Introduction
Pinacidil is an antihypertensive drug, which reduces
blood pressure by directly relaxing vascular smooth muscle,
resulting in vasodilation of peripheral arterioles. Pinacidil is
primarily a mitochondrial adenosine triphosphate (ATP)-sensitive
potassium channel (mito-KATP) opener (1). The structure and mechanism of action
of pinacidil are distinct from those of other commonly used
antihypertensive drugs (1).
Pinacidil pharmacology has been studied in numerous types of smooth
muscle, including cardiac vascular, tracheal, bladder and neuronal
tissues (2). In a previous study,
pinacidil prevented subarachnoid hemorrhage-induced vasospasm by
the restoration of large-conductance Ca2+-activated
K+ channel activities (3). Furthermore, pinacidil pre-treatment
was able to exert significant protective effects following
hemorrhagic shock, mainly by the activation of protein kinase C
(PKC)α and PKCɛ via adenosine receptor A1, which restored vascular
reactivity and calcium sensitivity and influenced the success of
hemorrhagic shock therapy (4).
Pinacidil was also demonstrated to enhance the survival of
cryopreserved human embryonic stem cells (hESCs) (5). Following plating of thawed hESCs,
pinacidil significantly enhanced cell attachment capacity. Previous
studies have focused on investigating the function of pinacidil in
the heart and coronary artery in order to elucidate the mechanism
underlying its protective effects (4). It was hypothesized that the
activation and opening of the mito-KATP (6) and the sarcolemmal KATP channel
(7) may be beneficial in the
restoration of mitochondrial energy metabolism by closing the
mitochondrial transition pore (MTP), which stabilizes the
mitochondrial membrane potential (MMP) and reduces intracellular
Ca2+ levels (8). KATP
channels function in the coupling of redox potential (9) and/or mediating free-radical release
in mitochondria (10). Activation
of mito-KATP produces an increase in the matrix volume of
mitochondria, which may result in the activation of the electron
transport chain (11).
Oxidative stress is caused by excessive reactive
oxygen species (ROS) formation; therefore, an increase in the
production of ROS and reactive nitrogen species results in
mitochondrial dysfunction, cellular damage and cell death in
numerous degenerative diseases and aging (12). Osteoporosis is one such
pathological condition in which markedly increased levels of
oxidative stress markers are detected in the blood (13). Osteoporosis is primarily associated
with aging and mitochondrial dysfunction is implicated in the
ageing process where the respiratory chain represents a potential
source of ROS (14).
Antimycin A inhibits mitochondrial electron
transport by binding to complex III (15). Inhibition of electron transport
results in disruption of the proton gradient across the
mitochondrial inner membrane, perturbing the MMP and inducing the
production of ROS. In a previous study by our group, it was
demonstrated that pinacidil stimulated osteoblast differentiation
and decreased the osteoclast differentiation-inducing factors
promoted by antimycin A (16).
However, the mechanism of action of the mito-KATP opener pinacidil
in osteoblastic cells remains to be elucidated. The
pinacidil-induced cellular protection may be produced via a
mitochondrial pathway and this effect may therefore influence the
total cellular energy turnover. Mitochondria are one of the key
cellular organelles involved in mediating energy metabolism and
oxidative stress; therefore, in the present study, the protective
effects of pinacidil against antimycin A-induced cytotoxicity were
investigated using osteoblastic MC3T3-E1 cells, a model frequently
used to study osteogenic development in vitro.
Materials and methods
Reagents
Pinacidil was purchased from Sigma-Aldrich (St.
Louis, MO, USA). α-modified minimal essential medium (α-MEM) and
fetal bovine serum (FBS) were purchased from Gibco-BRL (Invitrogen
Life Technologies, Carlsbad, CA, USA). Other reagents were
purchased from Sigma-Aldrich and were of the highest commercial
grade available.
Cell culture
Mouse osteoblastic MC3T3-E1 cells were obtained from
American Type Culture Collection (Manassas, VA, USA) and cultured
at 37°C in a 5% CO2 atmosphere in α-MEM. Unless
otherwise specified, the medium contained 10% heat-inactivated FBS,
100 U/ml penicillin and 100 μg/ml streptomycin. The cells were
treated, at confluence, with culture medium containing 5 mM
β-glycerophosphate and 50 μg/ml ascorbic acid to initiate
differentiation. Following three days of culture, the cells were
pre-incubated with pinacidil for 1 h prior to treatment with
antimycin A for 48 h (17).
Cell viability
To investigate the molecular pathway underlying the
protective effect of pinacidil on cell viability, 0.1 μM
glibenclamide (a KATP channel inhibitor), 0.1 μM LY294002 (a PI3K
inhibitor), 0.1 μM GSK690693 (an Akt inhibitor) and 0.2 μM
auranofin (a thioredoxin reductase inhibitor) were used. All
inhibitors were purchased from Sigma-Aldrich. Cell viability was
determined via reduction of MTT by nicotinamide adenine
dinucleotide-dependent dehydrogenase activity to form a colored
reaction product (18). MTT (0.5
mg/ml in PBS) was added to each well and the plates were incubated
for an additional 2 h. Following removal of the solutions in each
well, dimethyl sulfoxide (DMSO) was added to dissolve the formazan
products and the plates were agitated for 5 min. The absorbance of
each well was recorded using a microplate spectrophotometer (Zenyth
3100 multimode detector; Anthos Labtec Instruments GmbH, Salzburg,
Austria) at 570 nm.
Measurement of PI3K activity
PI3K activity in cell lysates was evaluated using a
PI3 kinase ELISA kit (Echelon Biosciences, Inc., Salt Lake City,
UT, USA) (18). The assay was a
competitive ELISA in which the signal was inversely proportional to
the amount of PI(3,4,5)P3 produced. Following completion of the
PI3K reactions, the reaction products were mixed and incubated with
a PI(3,4,5)P3 detector protein, and then added to the
PI(3,4,5)P3-coated microplate for competitive binding. A
peroxidase-linked secondary detector and colorimetric detection
were used to identify PI(3,4,5)P3 detector binding to the plate.
The colorimetric signal was inversely proportional to the amount of
PI(3,4,5)P3 produced by PI3K.
