Introduction
Epithelial ovarian cancer (EOC) is a lethal
gynecological malignancy, the cells of which have highly aggressive
and widely metastatic capabilities. The majority of patients with
EOC are diagnosed at an advanced stage with extensive peritoneal
metastases, leading to a high frequency of postoperative recurrence
and mortality within five years (1). However, little is currently known
about the underlying mechanisms of EOC tumor oncogenesis,
invasiveness and metastasis.
The epithelial-mesenchymal transition (EMT) is a
process constantly activated during tumor cell migration, invasion,
intravasation and angiogenesis (2,
3). It is the biological process
by which an epithelial cell transforms into a more motile
mesenchymal cell, through the induction of mesenchymal markers
including N-cadherin and Vimentin (4). Whilst undergoing EMT, the cells lose
their epithelial hallmarks, which are represented by
E-cadherin-regulated cell-cell junctions and apical polarity
(5). The cells start to exhibit a
scattered and spindle-shaped mesenchymal morphology, reorganizing
their cytoskeletons to facilitate migration and resist anoikis
(5). The process of EMT is thought
to be important to ovarian carcinogenesis and progression. It is
associated with ovarian cancer peritoneal metastasis, as a
consequence of the loosened contact of tumor cells with neighboring
cells and disruption of the tissue architecture. These cellular
changes enable the EOC cells to dissociate from the tumor mass and
migrate to the peritoneal cavity (6–9).
Furthermore, side population (SP) cells, which are considered to be
cancer stem-like cells (CSCs), were identified as having an
epithelial origin. Differentiation of SP cells into non-SP cells
was previously demonstrated to be concomitant to the process of EMT
and resulted in tumor cell heterogeneity, engraftment and
progression (10). The reverse
process of EMT, mesenchymal to epithelial transition (MET), has
also been observed during both neoplasm development and tumor
colonization (7,11,12).
It has been proposed that metastasizing tumor cells reverse the
process of EMT, in order to implant at distant sites where the
epithelial growth pattern is promoted (12–14).
During EMT, the TGF-β-Smad signaling pathway is
associated with transcriptomic reprogramming (15). Regulating transcripts of EMT
include: Smad1, Smad2, Smad3, Smad5 and Smad8, which are
receptor-activated molecules; Smad4, a complex of Smad2 and Smad3
and a common-mediator; and Smad6 and Smad7, which are inhibitory
proteins. It has previously been indicated that Smad7 represses the
TGF-β1 signaling pathway by combining with the activated TGF-β1
receptor (TβRI) and preventing the phosphorylation of Smad2 and
Smad3 (16). However, Smad7 has
other effects, beyond its well-known inhibition of the canonical
TβRI-Smad pathway. It has heterogeneous functions in the
progression of numerous cancers. In previous studies of
nasopharyngeal carcinoma and cervical cancer, TGF-β1 was shown to
induce the overexpression of Smad7, which resulted in the malignant
transformation and tumorigenicity (17,18).
In addition, the ectopic expression of Smad7 has been shown to
block the progression of cancer in endometriosis (19). However, the association between
Smad7 and TGF-β1 stimulation in ovarian cancer EMT remains largely
unknown.
In the present study, the expression of Smad7 during
the process of EMT in different ovarian cancer cell lines was
investigated. Analyzing the association between Smad7 and EOC
pathogenesis may advance the understanding of the mechanisms behind
the progression of EOC, and provide potential targets for
anti-cancer therapy.
Materials and methods
Cell lines and culture conditions
ES-2, SK-OV-3 and OVCAR-3, human EOC cell lines,
were obtained from American Type Culture Collection (Manassas, VA,
USA). The HO-8910 and highly metastasizing HO-8910PM daughter line
(20) EOC cells were purchased
from the Cell Bank of the Shanghai Institute of Biochemistry and
Cell Biology, (Shanghai, China). The cells were cultured in
RPMI-1640 media (HyClone Laboratories Inc., Logan, UT, USA),
supplemented with 10% fetal bovine serum (FBS; HyClone) and 1%
penicillin/streptomycin (Gibco Life Technologies, Carlsbad, CA,
USA). OVCAR-3 cells were maintained in RPMI-1640 containing 20% FBS
and 0.01 mg/ml bovine insulin (Sigma-Aldrich, St. Louis, MO, USA),
according to the culturing guidelines. For TGF-β1 stimulation, 10
ng/ml TGF-β1 (R&D Systems, Inc., Minneapolis, MN, USA) was
added and the media was refreshed every 12 h.
