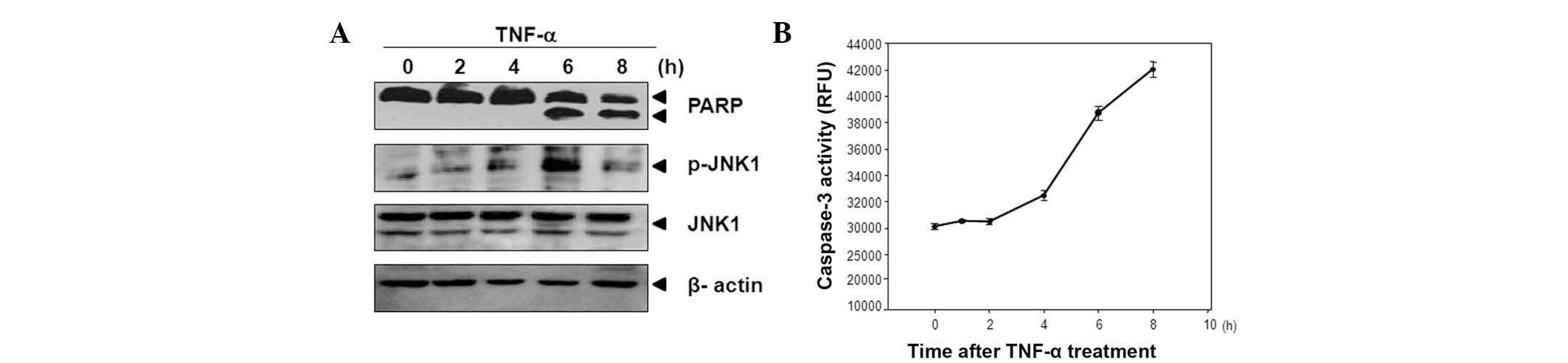
JNK1 is upregulated during TNF-α-induced
apoptosis in HeLa cells
To examine whether JNK1 activity is upregulated
during TNF-α-induced apoptosis, HeLa cells were treated with TNF-α
(5 ng/ml) and cycloheximide (5 μg/ml) for the indicated time
periods.
It was determined that JNK1 is phosphorylated at 6 h
and that subsequently the PARP cleavages occur (Fig. 1A). As shown in Fig. 1B, the levels of caspase-3 activity
also increased 6 h after TNF-α treatment. Using these data, it was
assumed that activation of JNK1 following TNF-α treatment may be
associated with the promotion of apoptosis during TNF-α-induced
apoptosis.
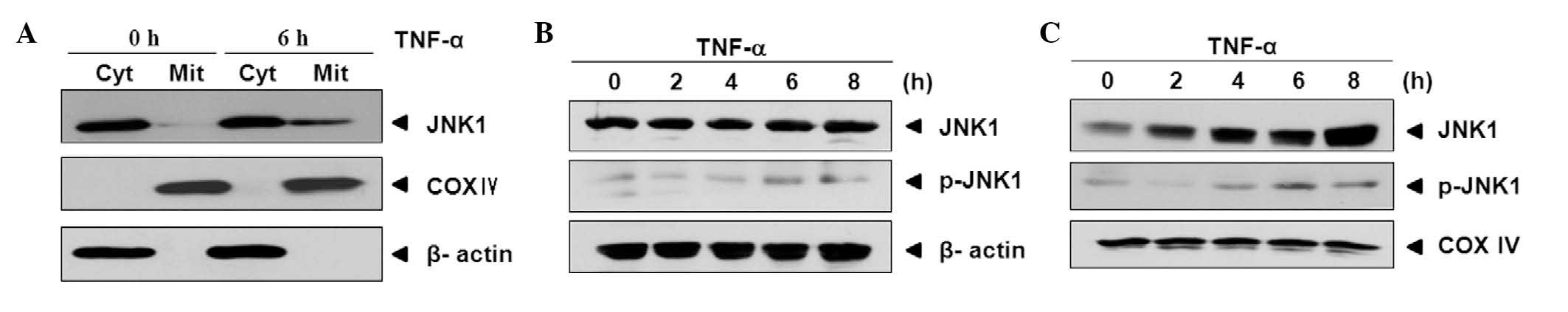
JNK1 is translocated from cytosol to
mitochondria by TNF-α treatment
It has been reported that JNK mediates the release
of Smac after translocation to mitochondria (16). Hence, it was examined whether JNK1
is translocated from cytosol to mitochondria during TNF-α-induced
apoptosis. As shown in Fig. 2, the
levels of JNK1 were increased in mitochondria after 6 h compared
with the levels of JNK1 at 0 h. To confirm this result, cells were
treated with TNF-α for 0, 2, 4, 6 and 8 h and cytosolic and
mitochondrial fractions were prepared. The same levels of JNK1 and
a slight increase in the levels of activated JNK1 (p-JNK1) were
observed in the cytosol (Fig. 2B).
