Introduction
The thioredoxin (Trx) system, consisting of Trx, Trx
reductase (TrxR) and nicotinamide adenine dinucleotide phosphate
(NADPH), is a major disulfide reductase system, which is important
in cellular redox balance (1).
While Trx1 and TrxR1 are usually localized in the cytoplasm, Trx2
and TrxR2 are located in the mitochondria (2). Trx is typically a small, 12 kDa
protein reductase with two active site cysteine residues, 32 and
35. It exists either as a dithiol in the reduced form or as a
disulfide in the oxidized form. When Trx is oxidized, it is reduced
back to the dithiol by the NADPH-dependent selenoprotein, TrxR
(3). TrxR not only controls
oxidative stress via Trx, but also affects a number of cellular
functions, including DNA repair, cell proliferation and
angiogenesis (4). Trx and TrxR1
are overexpressed in numerous cancer cells, including lung cancer
(5–7). Therefore, modulation of the Trx
system is a promising target for cancer therapy. It has been
reported that the inhibition of TrxR increases the sensitivity of
anti-cancer drugs in various cancer cells, including lung and colon
cancer (8,9).
Auranofin (Au), an inhibitor of TrxR, has an
anti-inflammatory effect (10) and
is used to treat rheumatoid arthritis (11). It has been reported that Au
suppresses the immune response in dendritic cells and macrophages
by inhibiting the major histocompatibility complex-restricted
antigen presentation and pro-inflammatory cytokines, including
interleukin-1β (12,13). However, several studies have
suggested that Au induces apoptosis in breast cancer and leukemia
cells (14,15). This agent also leads to
mitochondrial permeability transition and the generation of
reactive oxygen species (ROS) (16,17).
Cervical cancer is a major cause of mortality in
females worldwide and its occurrence results from multiple factors,
including papillomaviruses, cigarette smoking and nutrition
(18). Oxidative stress can also
contribute to the carcinogenesis of cervical cancer. Furthermore,
the level of TrxR2 is increased in dysplastic and neoplastic
cervical tissues (19). Previous
studies have suggested that Au is a good candidate anti-cancer drug
in various cancer cells (20–22).
However, the cellular effect of Au in cervical cancer cells remains
to be elucidated. Therefore, in the present study, the effects of
Au on cell growth and cell death in human cervical HeLa cells was
examined in association with changes in the levels of ROS and
glutathione (GSH).
Materials and methods
Cell culture
Human cervical adenocarcinoma HeLa cells from the
American Type Culture Collection (Manassas, VA, USA) were cultured
in RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA). This medium was
supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1%
penicillin-streptomycin (Gibco-BRL, Grand Island, NY, USA). The
HeLa cells were maintained in a humidified incubator containing 5%
CO2 at 37°C. The cells were routinely grown in 100 mm
plastic tissue culture dishes (Nunc, Roskilde, Denmark) and
harvested with a solution of trypsin-EDTA (Gibco-BRL).
Reagents
Au was purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA) and was dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich) at 10 mM as a stock solution. The pan-caspase
inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
(Z-VAD-FMK), caspase-3 inhibitor
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone (Z-DEVD-FMK),
caspase-8 inhibitor
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone (Z-IETD-FMK)
and the caspase-9 inhibitor
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone (Z-LEHD-FMK)
were obtained from R&D Systems, Inc. (Minneapolis, MN, USA) and
were dissolved in DMSO at 10 mM to serve as stock solutions.
N-acetyl cysteine (NAC) and L-buthionine sulfoximine (BSO),
obtained from Sigma-Aldrich, were dissolved in buffer (20 mM HEPES
at pH 7.0) and water at 100 mM as a stock solution, respectively.
Cells were pretreated with 15 μM caspase inhibitors, 2 mM NAC or 10
μM BSO for 1 h prior to Au treatment. DMSO (0.01%) was used as a
control vehicle and it did not affect cell growth or cell
death.