Measurement of Akt activity
Akt activity in cell lysates was examined using an
Akt Kinase Activity kit (Enzo Life Sciences, Inc., Farmingdale, NY,
USA) according to the manufacturer’s instructions. The substrate,
which was readily phosphorylated by Akt, was pre-coated onto the
wells of the Akt microtiter plate supplied. The samples for assay
were added to the appropriate wells, followed by the addition of
ATP to initiate the reaction. The kinase reaction was terminated
and a phosphospecific substrate antibody, which binds specifically
to the phosphorylated peptide substrate, was added to the wells.
The phosphospecific antibody was subsequently bound by a
peroxidase-conjugated secondary antibody and the assay was
developed with tetramethylbenzidine substrate. The color developed
in proportion to the level of Akt phosphotransferase activity.
Measurement of phosphorylated CREB
Phosphorylated CREB levels were evaluated using
Cell-Based Phospho-CREB (S133) immunoassay (R&D Systems, Inc.,
Mineappolis, MN, USA) (19). This
cell-based kit contained the components required to run an ELISA
using fluorogenic substrates to measure phosphorylated CREB (S133)
levels in whole cells. Following stimulation with agents, cells
were fixed and permeabilized in the wells. The target protein
phosphorylation was measured using a double immunoenzymatic
labeling procedure. The cells were simultaneously incubated with
two primary antibodies: A phospho-specific antibody and a
normalization antibody that recognizes the pan-protein regardless
of phosphorylation status. The primary antibodies were derived from
different species. Two secondary antibodies labeled with
horseradish peroxidase (HRP) or alkaline phosphatase (AP) and two
spectrally distinct fluorogenic substrates for HRP or AP were used
for detection using a Zenyth 3100 multimode detector (Anthos Labtec
Instruments GmbH). The fluorescence of the phosphorylated protein
was normalized to that of the pan-protein in that well in order to
correct well-to-well variations.
Measurement of thioredoxin reductase
(TrxR) activity
TrxR activity was measured using an enzyme activity
assay kit (TrxR ELISA kit; Cayman Chemical Co., Ann Arbor, MI,
USA). Cells were homogenized in lysis buffer [20 mM
3-(N-morpholino)propanesulfonic acid, 50 mM sodium fluoride,
1 mM sodium vanadate, 5 mM ethylene glycol tetraacetic acid, 2 mM
EDTA, 1% nonyl phenoxypolyethoxylethanol, 1 mM dithiothreitol, 1 mM
benzamidine, 1 mM phenylmethylsulfonyl fluoride and 1% protease
inhibitor cocktail (pH 7.2)]. Following centrifugation at 10,000 xg
for 15 min at 4°C, the supernatant was used as a cell lysate for
assay.
Measurement of mitochondrial
superoxide
Mitochondrial superoxide levels were detected using
MitoSOXTM Red mitochondrial superoxide indicator (Molecular
Probes®; Life Technologies) (18). MitoSOXTM Red (Excitation/Emission
(Ex/Em)=510/580 nm) is a fluorogenic dye for selective detection of
superoxide in the mitochondria of cells. Cells were incubated with
5 μM MitoSOXTM Red at 37°C for 20 min according to the
manufacturer’s instructions. Following washing of the cells, the
MitoSOXTM Red fluorescence was detected using a fluorescence
microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Determination of MMP
The JC-1 mitochondrial membrane potential assay kit
(Cayman Chemical Co.) was used to investigate changes in the MMP of
cells (19). JC-1 is a lipophilic
and cationic dye, which permeates plasma and mitochondrial
membranes. The dye fluoresces red when it aggregates in healthy
mitochondria with high membrane potential, whereas it appears in
monomeric form and therefore fluoresces green in mitochondria with
diminished membrane potential. Cells were incubated with JC-1 for
20 min at 37°C, washed twice in PBS and then the ‘red’
(Ex/Em=550/600 nm) and ‘green’ (Ex/Em=485/535 nm) fluorescence was
measured using a fluorescence microplate reader (Molecular Devices,
LLC). Mitochondrial depolarization (loss of MMP) was indicated by a
decrease in the red/green fluorescence ratio.
Measurement of ATP levels
The ATP concentration was determined by luciferase
reaction using the EnzyLight™ ATP Assay kit (BioAssay Systems,
Hayward, CA, USA). This ATP assay provided a rapid method for the
measurement of intracellular ATP. Protein concentrations were
determined using the BioRad protein assay reagent (Bio-Rad
Laboratories, Hercules, CA, USA) and detected using a Zenyth 3100
multimode detector (Anthos Labtec Instruments GmbH) according to
the manufacturer’s instructions.
Intracellular Ca2+
measurement
Osteoblastic MC3T3-E1 cells were cultured in media
containing pinacidil and/or antimycin A. Cells were subsequently
treated with 5 μM fura-2-acetoxymethyl ester (AM) for 1 h at 37°C.
Following washing with PBS, fluorescence emission (510 nm) was
monitored at the excitation wavelengths of 340 and 380 nm using a
Gemini XPS microplate reader (Molecular Devices, LLC) according to
the manufacturer’s instructions.
Statistical analysis
Experiments were performed in triplicate and values
are expressed as the mean ± standard error of the mean. Statistical
analysis was performed using SAS software 9.2 (SAS Institute, Inc.,
Cary, NC, USA) Statistical significance was determined by analysis
of variance and subsequently applying the Dunnett’s t-test.
P<0.05 was considered to indicate a statistically significant
difference between values.
Results
Pinacidil enhances the viability of
MC3T3-E1 cells following antimycin A treatment
Cell viability was evaluated by MTT assay. In the
absence of antimycin A, pinacidil had no effect on the viability of
MC3T3-E1 cells at concentrations ≤1 μM (Fig. 1A). Antimycin A (70 μM)
significantly decreased the cell viability of osteoblasts, whereas
pinacidil pretreatment (0.01–1 μM) increased cell viability
compared with that of antimycin A-treated cells (Fig. 1B). These results indicated that
pinacidil exerted a protective effect against antimycin A-induced
cytotoxicity. Furthermore, the protective effects of pinacidil on
cell viability were perturbed by the presence of LY294002 (a PI3K
inhibitor), GSK690693 (an Akt inhibitor) or auranofin (thioredoxin
reductase inhibitor), but not by glibenclamide (KATP channel
inhibitor), indicating that the effect of pinacidil may be mediated
by PI3K, Akt and/or thioredoxin reductase activity (Fig. 1C).