However the levels of JNK1 and p-JNK1 exhibited different patterns
of changes in the expression levels of the mitochondria compared
with those in the cytosol. Following TNF-α treatment, the levels of
JNK1 and p-JNK1 were increased gradually in a time-dependent manner
(Fig. 2C).
These results indicate that JNK1 is activated and
translocated into the mitochondria during TNF-α-induced
apoptosis.
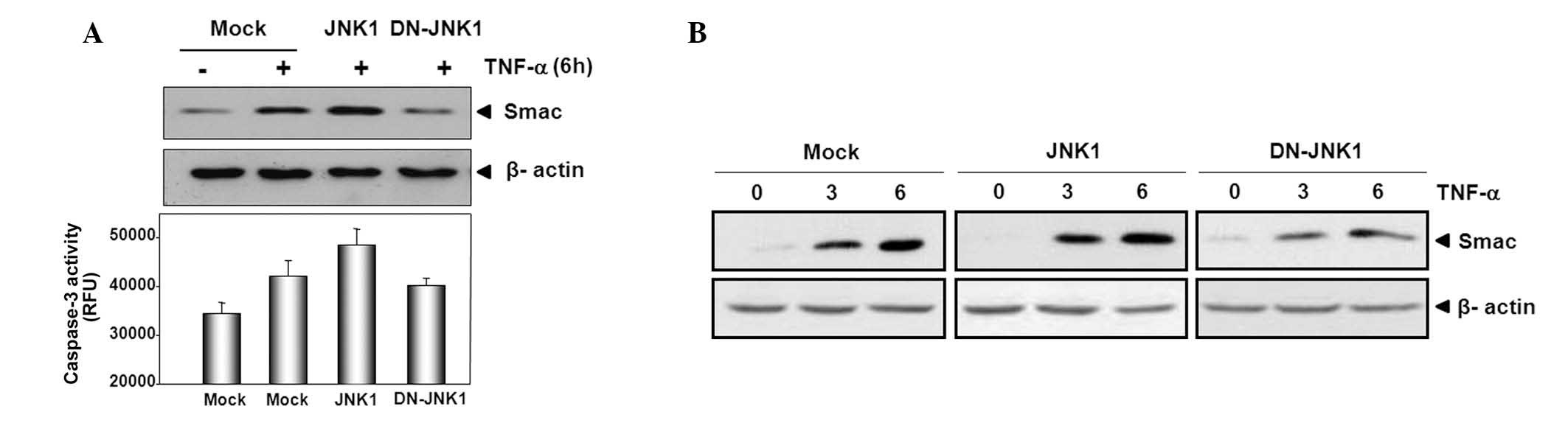
JNK1 activity is involved in Smac release
from mitochondria during TNF-α-induced apoptosis
The present study investigated the role of JNK1
translocation and activation in the release of Smac from
mitochondria during TNF-α-induced apoptosis. HeLa cells were
transfected with a control (mock), JNK1 or DN-JNK1 and then were
treated with TNF-α and cycloheximide 24 h later. Subsequently,
subcellular fractionation was performed and the caspase-3 activity
levels were measured. It was observed that the release of Smac from
mitochondria was promoted by transient expression of JNK1 compared
with release in the control, while DN-JNK1 suppressed the release
of Smac when compared with that in the control (Fig. 3A). The levels of caspase-3 activity
exhibited a similar pattern compared with that of Smac release;
activation of caspase-3 increased in HeLa cells overexpressing JNK1
as compared with that of those with mock transfectant, however
DN-JNK1 suppressed the activity of caspase-3.