Cell growth assay
To determine the effect of Au on cell growth, the
absorbance of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma-Aldrich) was measured in living cells, as
described previously (23).
Breifly, 5×103 cells were seeded in 96-well microtiter
plates (Nunc). After exposure to the indicated doses of Au with or
without 15 μM of each caspase inhibitor, 2 mM NAC or 10 μM BSO for
the designated times, MTT solution (20 μl: 2 mg/ml in phosphate
buffered saline; PBS) was added to each well of the 96-well plates.
The plates were incubated for 3 h at 37°C. Medium was removed from
the plates by pipetting and 200 μl DMSO was added to each well for
solubilizing the formazan crystals. The optical density was
measured at 570 nm using a microplate reader (Synergy™ 2; BioTek
Instruments Inc., Winooski, VT, USA).
Sub-G1 cell analysis
Sub-G1 cell analysis was determined by propidium
iodide (PI; Ex/Em=488/617 nm; Sigma-Aldrich) staining, as described
previously (24). In brief,
1×106 cells were incubated in a 60 mm culture dish
(Nunc) with the designated doses of Au for 24 h. Cells were washed
again with PBS and incubated with PI (10 mg/ml) with simultaneous
RNase treatment at 37°C for 30 min. Cellular DNA content was
measured using a FACStar flow cytometer (Becton Dickinson, Franklin
Lakes, NJ, USA) and analyzed using Lysis II and Cellfit software
(Becton Dickinson).
Annexin V-fluorescein isothiocyanate
(FITC)/PI staining for cell death detection
To determine apoptotic cell death, cells were
stained with annexin V-FITC (Invitrogen Life Technologies,
Carlsbad, CA, USA; Ex/Em=488/519 nm), as described previously
(24). In brief, 1×106
cells in a 60 mm culture dish (Nunc) were incubated with the
designated doses of Au, with or without 15 μM of each caspase
inhibitor, 2 mM NAC or 10 μM BSO for 24 h. Cells were washed twice
with cold PBS and then resuspended in 500 μl binding buffer (10 mM
HEPES/sodium hydroxide pH 7.4, 140 mM sodium chloride and 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Annexin V-FITC (5 μl) and PI (1 μg/ml) were then added and the
cells were analyzed with a FACStar flow cytometer (Becton
Dickinson).
Western blot analysis
The expression of proteins was evaluated using
western blot analysis, as previously described (25). In brief, 1×106 cells
were incubated in a 60 mm culture dish (Nunc) with the designated
doses of Au for 24 h. The cells were then washed in PBS and
suspended in five volumes of lysis buffer [20 mM HEPES, pH 7.9, 20%
glycerol, 200 mM potassium chloride, 0.5 mM EDTA, 0.5% NP40, 0.5 mM
DTT and 1% protease inhibitor cocktail (Sigma-Aldrich)]. The
concentration of supernatant proteins was determined using the
Bradford method. Supernatant samples containing 30 mg total protein
were resolved by 7.5 or 12.5% SDS-PAGE gels depending on the sizes
of target proteins, transferred onto Immobilon-P polyvinylidene
difluoride membranes (Millipore, Billerica, MA, USA) by
electroblotting and then probed with anti-poly ADP ribose
polymerase, monoclonal rabbit anti-B-cell lymphoma 2 (Bcl-2),
polyclonal rabbit anti-BCL2-associated X protein (Bax) purchased
from Cell signaling Technology, Inc. (Danvers, MA, USA), polyclonal
rabbit anti-Trx1, monoclonal mouse anti-TrxR1, polyclonal rabbit
anti-Trx2, polyclonal goat anti-TrxR2 and monoclonal goat
anti-β-actin antibodies (Santa Cruz Biotechnology, Inc.). Membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies and blots were developed using an ECL kit (Amersham,
Arlington Heights, IL, USA).