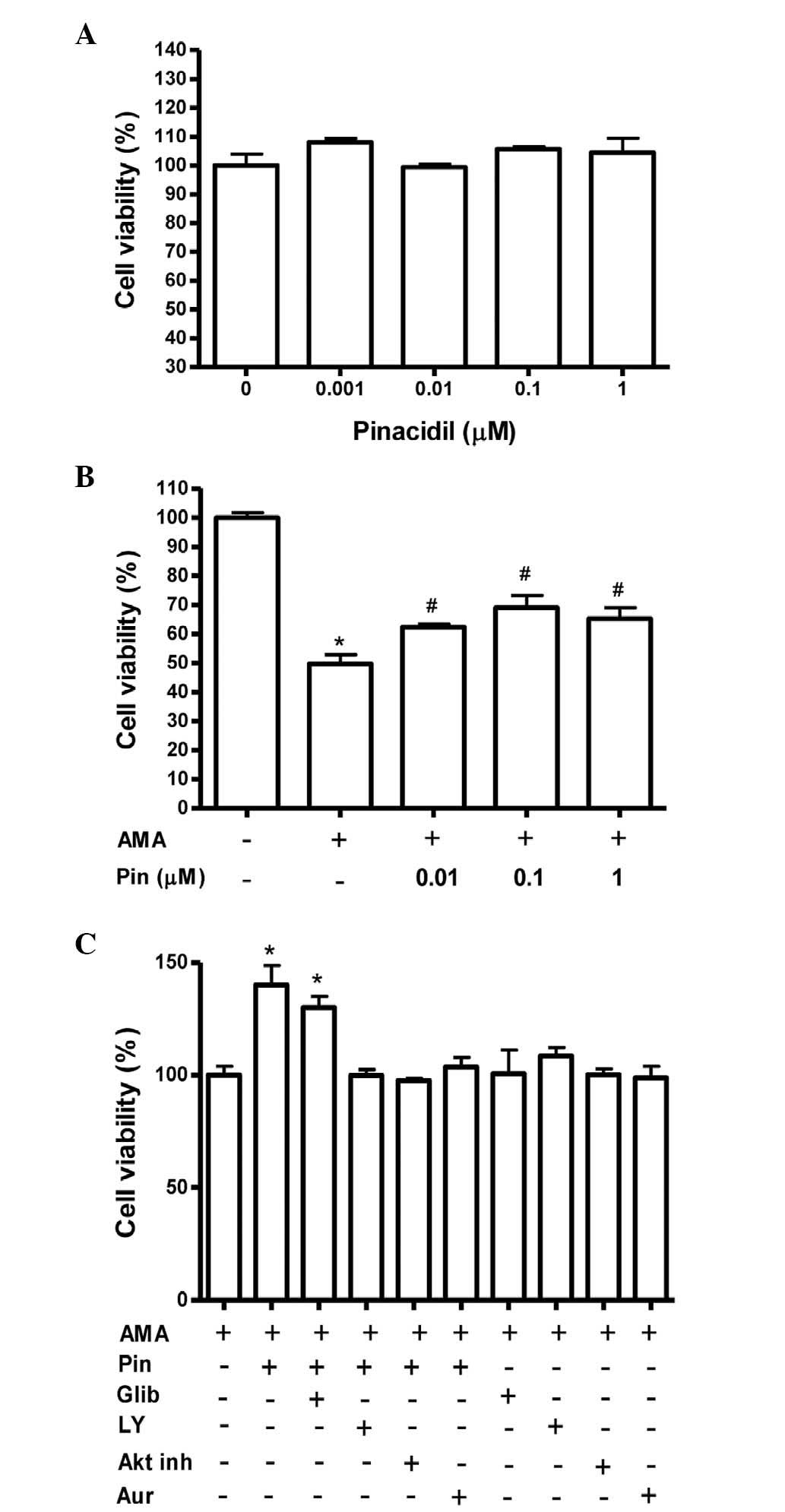 | Figure 1Effect of Pin on the cell viability of
osteoblastic MC3T3-E1 cells in the presence of AMA. Osteoblasts
were treated with Pin in the (A) absence or (B) presence of 70 μM
AMA for 48 h. Cell viability was subsequently assessed by MTT
assay. (C) One hour prior to the treatment with 0.1 μM Pin,
cultures were pre-treated with Glib (0.1 μM), LY (0.1 μM), Akt inh
(0.1 μM) or Aur (0.2 μM). Values are expressed as a percentage of
the control ± standard error of the mean. *P<0.05 vs.
control; #P<0.05, vs. AMA. Pin, pinacidil; AMA,
antimycin A; Glib, glibenclamide; LY, LY294002; Akt inh, Akt
inhibitor; Aur, auranofin. |
Pinacidil rescues the activation of PI3K,
Akt and CREB in the presence of antimycin A
The PI3K/Akt/CREB signaling pathway is known to
function as a pro-survival signal (18,19).
The role of PI3K/Akt/CREB activation in mediating the
cytoprotective effects of pinacidil in MC3T3-E1 cells was therefore
investigated. Following treatment of the cultures with 70 μM
antimycin A, in the presence or absence of pinacidil, PI3K and Akt
activities were measured. As shown in Fig. 2A and B, the results of these
experiments indicated that antimycin A decreased PI3K and Akt
activities and pretreatment with pinacidil prevented this decline
in activity in cells at concentrations of 0.01–0.1 μM and 0.1 μM,
respectively. Treatment with antimycin A inhibited phosphorylation
of CREB, while pinacidil pre-treatment (0.1 μM) rescued CREB
phophorylation levels (P<0.05; Fig.
2C).