SP cell detection and selection
The procedure of Hoechst 33342 staining was
performed as described by previous methods (21). Briefly, the cells were suspended at
1×106 cells/ml in Dulbecco’s modified Eagle’s medium,
containing 2% FBS, and stained with 5 μg/ml of the Hoechst 33342
fluorescent dye (Sigma-Aldrich) at 37°C for 90 minutes. Verapamil
was added prior to Hoechst 33342 staining, to reliably ensure the
identity and purity of the SP cells. Hoechst 33342-low cells were
then analyzed and sorted using an ultraviolet laser cytometer
(Beckman Coulter, Brea, CA, USA). The data analysis was performed
using FlowJo software (www.flowjo.com).
Polymerase chain reaction (PCR)
Total RNA was extracted from the EOC cells using
TRIzol® reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA). A total of 1 μg of RNA was used for each reverse
transcription (RT) reaction. RT and PCR reactions were performed
using the PrimeScript RT-PCR kit (Takara Bio, Inc., Otsu, Japan).
Quantitative PCR (qPCR) analysis was performed using the ABI
StepOne Plus Real-time PCR system (Applied Biosystems Life
Technologies, Foster City, CA, USA) and SYBR Green Real-time PCR
Master Mix (Takara Bio Inc.). The oligonucleotide primers used are
listed in Tables I and II.
 | Table IOligonucleotide primers used for
reverse transcription-polymerase chain reaction analysis. |
Table I
Oligonucleotide primers used for
reverse transcription-polymerase chain reaction analysis.
| Gene names | Accession | Size (bp) | Sequence | Tm (°C) |
|---|
| hE-cadherin (F) | NM_004360.3 | 425 |
TCCCTTCCCTTGAGATGA | 46 |
| hE-cadherin (R) | | |
GCCGATAGAATGAGACCCT | |
| hN-cadherin (F) | NM_001792.3 | 539 |
TCGGGTAATCCTCCCAAAT | 52 |
| hN-cadherin (R) | | |
CCACTGCCTTCATAGTCAAA | |
| hVimentin (F) | NM_003380.3 | 382 |
GCCAGGCAAAGCAGGAGTC | 44 |
| hVimentin (R) | | |
TGGGTATCAACCAGAGGGAG | |
| hSmad7 (F) | NM_005904.3 | 438 |
ACAACCGCAGCAGTTACCC | 44 |
| hSmad7 (R) | | |
AAACGAGGACGAGAAGAAGAA | |
| hSmad7 (F) | NM_005904.3 | 302 |
CCCTCCTTACTCCAGATACCC | 46 |
| hSmad7 (R) | | |
GCTGACTCTTGTTGTCCGAAT | |
| hSmad3 (F) | NM_005902.3 | 106 |
GCACCATCCGCATGAGCTTT | 44 |
| hSmad3 (R) | | |
TGCAAAGGCCCATTCAGGT | |
| hSmad4 (F) | NM_005359.5 | 692 |
AGCCATCGTTGTCCACTG | 48 |
| hSmad4 (R) | | |
GACCCAAACATCACCTTCAC | |
| hβ-actin (F) | NM_001614.3 | 159 |
GCCCTGAGGCACTCTTCCA | 52 |
| hβ-actin (R) | | |
TTGCGGATGTCCACGTCA | |
| Table IIOligonucleotide primers used for
real-time quantitative polymerase chain reaction analysis. |
Table II
Oligonucleotide primers used for
real-time quantitative polymerase chain reaction analysis.