Measurement of mitochondrial membrane
potentisal (MMP, ΔΨm)
To measure MMP levels, a rhodamine 123 fluorescent
dye (Sigma-Aldrich; Ex/Em=485/535 nm) was used as described
previously (24,26). In brief, 1×106 cells in
a 60 mm culture dish (Nunc) were incubated with the designated
doses of Au for 24 h. Cells were washed twice with PBS and
incubated with the rhodamine 123 (0.1 mg/ml) at 37°C for 30 min.
Rhodamine 123 staining intensity was determined using a FACStar
flow cytometer. Rhodamine 123 negative cells indicated a MMP loss
in cells.
Lactate dehydrogenase (LDH) assay for the
detection of necrosis
Necrosis in the cells treated with Au was evaluated
using an LDH kit (Sigma-Aldrich). In brief, 1×106 cells
were incubated in a 60 mm culture dish (Nunc) with the indicated
doses of Au, with or without 15 μM caspase inhibitors, 2 mM NAC or
10 μM BSO for 24 h. Following treatment, the culture media were
collected and centrifuged for 5 min at 211 × g. Media supernatant
(50 μl) was added to a fresh 96-well plate with LDH assay reagent
and incubated at room temperature for 30 min. The absorbance values
were measured at 490 nm using a microplate reader. LDH release was
expressed as the percentage of extracellular LDH activity compared
with the control cells.
Detection of intracellular
O2•− levels
The level of intracellular
O2•− was specifically detected using
dihydroethidium (DHE; Ex/Em=510/580 nm; Invitrogen Life
Technologies). In brief, 1×106 cells were incubated in a
60 mm culture dish (Nunc) with the designated doses of Au, with or
without 15 μM caspase inhibitors, 2 mM NAC or 10 μM BSO for 24 h.
Cells were then washed in PBS and incubated with 20 μM DHE at 37°C
for 30 min. DHE fluorescence was detected using a FACStar flow
cytometer (Becton Dickinson). Levels of mitochondrial
O2•− are expressed as mean fluorescence
intensity, which was calculated using CellQuest software (Becton
Dickinson).
Detection of intracellular GSH
Levels of cellular GSH were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA; Ex/Em=522/595 nm;
Invitrogen Life Technologies), as previously described (27). In brief, 1×106 cells
were incubated in a 60 mm culture dish (Nunc) with the designated
doses of Au, with or without 15 μM caspase inhibitors, 2 mM NAC or
10 μM BSO for 24 h. After washing with PBS, the cells were
incubated with 5 μM CMFDA at 37°C for 30 min. The intensity of
5-chloromethylfluorescein (CMF) fluorescence was determined using a
FACStar flow cytometer. The percentage of negative CMF cells
indicated GSH depletion of cells.
Detection of TrxR activity
The activity of TrxR was assessed using the
Thioredoxin Reductase assay kit according to the manufacturer’s
instructions (Sigma-Aldrich). In brief, 1×106 cells were
incubated in a 60 mm culture dish (Nunc) with the indicated doses
of Au for 24 h. The cells were then washed in PBS and suspended in
five volumes of lysis buffer (R&D systems, Inc.). Protein
concentrations were determined using the Bradford method.
Supernatant samples containing 30 μg total protein were used for
the determination of TrxR activity. These were added to each well
in 96-well microtiter plates (Nunc) with 5,5′-dithiobis
(2-nitrobenzoic) acid at 25°C for 1 h. The optical density of each
well was measured at 412 nm using a microplate reader (Synergy™2,
BioTek® Instruments Inc.).
Statistical analysis
The results are expressed as the mean of at least
three independent experiments (mean ± standard deviation). Data
were analyzed using Instat software (GraphPad Prism4; GraphPad
Software, San Diego, CA, USA). Student’s t-test or one-way analysis
of variance with post hoc analysis using Tukey’s multiple
comparison test were used for parametric data. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of Au on cell growth, cell death
and MMP in HeLa cells
Initially, the effect of Au on the growth inhibition
of HeLa cells was examined using MTT assays. Following exposure to
Au for 24, 48 and 72 h, the growth of HeLa cells dose and
time-dependently decreased with an IC50 of ~2, 1.5 and 1
μM at 24, 48 and 72 h, respectively (Fig. 1A).