To confirm these results, 24 h after transfection
with mock, JNK1 or DN-JNK1, cells were treated with TNF-α for 0, 3
or 6 h and cytosolic fractions were prepared. Smac release was
promoted slightly by transient expression of JNK1 in a
time-dependent manner when compared with that in cells transfected
with the mock. However, Smac release was suppressed in HeLa cells
transfected with DN-JNK1 compared with that in mock cells. These
results suggest that JNK1 has a pro-apoptotic role and that JNK1
activity is involved in Smac release from mitochondria during
TNF-α-induced apoptosis.
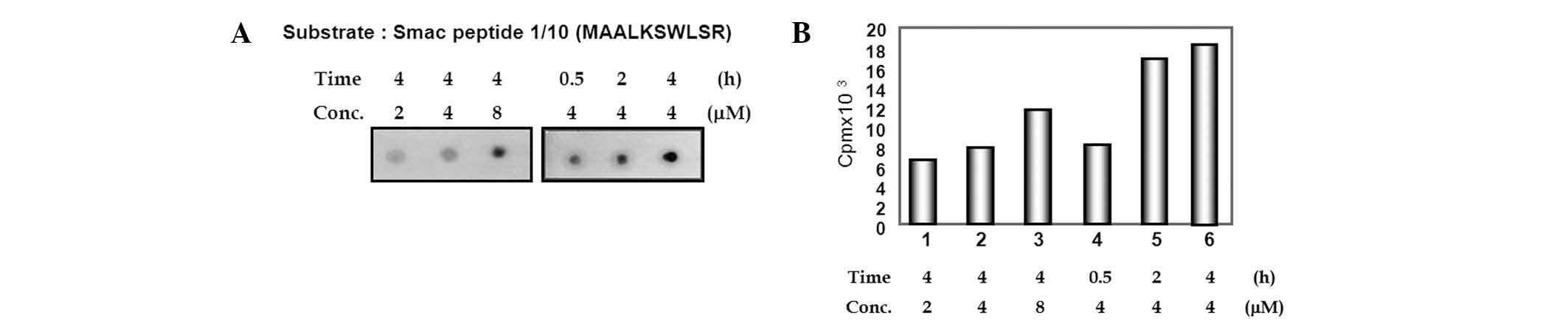
The N-terminus of Smac is phosphorylated
by JNK1
It has previously been reported that the
phosphorylation of Smac by JNK3 attenuates the apoptotic
progression induced by the anticancer drug etoposide (18). Subsequently, the present study
investigated the role of Smac phosphorylation in the release of
Smac by JNK1. The potential phosphorylation sites of full-length
Smac by JNK1 were studied using the public phosphorylation
prediction database NetPhos 2.0 server (http://www.cbs.dtu.dk/services/NetPhos/). When full
sequences of Smac were submitted to NetPhos 2.0, several serine and
threonine residues were predicted to be phosphorylated by protein
kinases. From the results of the prediction by NetPhos 2.0,
N-terminal serines 6 and 9 of Smac were suggested as putative
phosphorylation sites. To examine whether JNK1 could phosphorylate
this site, a peptide containing residues of serines 6 and 9 was
synthesized; pep0110 corresponded to amino acids 1–10 of
full-length Smac. A peptide filter binding assay was performed,
which increased the radioactivity of phosphorylated pep0110 in
proportion to the quantity of added peptide and the length of time
(Fig. 4).