| Gene names | Accession | Size (bp) | Sequence |
|---|
| hE-cadherin
(F) | NM_004360.3 | 114 |
TTCCCTCGACACCCGATTC |
| hE-cadherin
(R) | | |
TAGGTGGAGTCCCAGGCGTA |
| hN-cadherin
(F) | NM_001792.3 | 201 |
ACAGTGGCCACCTACAAAGG |
| hN-cadherin
(R) | | |
CCGAGATGGGGTTGATAATG |
| hVimentin (F) | NM_003380.3 | 91 |
CGAGGAGAGCAGGATTTCTC |
| hVimentin (R) | | |
GGTATCAACCAGAGGGAGTGA |
| hSmad7 (F) | NM_005904.3 | 150 |
CCCCATCACCTTAGCCGACTCTGC |
| hSmad7 (R) | | |
CCCAGGGGCCAGATAATT |
| hSmad3 (F) | NM_005902.3 | 106 |
GCACCATCCGCATGAGCTTT |
| hSmad3 (R) | | |
TGCAAAGGCCCATTCAGGT |
| hSmad4 (F) | NM_005359.5 | 221 |
CCATTTCCAATCATCCTGCT |
| hSmad4 (R) | | |
ACCTTTGCCTATGTGCAACC |
| hβ-actin (F) | NM_001614.3 | 159 |
GCCCTGAGGCACTCTTCCA |
| hβ-actin (R) | | |
TTGCGGATGTCCACGTCA |
Western blot analysis
The cells were lysed using radioimmunoprecipitation
assay buffer. The proteins were separated by denaturing
electrophoresis using a 10% SDS-containing polyacrylamide gel, and
then transferred onto polyvinyldene fluoride membranes (Invitrogen
Life Technologies). The following rabbit anti-human antibodies were
used: anti-Smad7 (1:700 dilution; Santa Cruz Biotechnology Inc.,
Dallas, TX, USA), anti-E-cadherin, anti-N-cadherin (1:1,000
dilutions; Cell Signaling Technology Inc., Danvers, MA, USA) and
anti-GAPDH (Cell Signaling Technology Inc.). The membranes were
incubated with the primary antibodies in Tris-buffered saline
Tween® 20 (TBST), containing 5% non-fat dry milk
overnight at 4°C, followed by an incubation with goat anti-rabbit
immunoglobulin G (1;1,000; GE Healthcare Life Sciences, Chalfont,
UK) in TBST, containing 2% nonfat dry milk at room temperature for
1 h. ImageQuant LAS 4000 and Analysis Software (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA) were used to quantify the
intensity of the bands.
Transwell invasion assay
A transwell membrane (8 μm pore size, 6.5 mm
diameter; Corning Costar, Corning, New York, NY, USA) was used.
Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA) was melted on
ice and diluted with cold RPMI-1640. The Matrigel™ (60 μl) was
added into the top chamber and was gelated, following an incubation
at 37°C for 5 hours. The cells were resuspended in RPMI-1640 at a
concentration of 500,000 cells/ml. A 100 μl cell suspension was
plated into the Matrigel™-coated top chambers and a total of 600 μl
RPMI-1640, containing 30% FBS, was added to the bottom well.
Following an incubation at 37°C for 48 hours, the membrane was
stained with 0.1% crystal violet and observed under a Nikon upright
microscope (Nikon Corporation, Tokyo, Japan). The experiments were
repeated in triplicate, and the data are expressed as the means ±
standard error of the mean.
Wound healing assay
SK-OV-3 cells were seeded in 6-well plates at
1×105 per well. Following an overnight starvation with
serum-free RPMI-1640, a single scratch wound was made on the
cellular confluent monolayers, using a micropipette tip. Following
incubation for 6, 12 and 24 h, the migratory status of the cells
was imaged using a Nikon microscope and analyzed with Image J
software (National Institutes of Health, Bethesda, MA, USA). The
migratory potential was determined by measuring the distance
between the wound edges. The results represent the percentage of
the distance, following incubation for the specified times,
relative to the original distance between the wound edges. Each
experiment was repeated in triplicate and the data are expressed as
the means ± standard error of the mean.
Lentiviral transduction and Smad7
silence
A lentivirus driving the expression of green
fluorescent protein (GFP) was transduced into SK-OV-3 cells to
generate fluorescence-expressing cells. A Smad7-specific small
hairpin RNA (shRNA) duplex (5′-ATTCGGACAACAAGAGTCA-3′) and a vector
control was cloned into U6-vshRNA-CMV-GFP lentiviral-based vectors
(Shanghai JiKai Company, Shanghai, China). GFP-positive cells were
detected using fluorescence-activated cell sorting (Beckman
Coulter). Smad7 expression was detected by qPCR and western blot
analysis.
Statistical analyses
The data are expressed as the means ± standard error
of the mean of ≥3 independent experiments. The significance of the
differences between the mean values was determined using a
two-tailed Student’s t-test. A P<0.05 was considered to indicate
a statistically significant difference.
