Introduction
Hearing impairment is the most common sensory
disorder worldwide (1) and genetic
inheritance presents a major source of the auditory system
dysfunction resulting in hearing loss (2). Presently, 54 gene loci associated
with an autosomal dominant mode of inheritance and 67 gene loci
associated with an autosomal recessive mode of inheritance have
been identified, of which seven are X chromosome-linked and four
are mitochondrial (3). The cochlea
is a complex organ in the ear, which is composed of several cell
types and specialized regions that are involved in the normal
process of hearing. A number of genes have been associated with
hearing loss and several corresponding proteins have been
identified as being expressed in the cochlea. Ionic homeostasis in
the cochlear duct is associated with a several genes associated
with deafness (4). In mice,
endolymph (the fluid surrounding the upper surface of the hair
cell) has a high concentration of potassium and a low concentration
of sodium, and is maintained at a high positive resting potential
of approximately +100 mV. This high resting potential is considered
to be essential for the normal functioning of hair cells as, when
its value is reduced to zero, deafness occurs (5).
Communication between the majority of cells in
animal tissues is mediated by unique intercellular cytoplasmic
channels, gap junctions, spanning across two cell membranes. These
cell-to-cell channels consist of assemblies of proteins termed
connexins (Cxs) or pannexins in vertebrates and innexins in
invertebrates (6). Cxs belong to a
protein family of >20 members, each of which is encoded by a
different gene and they are assigned a number which is associated
with their approximate molecular weights. Cxs share a common
structure of four transmembrane segments, which extend into two
extracellular and three cytoplasmic domains (7). Gap junction intercellular
communication has a range of functions in order to meet the
requirements of the organs, tissues and cell groups in which the Cx
genes are expressed (8), and the
importance of these gap junctions in auditory functions has been
confirmed by numerous studies (9–13).
In the sensory epithelia of the inner ear, gap junction channels
are important in the recycling of potassium ions that enter the
hair cells and are also involved in auditory signal transduction
(14).
Immunolabeling analysis has identified several types
of Cx product, including Cx26, Cx29, Cx30, Cx31 and Cx43, in the
mature cochlea (10–12,15–17).
Through immunohistochemiical and reverse transcription-quantitative
polymerase chain reaction analyses, our previous study indicated
that Cx30.3 is present and localized in the rat cochlea (18). In addition, a study of 555 deaf
patients revealed a common (4.1%; 23/555) frameshift mutation
(c.154del4) in gap junction β4 (GJB4, also termed Cx30.3) in
deaf individuals (18). In the
study, five amino acid variants (c.307 C>T, c.371 G>A, c.478
C>T, c.507 C>G and c.611 A>C) were detected in deaf
individuals without skin disorders (19). In our previous genetic survey of
373 individuals, including 253 with nonsyndromic deafness and 120
with normal hearing, 11 mutations were detected in the patients
with hearing loss (20). However,
the correlation between the GJB4 gene mutations and the
audiology phenotype in deaf patients was not examined. Therefore,
the present study investigated the phenotype-genotype correlation
in deaf patients with mutations in GJB4, the results of
which may provide assistance in the clinical evaluation and
effective management of care for families of children with
GJB4.
Materials and methods
Patient selection
A total of 253 individuals with hearing loss were
screened for GJB4 variants in the present study. For the
patients with hearing loss, a total of 173 school children were
selected from the National Tainan School for the Deaf (Tainan,
China) and 80 individuals with hearing loss, who were managed at
the Chang Gung Memorial Hospital (Chiayi, China), were selected.
The frequency range of hearing loss was between 250 and 8,000 Hz,
with a mean threshold (500, 1,000, 2,000 and 4,000 Hz) of >40 dB
in the right and left ears. All probands were 17 years old or
younger at the time of molecular diagnosis. In the present study,
the 11 patients with GJB4 missense and nonsense mutations
had complete audiograms and were used for analysis.
Patients with syndromic hearing loss or
environment-associated hearing loss were excluded from the present
study, as determined by an otorhinolaryngologist. The complete
medical history of each child was obtained to determine the age of
onset of deafness and to exclude the possibility of environmental
causes, including maternofetal infection, perinatal complications,
meningitis, mumps, prenatal or postnatal drug ototoxicity and
acoustic trauma. All procedures were approved by the Institutional
Review Board of Chung Gung Memorial Hospital (96-1294B). Written
informed consent was obtained from all patients.
Clinical evaluation
The genetic and audiological data were categorized
according to recommendations on geneotype-phenotype correlations by
the Genetic Deafness Study Group (21). According to these guidelines, the
groups were recognized as follows: Mild hearing loss (20–40 dB),
moderate hearing loss (41–70 dB), severe hearing loss (71–95 dB)
and profound hearing loss (>95 dB). The audiometric
configurations were determined for each ear by differences in
hearing level (HL) as follows: Ascending low frequency, >15 dB
difference in HL between the poorer low frequency thresholds and
the higher frequencies; U-shaped mid frequency, >15 dB
difference in HL between thresholds at the poorest mid-frequencies
and those at higher and lower frequencies; gently sloping high
frequency, 5–29 dB difference in HL between the mean thresholds at
0.5 and 1 kHz and at 4 and 8 kHz; steeply sloping high frequency,
>30 dB difference in HL between the above-mentioned frequencies;
and flat, <15 dB difference in HL between the mean thresholds at
0.25 and 0.5 kHz, 1 and 2 kHz and 4 and 8 kHz. Asymmetric HL was
defined as an interaural pure tone average (PTA) difference of
>10 dB in at least two frequencies.