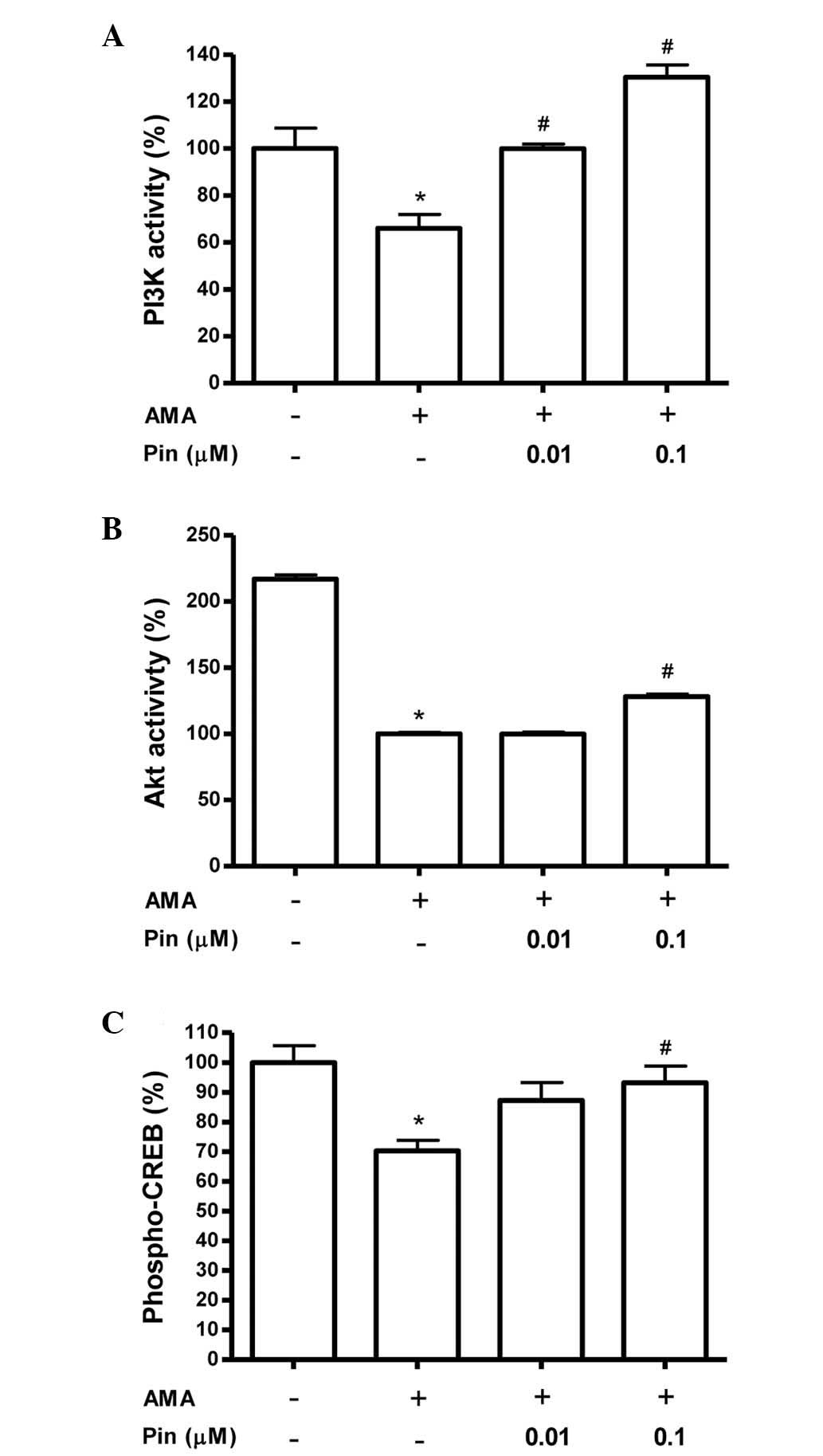 | Figure 2Effect of pinacidil on PI3K activity,
Akt activity and CREB phosphorylation of osteoblasts in the
presence of AMA. Values are expressed as a percentage of the
control ± standard error of the mean. The control values for (A)
PI3K, (B) Akt activities and (C) phosphorylated CREB were
6.99±0.607 nmol/mg, 2.632±0.037 OD/mg and 0.46±0.026
phospho-CREB/total CREB, respectively. *P<0.05,
control vs. AMA; #P<0.05, AMA vs. Pin. PI3K,
phosphoinositide 3-kinase; CREB, cyclic adenosine
monophosphate-responsive element-binding protein; AMA, antimycin A;
Pin, pinacidil. |
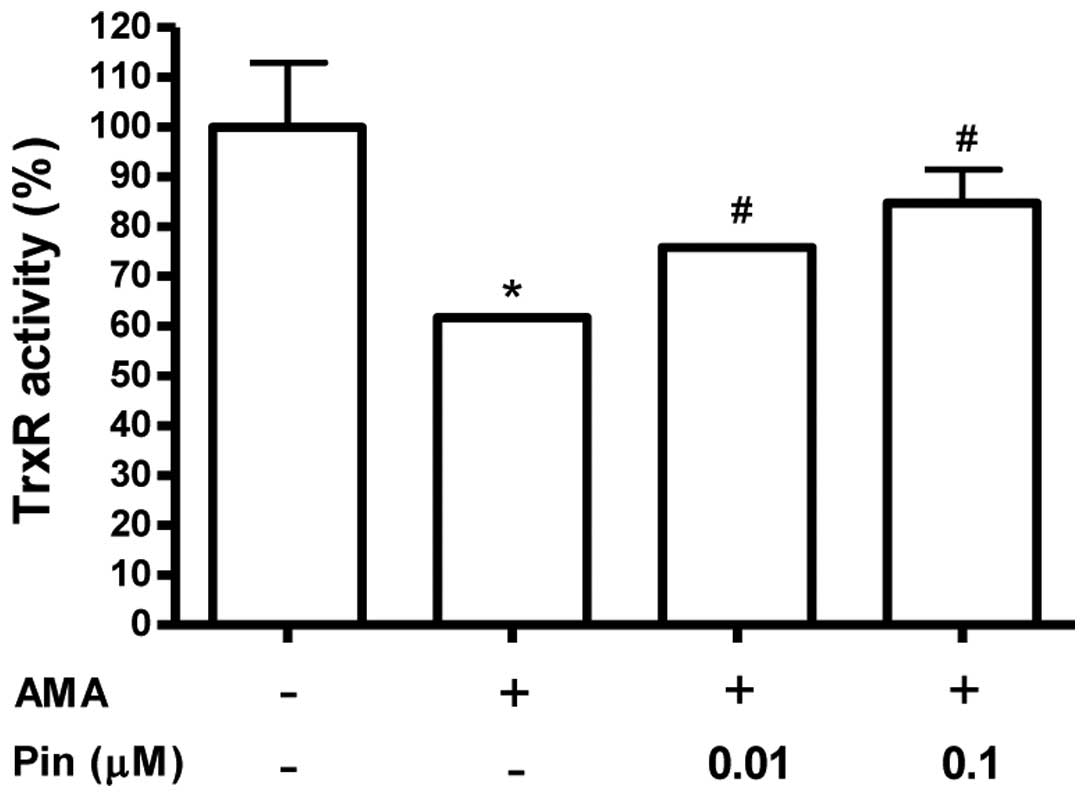
Pinacidil has an antioxidant effect in
osteoblastic MC3T3-E1 cells
Activation of thioredoxin reductase (TrxR) was also
considered as a potential mechanism underlying the cytoprotective
effects of pinacidil. The effect of pinacidil on TrxR activity was
evaluated using the 5,5′-dithiobis(2-nitrobenzoic) acid reduction
assay. Exposure of MC3T3-E1 cells to antimycin A resulted in a
significant decrease in TrxR activity (Fig. 3). This inhibitory effect of
antimycin A was counteracted by pre-treatment with pinacidil (0.01
and 0.1 μM).