Results
Smad7 is expressed at a higher level in
the epithelial growth-patterned SK-OV-3 cell line
To observe the functions of the TGF-β/Smad signaling
pathway in EOC, the relative mRNA expression levels of Smad3, Smad4
and Smad7 were determined in SK-OV-3, ES-2, OVCAR-3, HO-8910 and
HO-8910PM cells. As shown in Fig. 1A
and B, Smad3, Smad4 and Smad7 were more highly expressed in the
SK-OV-3 cells, as compared with the other ovarian cancer cell
lines. Among the Smads, Smad7 was the most elevated. These results
indicate that Smad7 may have a unique function in the TGF-β/Smad
signaling pathway and may exert an effect on the regulation of EMT
in EOC cells.
To further distinguish whether the expression of the
Smads was associated with an epithelial/mesenchymal phenotype, an
EMT-related gene expression profile, including E-cadherin,
N-cadherin and Vimentin, was compared between the five cell lines
(Fig. 1A and C). The epithelial
marker E-cadherin, was more highly expressed in the SK-OV-3 cells,
as compared with the other cell lines. The mesenchymal marker
N-cadherin, was more highly expressed in the ES-2 cells. In
addition, SK-OV-3 and OVCAR-3 cells exhibited a more distinct
epithelial round-clonal growth pattern and exhibited abundant
intercellular junctions, whereas ES-2 cells exhibited a sparse
fibroblast growth pattern and a branched cytoplasm.
These results indicate that, although the five cell
lines were all generated from the tissue samples of patients with
EOC, the SK-OV-3 cells had a more epithelial behavior and the ES-2
cells more mesenchymal. Furthermore, the relative expression level
of Smad7 was highest in the SK-OV-3 cells.
Smad7 is overexpressed in ovarian cancer
stem-like SP cells
Ovarian cancer tumors are heterogeneous and contain
CSCs that are able to self-renew and are known to be essential to
tumorigenesis and progression (22). EOC stem-like SP cells were shown to
exhibit an epithelial phenotype and a reduced expression of cell
adhesion molecules. Non-SP cells forfeited the CSC properties and
exhibited a more mesenchymal phenotype (10). To explore the underlying
association between Smad7 and ovarian CSCs, a Hoechst
33342-effluxing assay was used to detect and enrich SP cells from
EOC HO-8910PM cells (Fig. 2A). The
SP cells were shown to exhibit much higher relative mRNA expression