Computed tomography (CT) of the inner
ear
CT images of the inner ears were examined in 11
probands in the cohort of the present study. All the images
examined were high resolution 1-mm contiguous, axial and coronal
images of the temporal bones. Digital or printed images were
evaluated for abnormalities of the cochlea, vestibule, semicircular
canals and endolymphatic aqueduct.
Results
Severity and configuration of hearing
impairment and genotype
In our previous study, a total of nine different
GJB4 mutations were identified in 11 of the 253 probands
(19). Of these mutations, eight
were missense variants that led to amino acid substitution in the
encoded proteins and one was a nonsense mutation. No vestibular
symptoms or skin disorders were observed in any individual. Genetic
assessment facilitates the determination of the cause of deafness
and the prediction of the degree of hearing impairment and language
development (22). Therefore, the
present study investigated the phenotype-genotype correlation in
the 11 deaf patients with mutations of GJB4. The severity of
hearing impairment was assessed in the 11 patients with the
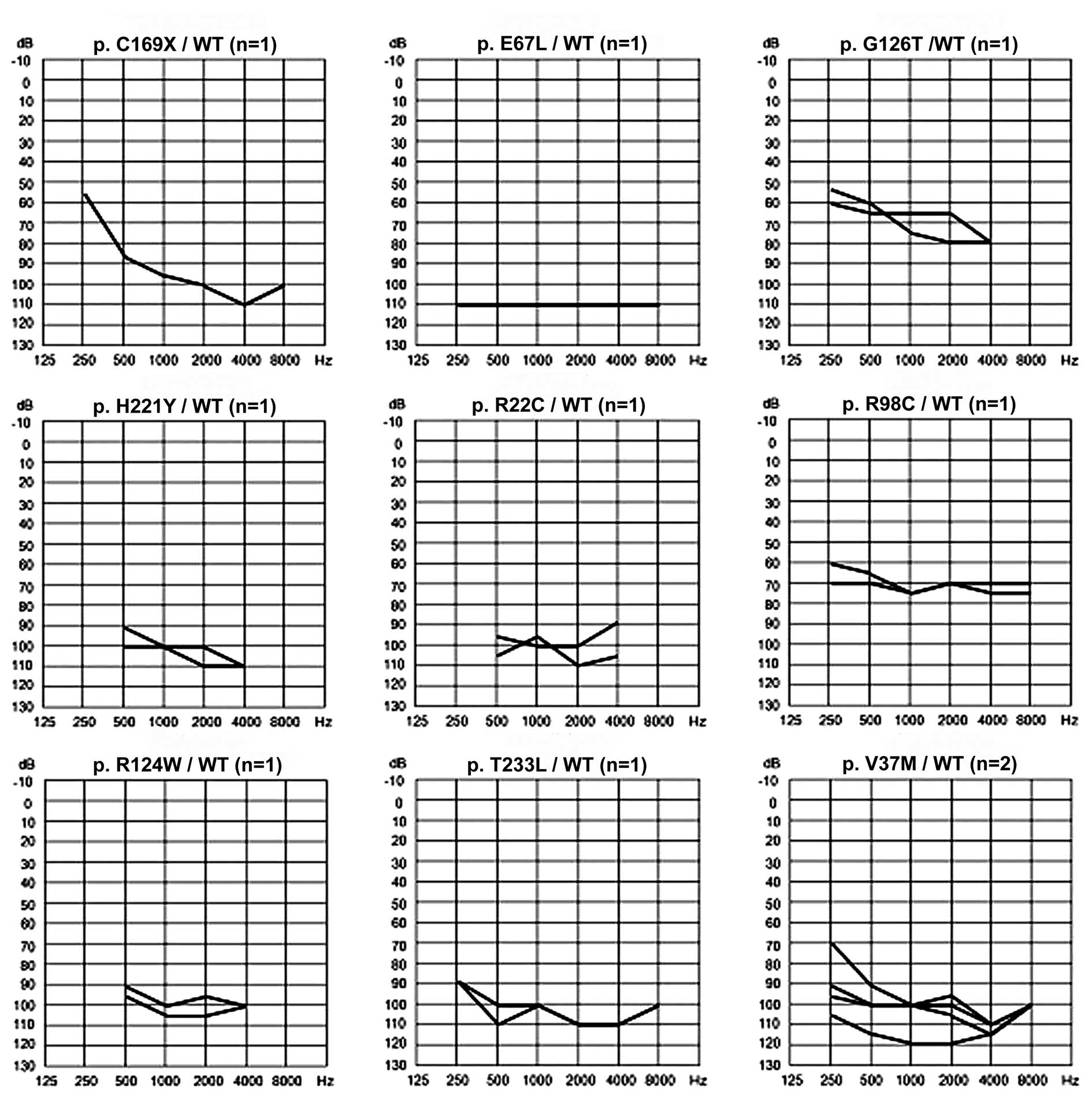