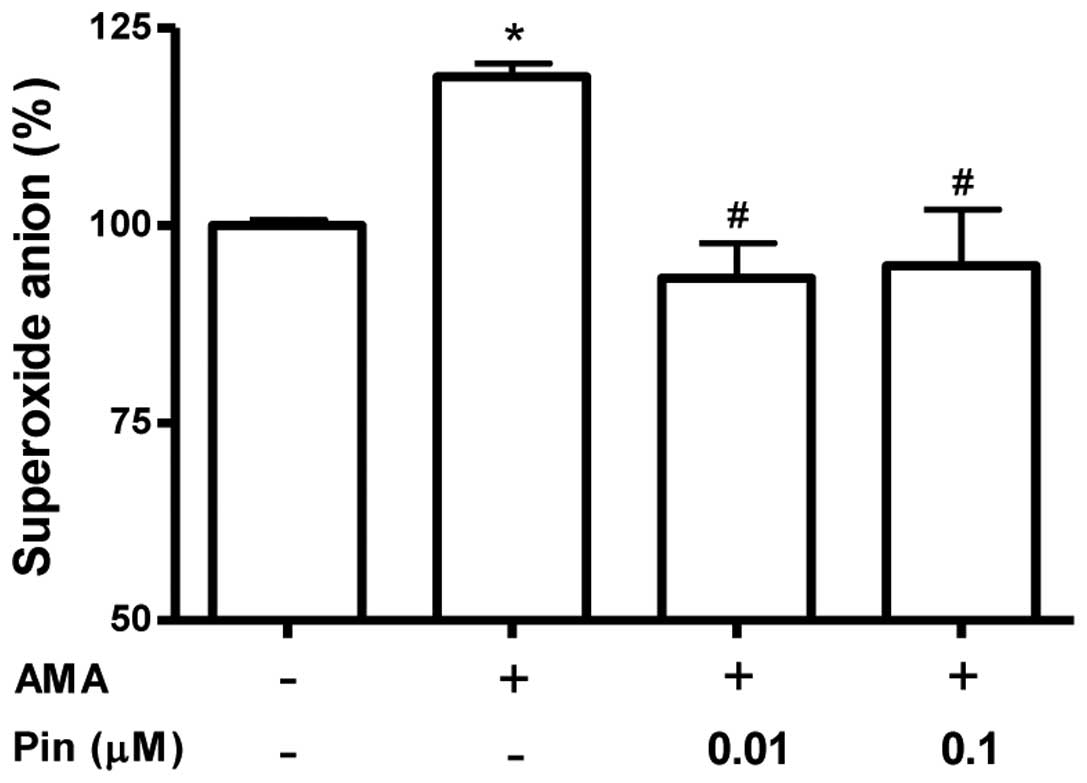
The effect of pinacidil on mitochondrial superoxide
production was measured using MitoSOX, a live-cell-permeable and
mitochondrial localizing superoxide indicator. MitoSOX is derived
from dihydroethidium, conjugated to a triphenylphosphonium moiety
and has a positive charge, which results in its accumulation within
the mitochondrial matrix (18). As
indicated in Fig. 4, mitochondrial
MitoSOX fluorescence was increased following antimycin A treatment;
however, pinacidil (0.01 and 0.1 μM) pre-treatment inhibited the
mitochondrial superoxide production induced by antimycin A.