levels of Smad7, as compared with the non-SP cells (Fig. 2B). Because of the epithelial origin
of ovarian cancer SP cells, Smad7 exhibited an increased expression
in the cells with an epithelial phenotype, either in ovarian CSCs
or in the EOC cell line. These results indicate that Smad7 may have
an important role in the maintenance of the epithelial
phenotype.
TGF-β1 upregulates the expression of
Smad7 in EOC cells
TGF-β1 is a fundamental determinant of cell
migration in wound healing, inflammation and tumorigenesis
(22). It has also been identified
as the main factor in the progression of an invasive phenotype in
epithelial tumors (23). To
determine the association between TGF-β1 stimulation and Smad7
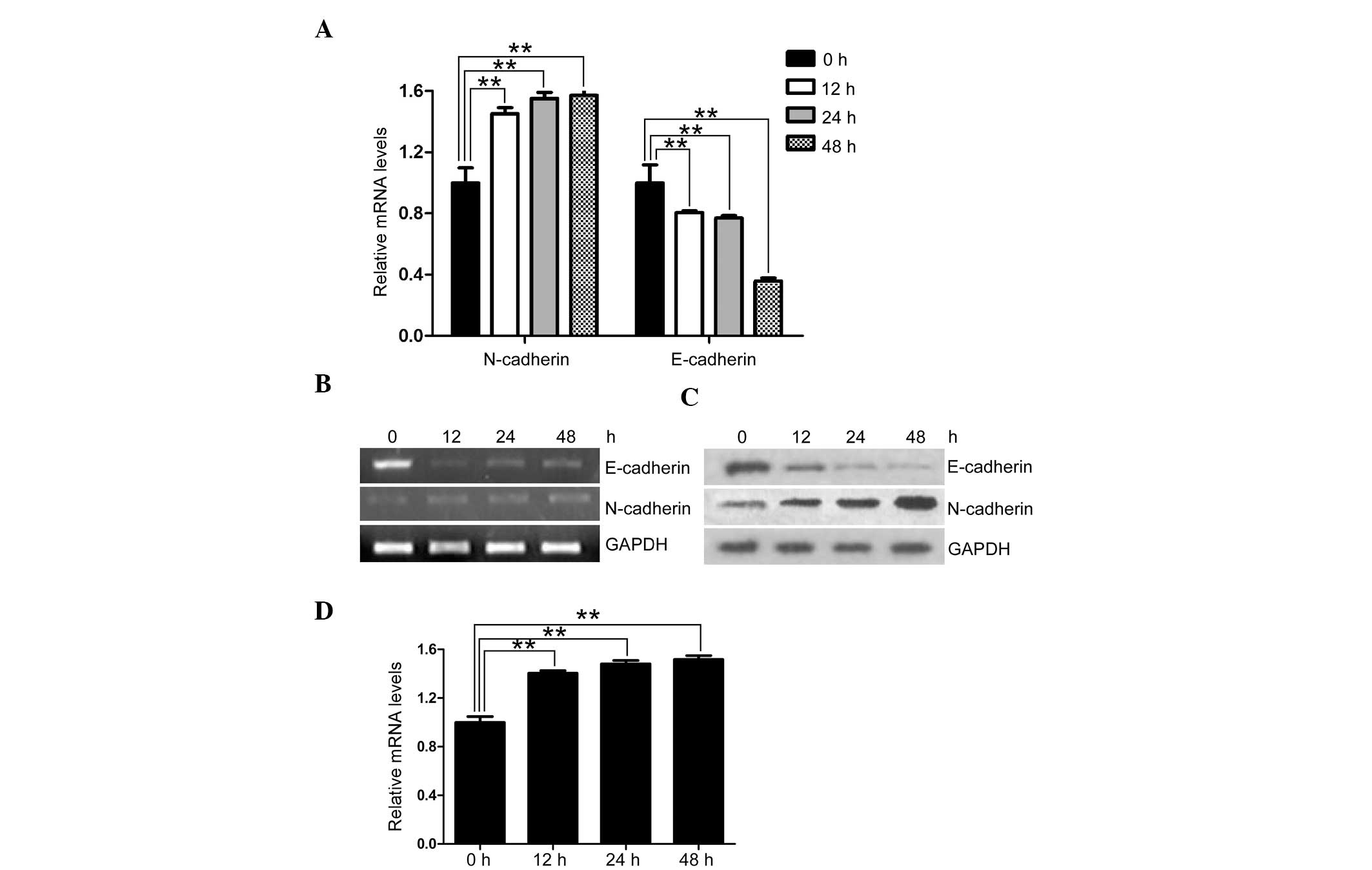
expression in EOC, SK-OV-3 cells were cultured in the presence of
TGF-β1 for two days. Following TGF-β1 stimulation, SK-OV-3 cells
exhibited a reduced relative expression of E-cadherin and an
increased relative expression of N-cadherin, at both the mRNA and
protein level (Fig. 3A–C). These
results imply an E-cadherin to N-cadherin switch (EN switch).
Furthermore, Smad7 was upregulated during TGF-β1 stimulation, in
accordance with the increase of the meshenchymal marker N-cadherin
(Fig. 3D). As a downstream
inhibitor of TGF-β1-induced EMT, the increased expression of Smad7
during TGF-β1 stimulation may be a result of a feedback mechanism
in EOC, reversing the EN switch to MET.
Silencing of Smad7 promotes EMT but
reduces the invasive and migratory capabilities of EOC cells
Both the process of EMT and the gene expression of
Smad7 were stimulated by TGF-β1, therefore the present study
further aimed to determine the specific operative function of Smad7
on EMT. A shRNA lentiviral construct specifically targeting the
human Smad7 gene was generated and transduced into SK-OV-3 cells.
The relative expression of Smad7 was efficiently inhibited at both
the mRNA and protein level (Fig. 4A
and B). As predicted, the process of EMT was accelerated by the
inhibition of Smad7. The mRNA encoding epithelial marker,
E-cadherin, was downregulated in Smad7 shRNA (shSmad7)-transduced
SKOV3 cells, whereas the mRNA encoding mesenchymal marker,
N-cadherin, was upregulated (Fig. 4A
and B). These results confirm that Smad7 inhibited EMT, and
repression of Smad7 may induce cellular mesenchymal transformation
in tumors.