GJB4 mutations (Table I;
Fig. 1), and the four-frequency
PTA was calculated as the average of air-conduction thresholds at
500, 1,000, 2,000 and 4,000 Hz. The mean (± standard deviation)
threshold of hearing for all GJB4 mutations was 97.16 dB (±
13.52 dB). In the present study, 10 probands with the GJB4
mutation were observed to have symmetrical HL. Asymmetric HL was
observed in only one proband (TDF547), with an interaural PTA
difference of 20 dB. This proband had a c.109G>A/WT heterozygous
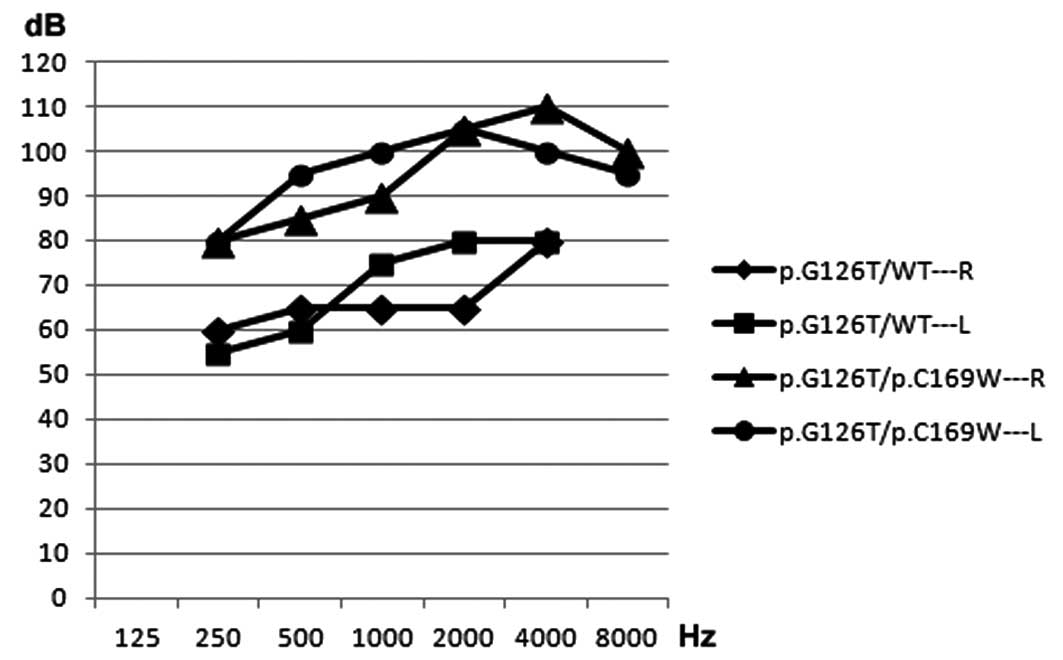
genotype. In addition, one proband (TDF521) was identified with a
compound missense heterzogous mutation (c.376G>A/c.507C>G) of
GJB4 and had more severe hearing loss, compared with the
proband exhibiting a heterozygous missense mutation
(c.376G>A/wt), in the right and left ears (Fig. 2).
 | Table IAudibility thresholds for air
conduction in pure tone audiometry of the 11 patients with gap
junction β4 missense and nonsense variants at frequencies between
250 and 8,000 Hz. |
Table I
Audibility thresholds for air
conduction in pure tone audiometry of the 11 patients with gap
junction β4 missense and nonsense variants at frequencies between
250 and 8,000 Hz.