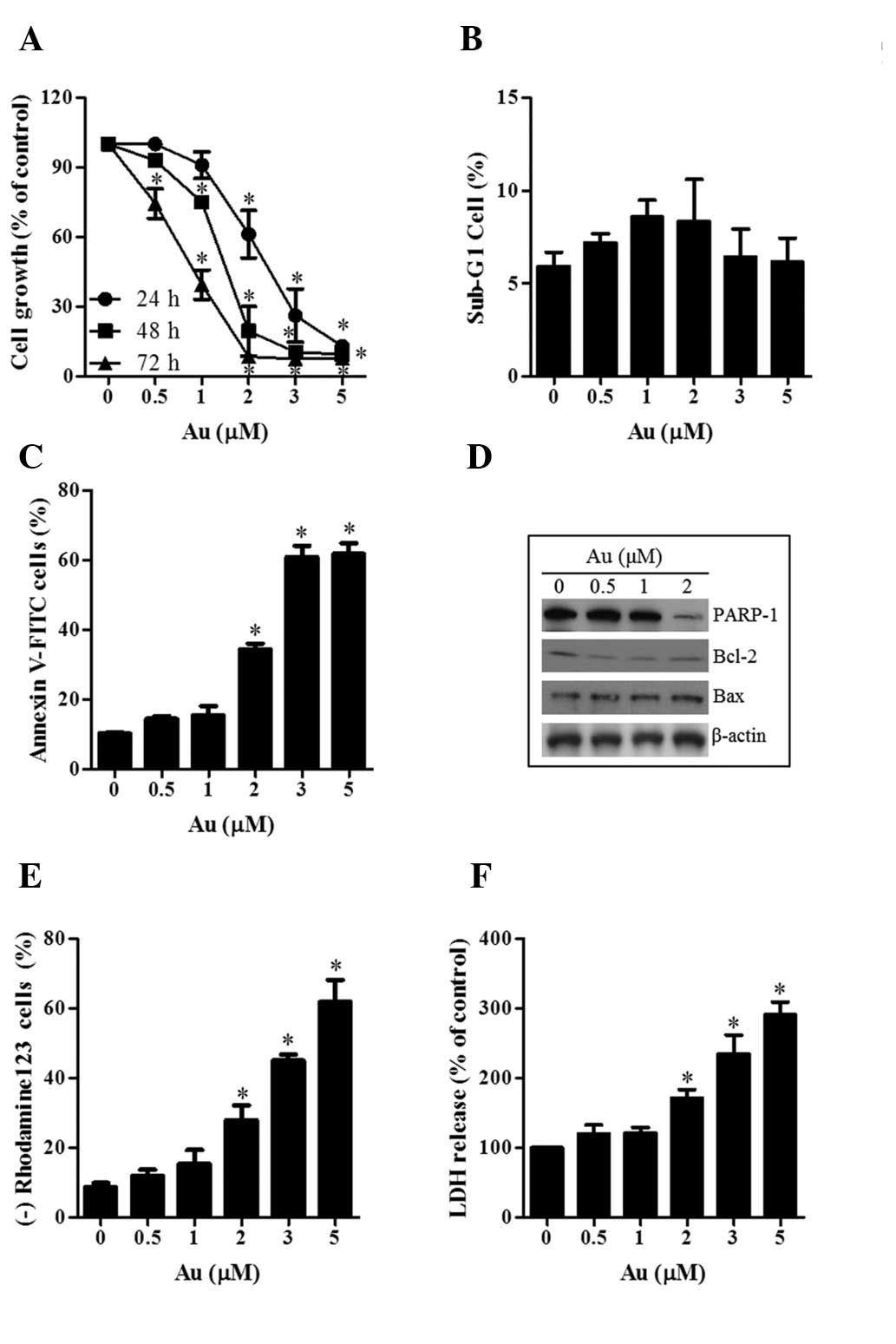 | Figure 1Effects of Au on cell growth and
death in HeLa cells. Exponentially growing cells were treated with
the designated concentrations of Au for the indicated durations.