To evaluate the variation of tumor biological
characteristics in shSmad7-SKOV3 cells, transwell invasion and
wound healing assays were performed to estimate the cellular
invasive and migratory capabilities, respectively. As compared with
the vector control, ectopic deletion of Smad7 led to a markedly
reduced number of infiltrating and migrating cells in shSmad7-SKOV3
cells (Fig. 4C–F). These results
indicate that Smad7 exerts its function on cellular motility and
metastatic capacity in EOC development. This is in contrast with
the previous findings that shSmad7-SKOV3 cells sustained a
mesenchymal phenotype (Fig. 4A and
B). The process of EMT is complex and is associated with
numerous signaling pathways (24),
therefore other EMT molecular mechanisms may be compensating for
the lack of Smad7 expression, in order to maintain the invasiveness
of the EOC cells.
Discussion
EMT has been indicated as a dynamic process required
for cellular remodeling during embryogenesis, wound healing and the
acquisition of malignant traits (25). It also leads to the invasion and
metastasis of tumor cells, as the result of alternation of
cell-cell adhesion, cell polarity and cell-extracellular matrix
interaction (5). Its reverse
process, MET, may accelerate neoplasm development and tumor
colonization once metastatic tumor cells have grown at the distant
sites (12–14). Numerous metastatic initiating
elements are required to induce tumor cell migration, invasion,
intravasation and activation of angiogenesis (4,6),
among which Smad7 has contradictory effects on cancer progression
and metastasis as previously described (17–19).
In the intracellular TGF-β signaling pathway, the polypeptide Smad7
serves to accommodate negative feedback (15,23).
As previously reported, Smad7 is the predominant inhibitor in
TGF-β-induced EMT, through the binding of the TGF-β1 receptor and
obstructing the phosphorylation of Smad2 and Smad3. A pre-existing
nuclear pool of Smad7 can also be mobilized by TGF-β1. Hence it
constitutes a delicate negative feedback loop, which is associated
with sustaining the balance of TGF-β/Smad-regulated EMT (15,26).
Tumor cell migration involves complex pathways which
are spatially and temporally associated with the transformation of
the cytoskeleton and morphology of cells (27). It has been previously suggested
that Smad7 is associated with the TGF-β-triggered cytoskeletal
responses of migrating cells, through local regulation of polarity
complexes (28). As a feedback
regulator in TGF-β/Smad-modulated EMT, the aberrant expression of
Smad7 can induce malignant cellular progression and is considered
to be oncogenic (22,29). However, it has been observed, in
numerous cases, that Smad7 may have an opposite effect, according
to the cancer type. Overexpression of Smad7 has previously been
shown to hinder the advancement of brain (30), breast (31), colorectal (32), nasopharyngeal (17), cervical (18), bone and lung (30,31)
cancers, both in vitro and in vivo. Conversely, Smad7
induced malignant processes of squamous cell (33) and colon carcinoma (34), especially tumorigenicity and
metastasis recurrence. Despite the identified status of Smad7 in
numerous cancers, the functions of Smad7 in EOC pathogenesis
remains undefined.
In the present study, the transcription of Smad3,
Smad4 and Smad7 were assessed in five EOC cell lines. The relative
expression of Smad7 was shown to be highest in the SK-OV-3 cells,
which presented a more epithelial and less mesenchymal phenotype.
EOC tumors are heterogeneous and whether ovarian cancer cells are
of a epithelial or mesenchymal origin remains unknown. A previous
study confirmed that ovarian cancer stem-like SP cells were of
epithelial origin (10).
Furthermore, Smad7 was identified as being more highly expressed in
the epithelial phenotypic SP cells, as compared with the
mesenchymal phenotypic non-SP cells. These data are consistent with
the hypothesis that Smad7 is associated with the epithelial ovarian
tumor initiator CSCs. Downregulation of Smad7 by a shSmad7
lentiviral vector, resulted in the decreased expression of
E-cadherin and increased expression of N-cadherin. These results
indicate that as an inhibitor in the process of EMT, Smad7 has an
important role in the maintenance of the epithelial phenotype in
ovarian cancer cells. The particular mechanism by which Smad7
affects the endogenous phenotypic transformation requires further
investigation.