| Patient | Genotype variant
(amino acid change) | Protein domain | Frequency (Hz) | Mean
thresholda |
|---|
|
|---|
| Ear | 250 | 500 | 1,000 | 2,000 | 4,000 | 8,000 |
|---|
| KDF026 | c.64 C>T/WT | M1 | R | AR | 95 | 100 | 100 | 90 | AR | 96.3±4.8 |
| (pR22C) | | L | AR | 105 | 95 | 110 | 105 | AR | 103.8±6.3 |
| TDF547 | c.109
G>A/WT | M1 | R | 70 | 90 | 100 | 100 | 110 | 100 | 100.0±8.2 |
| (p.V37M) | | L | 90 | 100 | 100 | 100 | 110 | 100 | 102.5±5.0 |
| TDF553 | c.109 G>
A/WT | M1 | R | 95 | 100 | 100 | 105 | 115 | 100 | 105.0±7.1 |
| (p.V37M) | | L | 105 | 115 | 120 | 120 | 115 | 100 | 117.5±2.9 |
| TDF067 | c.199G
>A/WT | El | R | 110 | 110 | 110 | 110 | 110 | 110 | 110.0±0.0 |
| (p.E 67L) | | L | 110 | 110 | 110 | 110 | 110 | 110 | 110.0±0.0 |
| CDF006 | c.292
C>T/WT | CL | R | 70 | 70 | 75 | 70 | 75 | 75 | 72.5±2.9 |
| (pR.98C) | | L | 60 | 65 | 75 | 70 | 70 | 70 | 70.0±4.1 |
| LDF011 | c.370
C>T/WT | CL | R | AR | 95 | 105 | 105 | 100 | AR | 101.3±4.8 |
| (p.R124W) | | L | AR | 90 | 100 | 95 | 100 | AR | 96.3±4.8 |
| TDF512 | c376G>A/WT | CL | R | 60 | 65 | 65 | 65 | 80 | AR | 68.8±7.5 |
| (p.G126T) | | L | 55 | 60 | 75 | 80 | 80 | AR | 73.8±9.5 |
| LDF014 | c.507
C>A/WT | E2 | R | 55 | 85 | 95 | 100 | 110 | 100 | 97.5±10.4 |
| (p.0 169X) | | L | 55 | 85 | 95 | 100 | 110 | 100 | 97.5±10.4 |
| KDF012 | c.661
C>T/WT | C | R | AR | 100 | 100 | 100 | 110 | AR | 102.5±5.0 |
| (p.H221Y) | | L | AR | 90 | 100 | 110 | 110 | AR | 102.5±9.6 |
| TD F035 | c.698C>A/WT | C | R | 90 | 100 | 100 | 110 | 110 | 100 | 105.0±5.8 |
| (p.T233L) | | L | 90 | 110 | 100 | 110 | 110 | 100 | 107.5±5.0 |
| TDF521 | c.376G>A/c.507
C>G | CL/E2 | R | 80 | 85 | 90 | 105 | 110 | 100 | 97.5±11.9 |
|
(p.G126T)/(p.C169W) | | L | 80 | 95 | 100 | 105 | 100 | 95 | 100.0±4.1 |
Cx30.3 protein structure and hearing
loss
Similar to other Cx proteins, Cx30.3 consists of
four transmembrane (TM) domains, TM1 (amino acid 21–40), TM2 (amino
acid 76–98), TM3 (amino acid 127–149) and TM4 (amino acid 188–210).
These are linked by one cytoplasmic and two extracellular loops
with cytoplasmic C- and N-terminal ends. The p.R22C and p.V37M
substitutions detected in the present study occurred in TM1 of
Cx30.3, and the p.E67L and p.C169X substitutions occurred in the
first extracellular loop (E1) and the second extracellular loop
(E2) of Cx30.3, respectively. In addition, three variants, p.R98C,
p.R124W and p.G126T, were located at the cytoplasmic domain and two
variants, p.H221Y and p.T233L, were located at the C-terminal
domain. The relative predictive values of PTA were then examined in
the right and left ears of the patients with the GJB4
mutations (Table I). The hearing
threshold results revealed that cytoplasmic linking (CL) domain
mutations of the Cx30.3 protein had a PTA of 68–72 dB, with the
exception of the p.R124W missense mutation. However, the mean PTA
was >96 dB in the other domains of the Cx30.3 protein (Table I), suggesting that the degree of
PTA was lower in CL domain mutations compared with mutations in
others domains of the Cx30.3 protein. In addition, in the proband
with the c.507C>A (p.C169X) mutation, the degree of hearing loss
was more marked at high frequencies compared with low frequencies
(Table I; Fig. 1).
Configuration of hearing loss
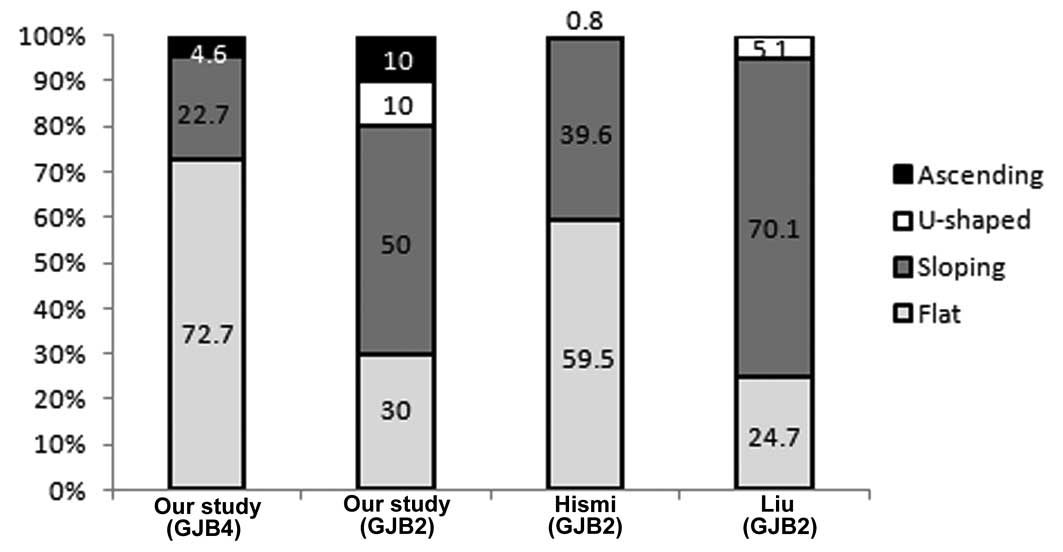
Furthermore, the relative frequencies of the
configuration of hearing impairment in patients with GJB4
and GJB2 genotype variants in the present study were
compared with those in previous studies by Hişmi et al
(23) and Liu et al
(24) (Table II; Fig. 3). The results indicated that the
frequency of a flat audiometric configuration in patients with
GJB4 variants was significantly higher compared with that in
patients with GJB2 variants (P=0.016). Similarly, a
significant difference was observed between the patients with
GJB4 variants in the present study and the patients with
GJB2 variants in the study by Liu et al (P<0.001).