(A) Graph of the cellular growth changes in HeLa cells at 24, 48
and 72 h as assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assays. (B and C) Graphs of the percentages of sub-G1 cells and
annexin V-positive cells at 24 h, respectively. (D) Western
blotting results showing the levels of PARP,-1 Bcl-2, Bax and
β-actin. (E and F) Graphs of the percentages of rhodamine
123-negative (loss of mitochondrial membrane potential) cells and
release of LDH compared with the control cells at 24 h.
*P<0.05, compared with the control group. Au,
auranofin; LDH, lactate dehydrogenase; FITC, fluorescein
isothiocyanate; PARP-1, poly ADP-ribose polymerase 1; Bcl-2, B-cell
lymphoma-2; Bax, BCL2-associated X protein. |
As shown in Fig.
1B, Au did not affect the proportion of sub-G1 cells in the
HeLa cells at 24 h. However, when HeLa cells were stained with
annexin V-FITC to evaluate the induction of apoptosis, the
percentages of annexin V-staining cells increased in a
dose-dependent manner in Au-treated HeLa cells (Fig. 1C). This agent decreased the
expression of PARP-1 and Bcl-2, whereas it increased the expression
of Bax (Fig. 1D). In addition, Au
triggered the loss of MMP and increased the release of LDH in HeLa
cells at 24 h in a dose-dependent manner (Fig. 1E and F). These results indicated
that Au induced necrosis as well as apoptosis in the HeLa
cells.
Effects of Au on levels of intracellular
O2•− and GSH in HeLa cells
Alterations in the levels of intracellular
O2•− and GSH in Au-treated HeLa cells were
also examined. As shown in Fig.
2A, Au significantly increased the levels of intracellular
O2•− (DHE) in HeLa cells at 24 h. Au, a known
inhibitor of TrxR, also decreased the activities of TrxR in HeLa
cell lysates and cells (Fig. 2B).
In addition, examination of the expression of Trx system-related
proteins demonstrated that the levels of TrxR1 and Trx2 were
downregulated by Au (Fig. 2B).
Additionally, Au markedly increased the percentage of GSH-depleted
cells in HeLa cells at 24 h (Fig.
2C).
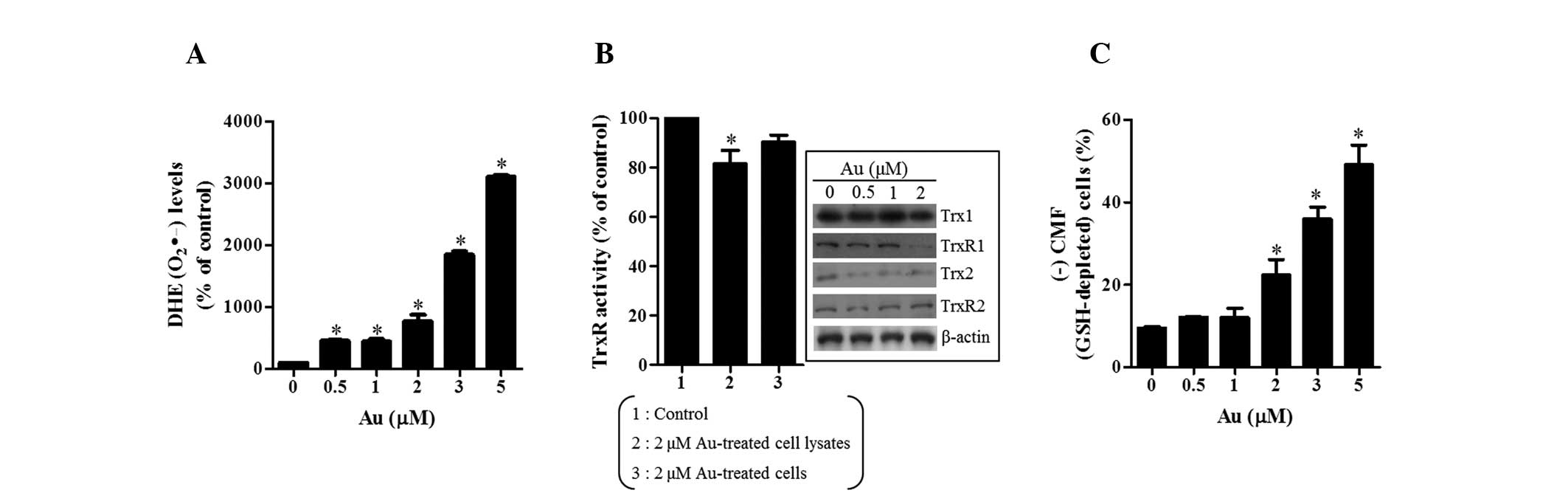 | Figure 2Effects of Au on the levels of
intracellular O2•− and GSH in HeLa cells.
Exponentially growing cells were treated with the designated
concentration of Au for 24 h. (A) Graph showing the levels of DHE