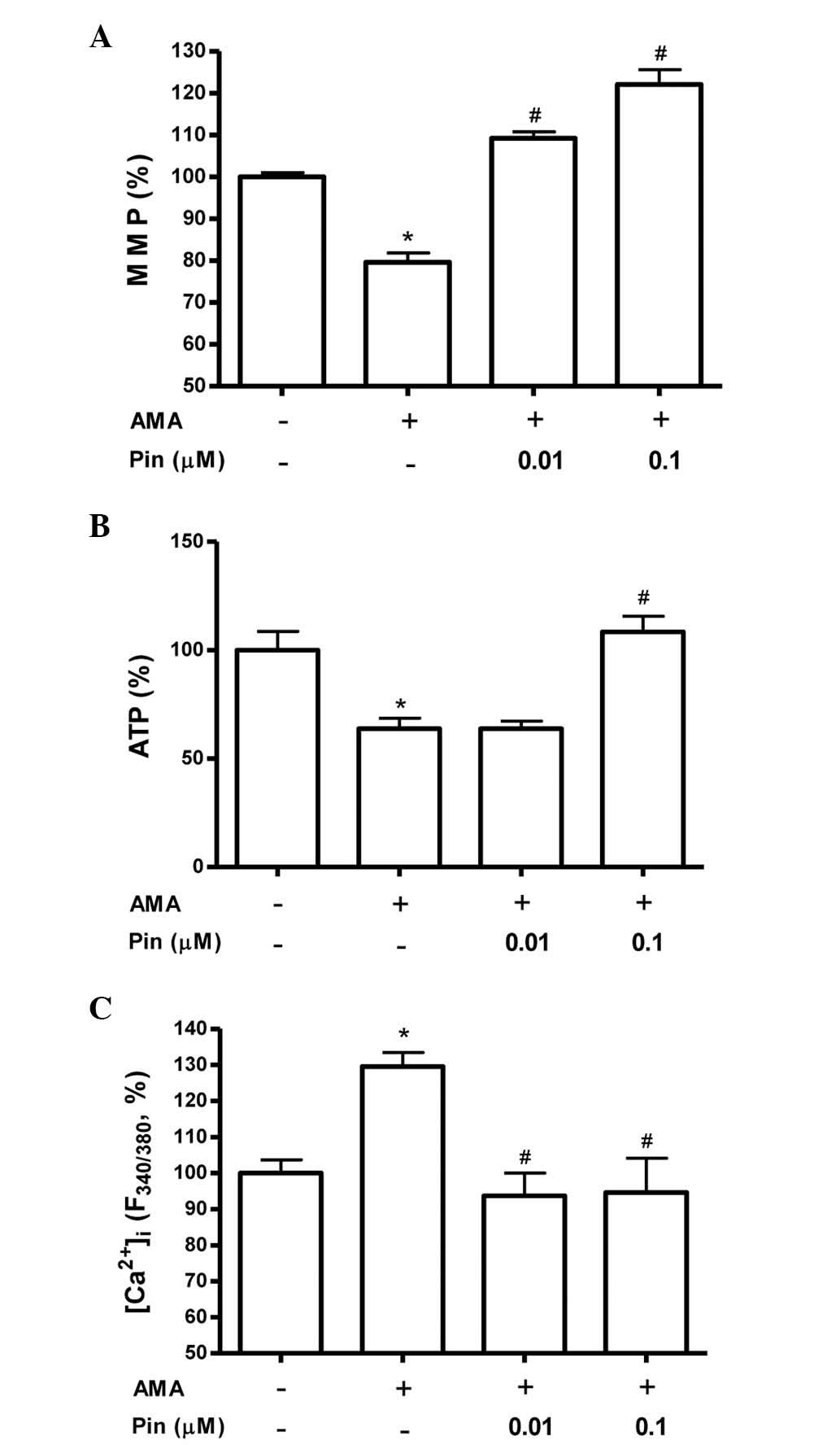
Pinacidil attenuates antimycin A-induced
mitochondrial dysfunction in osteoblastic MC3T3-E1 cells
When cells are subjected to oxidative stress,
mitochondrial membrane integrity is lost, which initiates a
signaling cascade resulting in cell death. To investigate the
mitochondrial function of osteoblasts, MMP and ATP content were
assayed following treatment of MC3T3-E1 cells with antimycin A in
the presence or absence of pinacidil. As shown in Fig. 5A, a decrease in MMP occurred
following exposure to antimycin A. Pinacidil (0.01 and 0.1 μM)
abrogated this reduction in MMP, suggesting that pinacidil
pre-treatment may be able to maintain the membrane potential of
these cells, thereby precluding this induced cell death. ATP levels
were evaluated using a standard luminescent luciferin/luciferase
assay. As illustrated in Fig. 5B,
the levels of ATP in osteoblasts decreased following antimycin A
treatment. However, pinacidil (0.1 μM) pre-treatment significantly
increased intracellular ATP levels when compared with those of the
antimycin A group. The intracellular calcium concentration
([Ca2+]i) has been demonstrated to have a
role in cell death, and alterations in
[Ca2+]i were therefore measured using the
Fura-2AM fluorescence technique. Osteoblastic MC3T3-E1 cells were
cultured in media containing pinacidil and/or antimycin A.
Subsequently, following incubation with Fura-2AM,
[Ca2+]i levels were measured. As depicted in
Fig. 5C, antimycin A markedly
increased [Ca2+]i, an effect that was
significantly attenuated by pinacidil (0.01 and 0.1 μM). In
conclusion, the results of the present study indicated that the
cytoprotective effect of pinacidil resulted from its function in
mitochondrial protection.
Discussion
The present study demonstrated the cytoprotective
effects of pinacidil against antimycin A-induced damage in
osteoblastic MC3T3-E1 cells. Pinacidil enhanced the antimycin
A-inhibited PI3K and Akt activity and CREB phosphorylation.
Activation of Akt has a crucial function in regulating fundamental
cellular functions, including cell proliferation and survival, by
phosphorylation of numerous substrates. PI3K/Akt signaling prevents
mitochondrial cytochrome c release by indirectly influencing
cellular metabolism, which sustains mitochondrial integrity
(20). CREB is a cellular
transcription factor, which is activated in part by the
phosphorylation of serine at position 133. Following activation,
CREB induces transcription of various downstream genes. Cammarota
et al (21) reported that
CREB was localized to the inner mitochondrial compartment, in
addition to localization in the nucleus. Mitochondrial CREB was
hypothesized to mediate pro-survival effects by regulating various
mitochondrial genes, the expression of which may be directed by
CRE-like elements located in the D-loop of the mitochondrial genome
(22). Furthermore, Lee et
al (23) demonstrated that the
binding of CREB to CRE sequences in the D-loop of mitochondrial DNA
enhanced the transcript levels of mitochondrial genes. Therefore,
the results of the present study suggested that the protective
effects of pinacidil in osteoblasts may be associated with PI3K,
Akt and CREB signaling.
Trx is a protein ubiquitously expressed in living
cells, which performs numerous functions associated with cell
proliferation and apoptosis (24)
and additionally confers cytoprotection against cellular damage by
oxidative stress (25). In the
present study, TrxR activity was demonstrated to be reduced
following antimycin A treatment, an effect which was reversed by
pinacidil pre-treatment. Previous in vitro studies indicated
that Trx was able to inhibit apoptosis-regulating kinase-1 (ASK-1)
activity. ASK-1 is a mitogen-activated protein kinase (MARK) that
activates pro-apoptotic protein kinases (24); therefore, ASK-1 inhibition has a
protective effect against damage resulting from oxidative stress
(26). The Trx system is the most
important factor in the regulation of the intracellular redox
environment, which influences the redox state, antioxidant defense
and redox regulation of multiple cellular processes (27). TrxR regulates the redox state of
cells via detoxification of hydrogen peroxide and lipid peroxides
(28), regeneration of ascorbic
acid (29) and by enhancing
superoxide dismutase activity (30). Therefore, the present study
hypothesized that the protective effect of pinacidil against cell
damage due to redox disruption may be mediated by an upregulation
of the Trx system.