However, the difference in the frequency of this configuration
between patients with GJB4 in the present study and with
GJB2 in the study by Hişmi et al was small (P=0.403).
This may be due to the difference in the point mutation site in the
GJB2 genotype, which was c.35delG in the Hişmi et al
study and c.235delC in the present study, or due to different
ethnicities resulting in different phenotypes. Therefore, in the
present study, the flat shape was more predominant in patients with
GJB4 variants compared with GJB2 variants, and this
data may be applied to direct the clinical evaluation of children
with GJB2 or GJB4.
| Table IIComparison of GJB4 and
GJB2 on the basis of audiogram shapes. |
Table II
Comparison of GJB4 and
GJB2 on the basis of audiogram shapes.
| Present study
GJB4 | Present study
GJB2 | Hişmi et al
(21) GJB2 | Liu et al
(22) GJB2 |
|---|
|
|
|
|
|
|---|
| Audiogram
shape | Ears (n) | % | Ears (n) | % | Ears (n) | % | Probands (n) | % |
|---|
| Flat | 16 | 72.7 | 12 | 30 | 75 | 59.5 | 48 | 24.7 |
| Sloping | 5 | 22.7 | 20 | 50 | 50 | 39.6 | 136 | 70.1 |
| U-shaped | - | 0.0 | 4 | 10 | 1 | 0.8 | 10 | 5.1 |
| Ascending | 1 | 4.6 | 4 | 10 | - | 0.0 | - | 0.0 |
| Total | 22 | 100.0 | 40 | 100 | 126 | 100.0 | 194 | 100.0 |
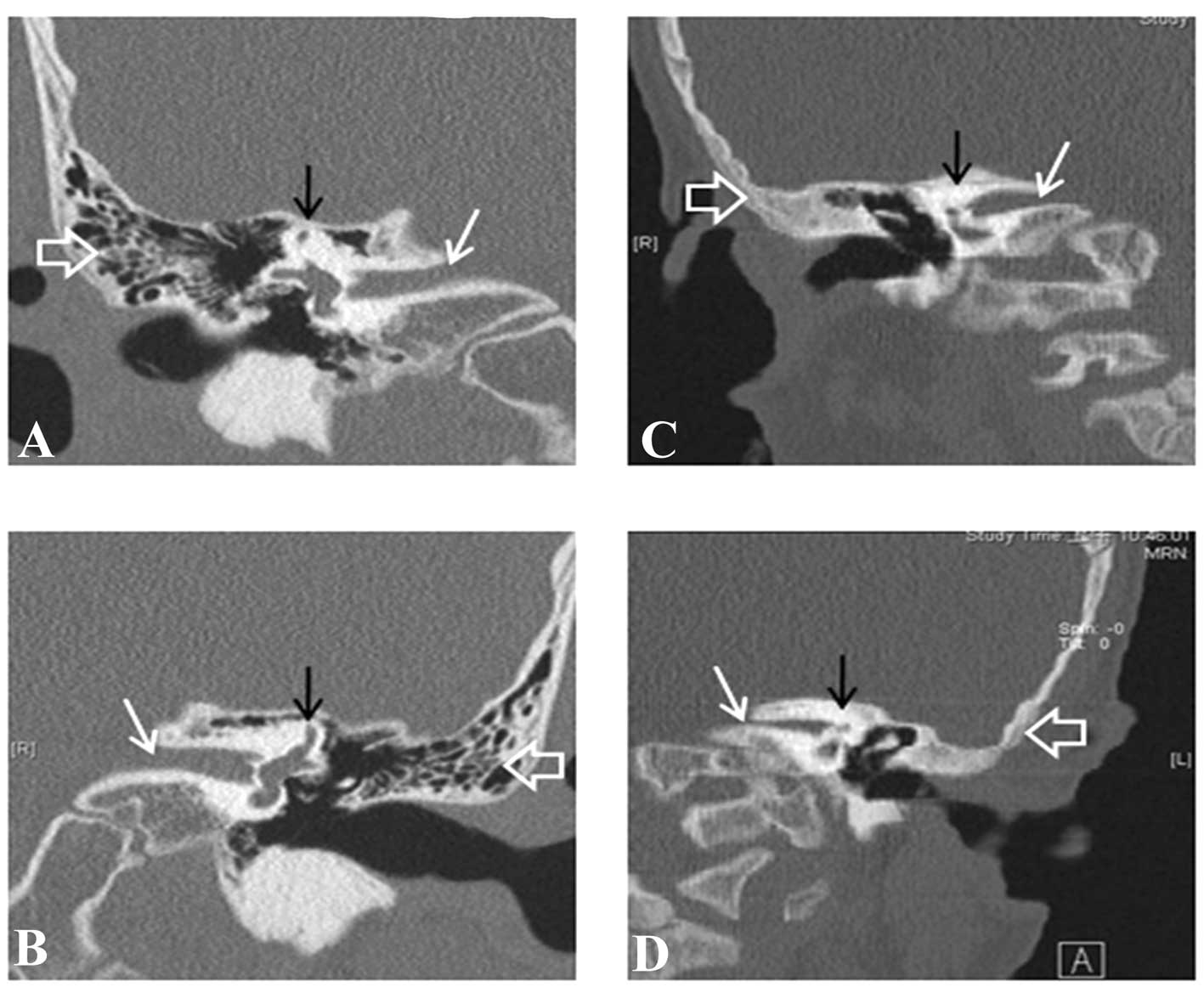
CT images of the 11 patients were also analyzed. A
total of 10 probands (20 ears) exhibited normal CT images of the
temporal bones. In one patient (LDF011) with a c.370 C>T
heterozygous genotype, inner ear and middle ear deformities were
observed (Fig. 4). The CT findings
included bilateral stenosis of the inner auditory canal, which was
greater on the left side, bilateral shortening of the superior and
lateral semicircular canals and bilateral non-pneumatization of the
mastoid air cells. In conclusion, only one of the 11 patients (9%)
with the GJB4 variant in the present study had a
morphological abnormality of the inner ear, as indicated on the CT
images. Therefore, the number of patients with morphological
abnormalities of the inner ear in the cohort was low.
Discussion
Several genetic studies have revealed the importance
of Cxs in normal cochlear function (Hereditary Hearing loss
Homepage; http://hereditaryhearingloss.org/). Few studies have
been conducted on the correlation of variants in the GJB4
gene and its phenotype in patients with nonsyndromic hearing loss.
These previous studies were compared and summarized in Table I (13,19,20).
In these results, the proportion of patients with GJB4
variants was determined to be 4.09% (21/513). The present study
identified that variation in GJB4 is the second most common
genetic risk factor in the Cx gene family for the development of
hearing loss in this population. In addition, the phenotype of
patients with variants of Cx30.3 included prelingual, bilateral,