JNK1-mediated phosphorylation of Smac at
N-terminal serine 6 residue is involved in Smac release during
TNF-α-induced apoptosis
Experiments were conducted to investigate whether
the phosphorylation of Smac at the N-terminus accounts for the
effect of JNK1 on Smac. To examine whether the JNK1-mediated
phosphorylation of Smac at the serine residues 6 or 9 is associated
with the mitochondrial release of Smac, an Smac which could not be
phosphorylated by JNK1 was created by substituting serine residues
6 (Smac S6A) and 9 (Smac S9A) with alanine. A cotransfection study
was then performed with wild-type (WT) Smac and the mutant
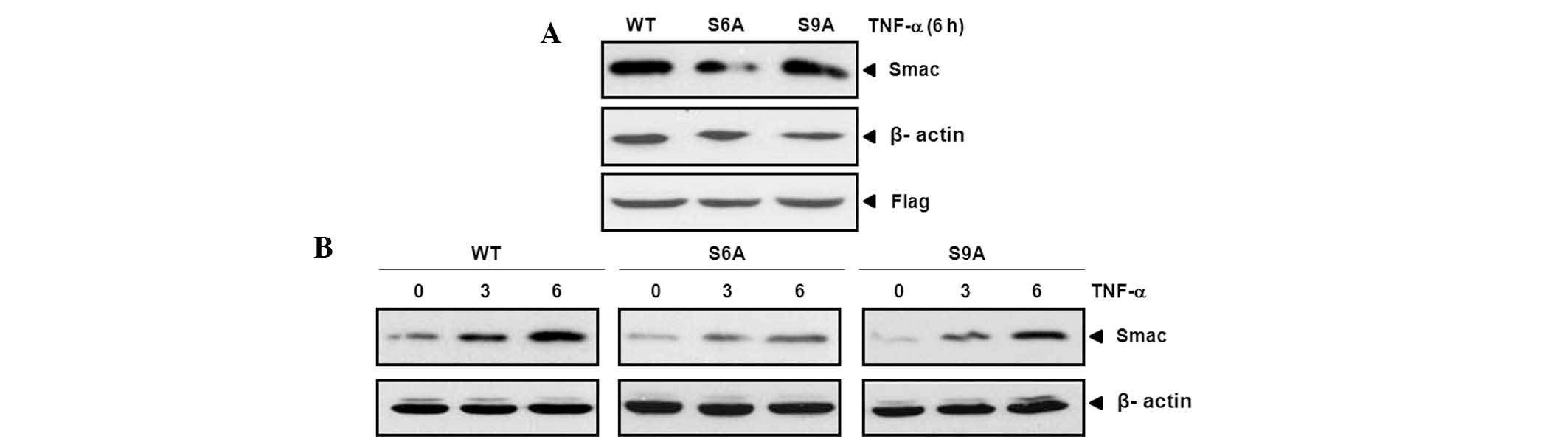
versions, Smac S6A and S9A. It was revealed that Smac release was
suppressed by cotransfection of JNK1 with Smac S6A in comparison
with other cotransfectants (Fig.
5A). To confirm these results, the same experiments were
performed over time. The release of Smac was also revealed to be
suppressed by cotransfection of JNK1 with Smac S6A in a
time-dependent manner (Fig.
5B).
Discussion
JNK regulates a number of important cellular events,
ranging from gene expression to apoptosis. JNK is activated in
response to a variety of extracellular stimuli, including TNF-α
(3) and UV (4). The involvement of JNK in
TNF-α-mediated apoptosis has been debated (8). Sustained activation of JNK may
directly promote TNF-α-mediated apoptosis (9), however activation of NF-κB and JNK
together is pivotal in cell proliferation and survival (7). Despite the fact that sustained
activation of JNK promotes cell death, the molecular basis of how
JNK contributes to TNF-α-mediated apoptosis remains to be
addressed.
The intrinsic apoptotic pathway is the result of
changes in mitochondrial membrane permeability and subsequent
release of proapoptotic factors (10). Notably, previous studies have shown
that JNK mediates the release of cytochrome c (15) and Smac (16). However, the mechanism of action by
which Smac is released from the mitochondria is yet to be
determined.
In the present study, it was demonstrated that JNK1
is activated and translocated from cytosol to mitochondria in HeLa
cells and that it can regulate Smac release from mitochondria by
phosphorylation of Smac at the N-terminal serine 6 residue during
TNF-α-induced apoptosis.
Different patterns of change in expression were
observed in the intracellular levels of JNK1 and its active
phospho-forms using immunoblotting assay. The levels of activated
JNK1 were sharply increased at 6 h, followed by PARP cleavage and
caspase-3 activation. From these results it was deduced that
increased levels of JNK1 activity may be associated with the
promotion of apoptosis during TNF-α-induced apoptosis.