Inhibition of enzymes in the mitochondrial
respiratory chain may result in aberrant single-electron reduction
of molecular oxygen to superoxide (13). It is therefore critical that
superoxide-induced redox signaling in the mitochondria is
prevented. Increased mitochondrial superoxide levels lead to
enhanced transferrin receptor expression, which results in
increases in iron uptake and generation of various lipid aldehyde
signaling molecules (31).
Mitochondrial-generated superoxide was demonstrated to activate
uncoupling proteins via the formation of lipid peroxidation
breakdown products (32). In the
present study, pinacidil was demonstrated to markedly attenuate the
production of superoxide in the mitochondria induced by antimycin A
treatment, which indicated that the cytoprotective effect of
pinacidil may be associated with its antioxidative functions.
ATP synthesis, the primary function of mitochondria,
is mediated by oxidative phosphorylation and associated with cell
redox status (33). The transfer
of electrons from complex I to V generates a proton concentration
gradient across the inner mitochondrial membrane, sustaining the
MMP and driving ATP synthesis (34). A failure of efficient electron
transport and ATP synthesis induced by oxidative stress results in
the accumulation of free radicals and leads to mitochondrial
dysfunction (35). In the present
study, pre-treatment with pinacidil was found to increase ATP
synthesis. This result suggested that pinacidil pre-treatment
normalized oxidative phosphorylation rates, thereby increasing
mitochondrial energy metabolism. This led to the hypothesis that
one of the mechanisms by which pinacidil confers protection against
oxidative stress may be by directly enhancing the rate of ATP
synthesis.
The integrity of the mitochondrial membrane
structure is also important in maintaining normal cell function, as
it is associated with mitochondrial functions, including the
transmembrane proton gradient and ATP energy production (36). It has been suggested that
mitochondrial dysfunction may be responsible for numerous human
disorders (37). The majority of
the proton motive force of mitochondria is in the form of the MMP;
therefore, the MMP may be used as a measure of mitochondrial
function in cellular homeostasis. A decrease in MMP results in a
reduction in mitochondrial dehydrogenase activity, depressed
oxidative phosphorylation enzyme complex function and therefore a
decrease in ATP synthesis. In the present study, pretreatment of
MC3T3-E1 cells with pinacidil reversed the antimycin A-induced
decline in MMP, which suggested that pinacidil may inhibit the
opening of the MTP and prevent mitochondrial depolarization,
averting osteoblast death. Therefore, mitochondria had an important
role in mediating the protective effects of pinacidil in damaged
MC3T3-E1 cells.
Antimycin A increased the cytosolic Ca2+
concentration, an effect that was attenuated by pinacidil
treatment. Ca2+ enhances cytochrome c
displacement from the mitochondrial inner membrane by competing for
cardiolipin binding sites or inducing the cytochrome c
releasing process during MTP opening (38). MTP opening allows the passage of
protons through the mitochondrial membrane and is therefore
associated with a rapid depolarization of the mitochondria and
subsequent uncoupling of oxidative phosphorylation (39). It was therefore hypothesized that
pinacidil may inhibit cytochrome c release by mediating a
reduction in intracellular calcium concentration.
In conclusion, pinacidil protected osteoblastic
cells against antimycin A-induced cell damage by enhancing
mitochondrial function and rescuing activation of PI3K, Akt and
CREB. Furthermore, pinacidil may potentially be used to protect
mitochondria against bursts of oxidative stress in osteoblastic
cells.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea,
funded by the Ministry of Education (no.
NRF-2013R1A1A2A10004361).
References
|
1
|
Friedel HA and Brogden RN: Pinacidil. A
review of its pharmacodynamic and pharmacokinetic properties, and
therapeutic potential in the treatment of hypertension. Drugs.
39:929–967. 1990.PubMed/NCBI
|
|
2
|
Jahangir A and Terzic A: K(ATP) channel
therapeutics at the bedside. J Mol Cell Cardiol. 39:99–112. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen JY, Cheng KI, Tsai YL, Hong YR, Howng
SL, Kwan AL, Chen IJ and Wu BN: Potassium-channel openers KMUP-1
and pinacidil prevent subarachnoid hemorrhage-induced vasospasm by
restoring the BKCa-channel activity. Shock. 38:203–212. 2012.
View Article : Google Scholar
|
|
4
|
Xu J, Li T, Yang G and Liu L: Pinacidil
pretreatment improves vascular reactivity after shock through PKCα
and PKCɛ in rats. J Cardiovasc Pharmacol. 59:514–522.
2012.PubMed/NCBI
|
|
5
|
Barbaric I, Jones M, Buchner K, Baker D,
Andrews PW and Moore HD: Pinacidil enhances survival of
cryopreserved human embryonic stem cells. Cryobiology. 63:298–305.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hanley PJ and Daut J: K(ATP) channels and
preconditioning: a re-examination of the role of mitochondrial
K(ATP) channels and an overview of alternative mechanisms. J Mol
Cell Cardiol. 39:17–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang L and Yu T: Prolonged donor heart
preservation with pinacidil: the role of mitochondria and the
mitochondrial adenosine triphosphate-sensitive potassium channel. J
Thorac Cardiovasc Surg. 139:1057–1063. 2010. View Article : Google Scholar
|
|
8
|
Downey JM, Davis AM and Cohen MV:
Signaling pathways in ischemic preconditioning. Heart Fail Rev.
12:181–188. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O’Rourke B, Ramza BM and Marban E:
Oscillations of membrane current and excitability driven by
metabolic oscillations in heart cells. Science. 265:962–966.
1994.
|
|
10
|
Hoppeler H, Vogt M, Weibel ER and Flück M:
Response of skeletal muscle mitochondria to hypoxia. Exp Physiol.
88:109–119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Das M, Parker JE and Halestrap AP: Matrix
volume measurements challenge the existence of
diazoxide/glibencamide-sensitive KATP channels in rat mitochondria.