severe-to-profound hearing loss. A flat audiometric configuration
was also more frequently detected in patients with GJB4
(Cx30.3) variants compared with patients with GJB2
variants.
In total, >20 different Cx proteins have been
identified in mammals. They all share a common structure, however,
each has its own tissue distribution-specificity,
electrophysiological characteristics and regulatory properties
(25). Electrophysiological
studies have indicated that gap junctions have multiple gating
mechanisms. At least two regulation mechanisms respond to
transjunctional voltage (Vj), including Vj gating (fast) and loop
gating (slow) (26). In addition,
membrane voltage (Vm) can also gate gap junctions, termed
Vm-gating, and by chemical factors, including the phosphorylation,
pH and Ca2+, which is termed chemical gating (27). Therefore, patients may exhibit
different phenotypes between mutations in different functional
domains of the Cx protein.
A three-dimensional (3D) characterization of protein
structures can be used to explain the functions of proteins and
their disease formation associations (28,29).
High-resolution characterization of proteins can be provided by
either experimental methods, including X-ray crystallography,
nuclear magnetic resonance or computational analysis (29). However, there is a significantly
higher number of known protein sequences compared with
experimentally solved protein structures. Use of a method
comprising reliable models of proteins, which share ≥30% sequence
identity between known structures and target proteins (28,30),
may assist in understanding the function of target proteins in the
absence of crystallographic structures. The crystalline structure
of the gap junction channel, which is formed by human Cx26, has
been previously described (31,32)
and the N-terminal and TM13 domains have been identified as
important in the permeation pathway of a gap junction channel with
an intracellular channel entrance, pore funnel and extracellular
cavity (31,32). In addition, analysis of the
crystalline structure revealed that the TM2, TM4, E1 and E2 domains
of Cx are associated with the structural organization of the
hexameric connexon, and two neighbor connexons of the gap junction
channel interact with the E1 and E2 domains (31). In classifying the Cx protein, human
Cx26 and Cx30.3 are referred to as the same subgroup, termed group
I or the β group, in phylogenetic tree analysis (33). Therefore, the Cx26 crystalline
structure may assist in explaining why, in the present study,
mutants in the CL domain of Cx30.3 affected the degree of hearing
loss compared with the other functional domains of Cx30.3. However,
in the present study, the functional effect of Cx30.3 was a
prediction and the real functional effect remains to be elucidated.