Subsequently, whether JNK1 is translocated to
mitochondria when apoptosis is induced by TNF-α treatment in HeLa
cells was investigated and confirmed in the present study. Chauhan
et al (16) have reported
that JNK is involved in the release of the mitochondrial protein
Smac. The present study examined the role of JNK1 in the promotion
of Smac release during TNF-α-induced apoptosis. The results
revealed that the levels of Smac release were increased by JNK1,
but suppressed by DN-JNK1. These results indicate that JNK1
influences TNF-α-induced apoptosis by regulating Smac release.
It has previously been reported that phosphorylation
of Smac by JNK3 attenuates its interaction with XIAP (18), so the present study investigated
whether Smac release by JNK1 is due to phosphorylation. Following
prediction analysis using NetPhos 2.0, JNK1 was revealed to
phosphorylate Smac peptide0110; a Smac peptide containing residues
of serines 6 and 9.
To verify whether the JNK1-mediated phosphorylation
of Smac at the serine residues 6 or 9 is involved in the
mitochondrial release of Smac, a cotransfection study was performed
with Smac WT and the mutant versions, Smac S6A and S9A. It was
demonstrated that Smac release was suppressed by cotransfection of
JNK1 with SmacS6A in comparison with that of the other
cotransfectants. These results indicate that JNK1-mediated
phosphorylation of Smac at the N-terminal serine 6 residue is
involved in Smac release.
In conclusion, the data presented in this study
revealed that the phosphorylation of Smac at serine 6 residue by
JNK1 is involved in promoting Smac release from mitochondria. These
results suggest that Smac is a major physiological substrate of
JNK1 in the regulation of apoptosis, particularly in
mitochondria.
Acknowledgements
This study was supported by the College of
Pharmacy-Specialized Research Fund (from the Institute for New Drug
Development) of Keimyung University.
References
|
1
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng Y, Ren X, Yang L, Lin Y and Wu X: A
JNK-dependent pathway is required for TNFalpha-induced apoptosis.
Cell. 115:61–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanos T, Marinissen MJ, Leskow FC, et al:
Phosphorylation of c-Fos by members of the p38 MAPK family. Role in
the AP-1 response to UV light. J Biol Chem. 280:18842–18852. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Genet Dev. 12:14–21. 2002.
View Article : Google Scholar
|
|
6
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SY, Reichlin A, Santana A, Sokol KA,
Nussenzweig MC and Choi Y: TRAF2 is essential for JNK but not
NF-kappaB activation and regulates lymphocyte proliferation and
survival. Immunity. 7:703–713. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Smaele E, Zazzeroni F, Papa S, et al:
Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic
JNK signalling. Nature. 414:308–313. 2001.PubMed/NCBI
|
|
9
|
Tang G, Yang J, Minemoto Y and Lin A:
Blocking caspase-3-mediated proteolysis of IKKbeta suppresses
TNF-alpha-induced apoptosis. Mol Cell. 8:1005–1016. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
11
|
Roy S and Nicholson DW: Cross-Talk in Cell
Death Signaling. J Exp Med. 192:f21–f26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chai J, Du C, Wu JW, Kyin S, Wang X and
Shi Y: Structural and biochemical basis of apoptotic activation by
Smac/DIABLO. Nature. 406:855–862. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu G, Chai J, Suber TL, et al: Structural
basis of IAP recognition by Smac/DIABLO. Nature. 408:1008–1012.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilkinson JC, Wilkinson AS, Scott FL,
Csomos RA, Salvesen GS and Duckett CS: Neutralization of
Smac/Diablo by inhibitors of apoptosis (IAPs). A
caspase-independent mechanism for apoptotic inhibition. J Biol
Chem. 279:51082–51090. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tournier C, Hess P, Yang DD, et al:
Requirement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chauhan D, Li G, Hideshima T, et al:
JNK-dependent release of mitochondrial protein, Smac, during
apoptosis in multiple myeloma (MM) cells. J Biol Chem.
278:17593–17596. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dérijard B, Hibi M, Wu IH, et al: JNK1: a
protein kinase stimulated by UV light and Ha-Ras that binds and
phosphorylates the c-Jun activation domain. Cell. 76:1025–1037.
1994.
|
|
18
|
Park BD, Ham YM, Jeong HJ, et al:
Phosphorylation of Smac by JNK3 attenuates its interaction with
XIAP. Biochem Biophys Res Commun. 361:994–999. 2007. View Article : Google Scholar : PubMed/NCBI
|