To induce EMT, SK-OV-3 cells were stimulated with
TGF-β1. The cells gradually converted from a round epithelial
pattern to a long fusiform mesenchymal shape; reducing epithelial
marker expression levels and fortifying the mesenchymal phenotype.
Meanwhile, the expression of Smad7 was also upregulated. It may be
hypothesized that during TGF-β-stimulated EMT, the increased
expression of Smad7 increasingly expressed at a notable level, may
be the result of a negative feedback mechanism (35).
In addition, the shSmad7-SKOV3 cells developed a
mesenchymal profile of cell surface makers. They exhibited a more
mesenchymal circular shape, with simultaneous N-cadherin
upregulation and E-cadherin downregulation. However, the invasive
and metastatic capacity of the shSmad7-SKOV3 cells was markedly
reduced. These results indicate that the Smad-associated pathway is
not the sole molecular mechanism participating in the regulation of
EMT. Numerous other signaling pathways, including NF-κB, Wnt and
Notch (24), may compensate for
the suppression of Smad7, in order to maintain the invasive
behavior of the cancer cells. By restoring the epithelial phenotype
and invasive behavior of ovarian cancer cells, Smad7 may contribute
to the MET process in EOC, which facilitates the growth of
epithelial tumor cells at metastatic sites.
In conclusion, the findings of the present study
indicate a negative regulatory mechanism between Smad7 and EMT in
EOC. Smad7 has an important role in retaining the epithelial
phenotype of ovarian cancer cells, and may induce tumor cell
invasion and migration. This bilateral behavior may exert its
function when ovarian cancer cells reverse EMT to MET, which is
necessary for the colonization of metastatic sites. The underlying
mechanisms that regulate TGF-β/Smad signaling in EOCs require
further elucidation.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 31371452; Hua Jiang) and the
Foundations from Science and Technology Commission of Shanghai
Municipality (nos. 11JC1401501, 12410710100; Hua Jiang).
References
|
1
|
Lengyel E: Ovarian cancer development and
metastasis. Am J Pathol. 177:1053–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwatsuki M, Mimori K, Yokobori T, et al:
Epithelial-mesenchymal transition in cancer development and its
clinical significance. Cancer Sci. 101:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Condeelis J and Segall JE: Intravital
imaging of cell movement in tumours. Nat Rev Cancer. 3:921–930.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nisticò P, Bissell MJ and Radisky DC:
Epithelial-mesenchymal transition: general principles and
pathological relevance with special emphasis on the role of matrix
metalloproteinases. Cold Spring Harb Perspect Biol. 4:2012.
|
|
6
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:11–26. 1994.PubMed/NCBI
|
|
7
|
Elloul S, Elstrand MB, Nesland JM, et al:
Snail, Slug, and Smad-interacting protein 1 as novel parameters of
disease aggressiveness in metastatic ovarian and breast carcinoma.
Cancer. 103:1631–1643. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin H, Yu Y, Zhang T, et al: Snail is
critical for tumor growth and metastasis of ovarian carcinoma. Int
J Cancer. 126:2102–2111. 2010.PubMed/NCBI
|
|
9
|
Ahmed N, Thompson EW and Quinn MA:
Epithelial-mesenchymal interconversions in normal ovarian surface
epithelium and ovarian carcinomas: an exception to the norm. J Cell
Physiol. 213:581–588. 2007. View Article : Google Scholar
|
|
10
|
Jiang H, Lin X, Liu Y, et al:
Transformation of epithelial ovarian cancer stemlike cells into
mesenchymal lineage via EMT results in cellular heterogeneity and
supports tumor engraftment. Mol Med. 18:1197–1208. 2012. View Article : Google Scholar
|
|
11
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Elloul S, Vaksman O, Stavnes HT, Trope CG,
Davidson B and Reich R: Mesenchymal-to-epithelial transition
determinants as characteristics of ovarian carcinoma effusions.