Therefore, in order to further investigate the effect of these
variants at the protein level, the 3D structure of the Cx30.3
protein requires investigation.
The c.507C>G (p.C169W) missense mutation has been
found in patients with nonsyndromic hearing loss (13,20).
The results of the present study revealed that the heterozygous
c.507C>G mutation was present in the normal hearing control
group. In addition, the proband containing the homozygous
c.507C>G mutation was inherited from parents with normal
hearing, suggesting that the c.507C>G missense mutation had a
recessive inheritance pattern (13). In the present study, a patient
carrying the compound heterozygous mutation, c.376G>A/c.507
C>G (p.G126T)/p.C169W), had more serious hearing loss in the
right and left ears compared with a patient carrying only a
heterozygous mutation (c.376G>A/wt) (Fig. 2). This result demonstrated that the
combination of two genetic mutations leads to a disease phenotype,
however, this phenotype is not present or is present in a mild form
when only one of these gene mutations is present. Analysis using
the ConSeq server (34), a web
server for the identification of structurally and functionally
important residues in protein sequences, determined that the
location of position 169 in the Cx30.3 protein was at E2, which is
exposed and highly conserved throughout evolution. The variants of
p.C169 at E2 may result in incompatibility between the different
species of connexin proteins to form heterotypic functional
channels (35). Therefore, in the
present study, it was hypothesized that the c.507C>G mutant of
GJB4 is a modifier and risk factor in the development of
hearing loss.
In addition, no vestibular symptoms or skin
disorders were found in patients with GJB4 gene variants.
Notably, one patient with the c.370 C>T heterozygous genotype
had inner ear and middle ear deformities on CT analysis, whereas
the other patients with Cx30.3 variants were normal. Therefore, it
was suggested that c.370 C>T heterozygous variants of
GJB4 provide an important base for improving the clinical
diagnosis of deaf patients with inner ear and middle ear
deformities.
The present study demonstrated that GJB4 may
be genetic risk factor for the development of nonsyndromic hearing
loss, and the data can be applied for the effective clinical
evaluation and management of care for families of children with
GJB4. Further investigation will be required to understand
how interference of the mutation contributes to hearing loss. In
addition, it may used in future prenatal genetic analysis.
Acknowledgements
This study was supported by the National Science
Council, Republic of China (nos. NSC 98-2320-B-040-016-MY3 and NSC
101-2320-B-040-014) and the Ministry of Science and Technology
(MOST 103-2320-B-040-021-MY3).
References
|
1
|
Bitner-Glindzicz M: Hereditary deafness
and phenotyping in humans. Br Med Bull. 63:73–94. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Apps SA, Rankin WA and Kurmis AP: Connexin
26 mutations in autosomal recessive deafness disorders. A review.
Int J Audiol. 46:75–81. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Birkenhäger R, Aschendorff A, Schipper J
and Laszig R: Non-syndromic hereditary hearing impairment.
Laryngorhinootologie. 86:299–309. 2007.
|
|
4
|
Resendes BL, Williamson RE and Morton CC:
At the speed of sound: gene discovery in the auditory system. Am J
Hum Genet. 69:923–935. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steel KP, Barkway C and Bock GR: Strial
dysfunction in mouse with cochleo-saccular abnormalities. Hear Res.
27:11–26. 1987. View Article : Google Scholar
|
|
6
|
Phelan P: Innexins: members of an
evolutionary conserved family of gap-junction proteins. Biochim
Biophys Acta Biomem. 1711:225–245. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bruzzone R, White TW and Paul DL:
Connections with connexins the molecular-basis of direct
intercellular singnalling. Eur J Biochem. 238:1–27. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spicer SS and Schulte BA: The fine
structure of spiral ligament cells relates to ion return to the
stria and varies with place-frequency. Hear Res. 100:80–100. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kelley PM, Abe S, Askew JW, Smith SD,
Usami S and Kimberling WJ: Human connexin 30 (GJB6), a candidate
gene for nonsyndromic hearing loss: molecular cloning,
tissue-specific expression, and assignment to chromosome 13q12.
Genomics. 62:172–176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lautermann J, ten Cate WJ, Altenhoff P,
Grummer R, Traub O, Jahnke K and Winterhager E: Expression of the
gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue
Res. 294:415–420. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia AP, Ikeda K, Katori Y, Oshima T,
Kikuchi T and Takasaka T: Expression of connexin 31 in the
developing mouse cochlea. Neuro Report. 11:2449–2453.
2000.PubMed/NCBI
|
|
12
|
Liu XZ, Xia XJ, Adams J, Chen ZY, Welch
KO, Tekin M, Ouyang XM, Kristiansen A, Pandya A, Balkany T, Arnos
KS and Nance WE: Mutations in GJA1 (connexin 43) are associated
with non-syndromic autosomal recessive deafness. Hum Mol Genet.