J Physiol. 547(Pt 3): 893–902. 2003.PubMed/NCBI
|
|
12
|
Beal MF: Mitochondria, free radicals, and
neurodegeneration. Curr Opin Neurobiol. 6:661–666. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maggio D, Barabani M, Pierandrei M,
Polidori MC, Catani M, Mecocci P, Senin U, Pacifici R and Cherubini
A: Marked decrease in plasma antioxidants in aged osteoporotic
women: results of a cross-sectional study. J Clin Endocrinol Metab.
88:1523–1527. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: a mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pham NA, Robinson BH and Hedley DW:
Simultaneous detection of mitochondrial respiratory chain activity
and reactive oxygen in digitonin-permeabilized cells using flow
cytometry. Cytometry. 41:245–251. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suh KS, Lee YS and Choi EM: Pinacidil
stimulates osteoblast function in osteoblastic MC3T3-E1 cells.
Immunopharm Immunotoxicol. 35:359–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choi EM: Protective effect of diazoxide
against antimycin A-induced mitochondrial dysfunction in
osteoblastic MC3T3-E1 cells. Toxicol In Vitro. 25:1603–1608. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi EM, Suh KS and Lee YS: Liquiritigenin
restores osteoblast damage through regulating oxidative stress and
mitochondrial dysfunction. Phytother Res. 28:880–886. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suh KS, Lee YS, Kim YS and Choi EM:
Sciadopitysin protects osteoblast function via its antioxidant
activity in MC3T3-E1 cells. Food Chem Toxicol. 58:220–227. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kennedy SG, Kandel ES, Cross TK and Hay N:
Akt/protein kinase B inhibits cell death by preventing the release
of cytochrome c from mitochondria. Mol Cell Biol. 19:5800–5810.
1999.PubMed/NCBI
|
|
21
|
Cammarota M, Paratcha G, Bevilaqua LR,
Levi de Stein M, Lopez M, Pellegrino de Iraldi A, Izquierdo I and
Medina JH: Cyclic AMP-responsive element binding protein in brain
mitochondria. J Neurochem. 72:2272–2277. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ryu H, Lee J, Impey S, Ratan RR and
Ferrante RJ: Antioxidants modulate mitochondrial PKA and increase
CREB binding to D-loop DNA of the mitochondrial genome in neurons.
Proc Natl Acad Sci USA. 102:13915–13920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee J, Kim CH, Simon DK, Aminova LR,
Andreyev AY, Kushnareva YE, et al: Mitochondrial cyclic AMP
response element-binding protein (CREB) mediates mitochondrial gene
expression and neuronal survival. J Biol Chem. 280:40398–40401.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tao L, Gao E, Bryan NS, Qu Y, Liu HR, Hu
A, Christopher TA, Lopez BL, Yodoi J, Koch WJ, Feelisch M and Ma
XL: Cardioprotective effects of thioredoxin in myocardial ischemia
and reperfusion: role of S-nitrosation. Proc Natl Acad Sci USA.
101:11471–11476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoshida T, Oka S, Masutani H, Nakamura H
and Yodoi J: The role of thioredoxin in the aging process:
involvement of oxidative stress. Antioxid Redox Signal. 5:563–570.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Y and Min W: Thioredoxin promotes ASK1
ubiquitination and degradation to inhibit ASK1-mediated apoptosis
in a redox activity-independent manner. Circ Res. 90:1259–1266.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lemarechal H, Anract P, Beaudeux JL,
Bonnefont-Rousselot D, Ekindjian OG and Borderie D: Impairment of
thioredoxin reductase activity by oxidative stress in human
rheumatoid synoviocytes. Free Radic Res. 41:688–698. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Björnstedt M, Hamberg M, Kumar S, Xue J
and Holmgren A: Human thioredoxin reductase directly reduces lipid
hydroperoxides by NADPH and selenocystine strongly stimulates the
reaction via catalytically generated selenols. J Biol Chem.
270:11761–11764. 1995.
|
|
29
|
May JM, Mendiratta S, Hill KE and Burk RF:
Reduction of dehydroascorbate to ascorbate by the selenoenzyme
thioredoxin reductase. J Biol Chem. 272:22607–22610. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Das KC, Lewis-Molock Y and White CW:
Elevation of manganese superoxide dismutase gene expression by
thioredoxin. Am J Respir Cell Mol Biol. 17:713–726. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jouihan HA, Cobine PA, Cooksey RC,
Hoagland EA, Boudina S, Abel ED, Winge DR and McClain DA:
Iron-mediated inhibition of mitochondrial manganese uptake mediates
mitochondrial dysfunction in a mouse model of hemochromatosis. Mol
Med. 14:98–108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Echtay KS, Esteves TC, Pakay JL, Jekabsons
MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S,
Roebuck SJ and Brand MD: A signalling role for 4-hydroxy-2-nonenal
in regulation of mitochondrial uncoupling. EMBO J. 22:4103–4110.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thorburn DR: Diverse powerhouses. Nature
Genet. 36:13–14. 2004. View Article : Google Scholar
|
|
34
|
Mourier A and Larsson NG: Tracing the
trail of protons through complex I of the mitochondrial respiratory
chain. PLoS Biol. 9:e10011292011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang DX and Gutterman DD: Mitochondrial
reactive oxygen species-mediated signaling in endothelial cells. Am
J Physiol Heart Circ Physiol. 292:H2023–H2031. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsujimoto Y and Shimizu S: Role of the
mitochondrial membrane permeability transition in cell death.
Apoptosis. 12:835–840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weissig V: Mitochondrial-targeted drug and
DNA delivery. Crit Rev Ther Drug Carrier Syst. 20:1–62. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hajnóczky G, Csordás G, Das S,
Garcia-Perez C, Saotome M, Sinha Roy S and Yi M: Mitochondrial
calcium signaling and cell death: approaches for assessing the role
of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium.
40:553–560. 2006.PubMed/NCBI
|
|
39
|
Tonin AM, Amaral AU, Busanello EN, Grings
M, Castilho RF and Wajner M: Long-chain 3-hydroxy fatty acids
accumulating in long-chain 3-hydroxyacyl-CoA dehydrogenase and
mitochondrial trifunctional protein deficiencies uncouple oxidative
phosphorylation in heart mitochondria. J Bioenerg Biomemb.
45:47–57. 2013. View Article : Google Scholar
|