Clin Exp Metastasis. 27:161–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wells A, Yates C and Shepard CR:
E-cadherin as an indicator of mesenchymal to epithelial reverting
transitions during the metastatic seeding of disseminated
carcinomas. Clin Exp Metastasis. 25:621–628. 2008. View Article : Google Scholar
|
|
15
|
Valcourt U, Kowanetz M, Niimi H, Heldin CH
and Moustakas A: TGF-beta and the Smad signaling pathway support
transcriptomic reprogramming during epithelial-mesenchymal cell
transition. Mol Biol Cell. 16:1987–2002. 2005. View Article : Google Scholar
|
|
16
|
Ikushima H and Miyazono K: Biology of
transforming growth factor-β signaling. Curr Pharm Biotechnol.
12:2099–2107. 2011.
|
|
17
|
Xiao J, Xiang Q, Xiao YC, et al: The
effect of transforming growth factor-beta1 on nasopharyngeal
carcinoma cells: insensitive to cell growth but functional to
TGF-beta/Smad pathway. J Exp Clin Cancer Res. 29:352010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hariharan R, Babu JM, PR and Pillai MR:
Mutational analysis of Smad7 in human cervical cancer. Oncol Rep.
21:1001–1004. 2009.PubMed/NCBI
|
|
19
|
Luo X, Xu J and Chegini N: The expression
of Smads in human endometrium and regulation and induction in
endometrial epithelial and stromal cells by transforming growth
factor-beta. J Clin Endocrinol Metab. 88:4967–4976. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shenhua X, Lijuan Q, Hanzhou N, et al:
Establishment of a highly metastatic human ovarian cancer cell line
(HO-8910PM) and its characterization. J Exp Clin Cancer Res.
18:233–239. 1999.PubMed/NCBI
|
|
21
|
Goodell MA, Brose K, Paradis G, Conner AS
and Mulligan RC: Isolation and functional properties of murine
hematopoietic stem cells that are replicating in vivo. J Exp Med.
183:1797–1806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Drabsch Y and ten Dijke P: TGF-β
signalling and its role in cancer progression and metastasis.
Cancer Metastasis Rev. 31:553–568. 2012.
|
|
23
|
Horiguchi K, Shirakihara T, Nakano A,
Imamura T, Miyazono K and Saitoh M: Role of Ras signaling in the
induction of snail by transforming growth factor-beta. J Biol Chem.
284:245–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar
|
|
25
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: an emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Itóh S, Landström M, Hermansson A, et al:
Transforming growth factor beta1 induces nuclear export of
inhibitory Smad7. J Biol Chem. 273:29195–29201. 1998.PubMed/NCBI
|
|
27
|
Ridley AJ, Schwartz MA, Burridge K, et al:
Cell migration: integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ekman M, Mu Y, Lee SY, et al: APC and
Smad7 link TGFβ type I receptors to the microtubule system to
promote cell migration. Mol Biol Cell. 23:2109–2121.
2012.PubMed/NCBI
|
|
29
|
Pittman AM, Naranjo S, Webb E, et al: The
colorectal cancer risk at 18q21 is caused by a novel variant
altering SMAD7 expression. Genome Res. 19:987–993. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Javelaud D, Mohammad KS, McKenna CR, et
al: Stable overexpression of Smad7 in human melanoma cells impairs
bone metastasis. Cancer Res. 67:2317–2324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Azuma H, Ehata S, Miyazaki H, et al:
Effect of Smad7 expression on metastasis of mouse mammary carcinoma
JygMC(A) cells. J Natl Cancer Inst. 97:1734–1746. 2005. View Article : Google Scholar
|
|
32
|
Rizzo A, Waldner MJ, Stolfi C, et al:
Smad7 expression in T cells prevents colitis-associated cancer.
Cancer Res. 71:7423–7432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu X, Lee J, Cooley M, Bhogte E, Hartley
S and Glick A: Smad7 but not Smad6 cooperates with oncogenic ras to
cause malignant conversion in a mouse model for squamous cell
carcinoma. Cancer Res. 63:7760–7768. 2003.PubMed/NCBI
|
|
34
|
Halder SK, Beauchamp RD and Datta PK:
Smad7 induces tumorigenicity by blocking TGF-beta-induced growth
inhibition and apoptosis. Exp Cell Res. 307:231–246. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gratchev A, Kzhyshkowska J, Kannookadan S,
et al: Activation of a TGF-beta-specific multistep gene expression
program in mature macrophages requires glucocorticoid-mediated
surface expression of TGF-beta receptor II. J Immunol.
180:6553–6565. 2008. View Article : Google Scholar
|