10:2945–2951. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang JJ, Huang SH, Chou KH, Liao PJ, Su CC
and Li SY: Identification of mutations in members of the connexin
gene family as a cause of nonsyndromic deafness in Taiwan. Audiol
Neurootol. 12:198–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Steel KP and Kros CJ: A genetic approach
to understanding auditory function. Nate Genet. 27:143–149. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang JJ, Liao PJ, Su CC and Li SY:
Expression patterns of connexin 29 (GJE1) in mouse and rat cochlea.
Bioche Biophy Res Comm. 338:723–728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kikuchi T, Kimura RS, Paul DL and Adams
JC: Gap junctions in the rat cochlea immunohistochemical and
ultrastructural analysis. Anat Embryol. 191:101–118. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Forge A, Becker D, Casalotti S, Edwards J,
Marziano N and Nevill G: Gap junctions in the inner ear: comparison
of distribution patterns in different vertebrates and assessement
of connexin composition in mammals. J Comp Neurol. 467:207–231.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang WH, Yang JJ, Lin YC, Yang JT and Li
SY: Novel expression patterns of connexin 30.3 in adult rat
cochlea. Hear Res. 265:77–82. 2010. View Article : Google Scholar
|
|
19
|
López-Bigas N, Melchionda S, Gasparini P,
Borragán A, Arbonés ML and Estivill X: A common frameshift mutation
and other variants in GJB4 (connexin 30.3) analysis of hearing
impairment families. Hum Mutat. 19:4582002.PubMed/NCBI
|
|
20
|
Yang JJ, Wang WH, Lin YC, Weng HH, Yang
JT, Hwang CF, Wu CM and Li SY: Prospective variants screening of
connexin genes in children with hearing impairment:
genotype/phenotype correlation. Human Genetics. 128:303–313. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mazzoli M, Van Camp G, Newton V, Giarbini
N, Declau F and Parving A: Recommendations for the description of
genetic and audiological data for families with nonsyndromic
hearing impairment. 2003, Available at Hereditary Hearing Loss
Homepage, http://hereditaryhearingloss.org/main.aspx?c=.HHH&n=86638.
Accessed, 2014
|
|
22
|
American College of Medical Genetics.
Genetics Evaluation Guidelines for the Etiologic Diagnosis of
Congenital Hearing Loss. Genetic Evaluation of Congenital Hearing
Loss Expert Panel ACMG statement. Genet Med. 4:162–171. 2002.
View Article : Google Scholar
|
|
23
|
Hişmi BO, Yilmaz ST, Incesulu A and Tekin
M: Effects of GJB2 genotypes on the audiological phenotype:
variability is present for all genotypes. Int J Pediatr
Otorhinolaryngol. 70:1687–1694. 2006.PubMed/NCBI
|
|
24
|
Liu XZ, Pandya A, Angeli S, Telischi FF,
Arnos KS, Nance WE and Balkany T: Audiological features of GJB2
(connexin 26) deafness. Ear Hear. 26:361–369. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Evans WH and Martin PE: Gap junctions:
structure and function (Review). Mol Membr Biol. 19:121–136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bukauskas FF, Bukauskiene A, Bennett MV
and Verselis VK: Gating properties of gap junction channels
assembled from connexin 43 and connexin 43 fused with green
fluorescent protein. Biophys J. 81:137–152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bukauskas FF and Verselis VK: Gap junction
channel gating. Biochim Biophys Acta. 1662:42–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bordoli L, Kiefer F, Arnold K, Benkert P,
Battey J and Schwede T: Protein structure homology modeling using
SWISS-MODEL workspace. Nat Protoc. 4:1–13. 2009. View Article : Google Scholar
|
|
29
|
Sasin JM and Bujnicki JM: COLORADO3D, a
web server for the visual analysis of protein structures. Nucleic
Acids Research. 32:W586–W589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Melo F and Feytmans E: Assessing protein
structures with a non-local atomic interaction energy. J Mol Biol.
277:1141–1152. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maeda S, Nakagawa S, Suga M, Yamashita E,
Oshima A, Fujiyoshi Y and Tsukihara T: Structure of the connexin 26
gap junction channel at 3.5° A resolution. Nature. 458:597–604.
2009.
|
|
32
|
Nakagawa S, Maeda S and Tsukihara T:
Structural and functional studies of gap junction channels. Curr
Opin Struct Biol. 20:423–430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Beyer EC and Berthoud VM: The family of
connexin gene. Connexin: A Guide. Harris A and Locke D: Humana
Press, Springer; USA: pp. 3–26. 2009, View Article : Google Scholar
|
|
34
|
Berezin C, Glaser F, Rosenberg J, Paz I,
Pupko T, Fariselli P, Casadio R and Ben-Tal N: ConSeq: the
identification of functionally and structurally important residues
in protein sequences. Bioinformatics. 20:1322–1324. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krutovskikh V and Yamasaki H: Connexin
gene mutations in human genetic diseases. Muta Res. 462:197–207.
2000. View Article : Google Scholar : PubMed/NCBI
|