(O2•−) compared with those in the control
cells. (B) Graph showing the activity of TrxR in HeLa cells. The
inside figures demonstrate the levels of Trx1, TrxR1, Trx2, TrxR2
and β-actin. (C) Graph of the percentage of (−) CMF (GSH-depleted)
cells in HeLa cells. *P<0.05, compared with the
control group. Au, auranofin; DHE, dihydroethidium; GSH,
glutathione; CMF, 5-chloromethylfluorescein; Trx, thioredoxin;
TrxR, thioredoxin reductase; CMF, 5-chloromethylfluorescein. |
Effects of caspase inhibitors on cell
death, O2•− and GSH levels in Au-treated HeLa
cells
To determine which caspases were involved in the
death of Au-treated HeLa cells, the effects of caspase inhibitors
in those cells were assessed. For this experiment, 2 μM Au was
selected as a suitable dose to differentiate the level of cell
death in the presence or absence of each of the following caspase
inhibitors: pan-caspase inhibitor (Z-VAD), caspase-3 inhibitor
(Z-DEVD), caspase-8 inhibitor (Z-IELD) and the caspase-9 inhibitor
(Z-LEHD). The concentration of each caspase inhibitor, 15 μM, was
selected as the optimal dose for the present study as it did not
significantly affect cell death in the control cells (28). Among the caspase inhibitors, Z-VAD
significantly attenuated apoptosis induced by Au in HeLa cells at
24 h (Fig. 3A). Although Z-DEVD
and Z-IETD also partially prevented apoptotic cell death in
Au-treated HeLa cells, Z-LEHD did not have an effect (Fig. 3A). Furthermore, all caspase
inhibitors, particularly Z-VAD, decreased the release of LDH
triggered by Au in HeLa cells (Fig.
3B). Therefore, a variety of caspases appeared to be involved
in apoptotic and necrotic cell death in Au-treated HeLa cells.
 | Figure 3Effects of caspase inhibitors on cell
death and levels of O2•− and GSH in
Au-treated HeLa cells. Exponentially growing cells were treated
with 2 μM Au and/or 15 μM caspase inhibitors for 24 h. (A and B)
Graphs of the percentages of annexin V-positive cells and release
of LDH, respectively. (C and D) Graphs showing the levels of DHE
(O2•−) and the percentages of (−) CMF
(GSH-depleted) cells in the HeLa cells. *P<0.05,
compared with the control group. #P<0.05, compared
with cells treated with Au only. Au, auranofin; DHE,
dihydroethidium; GSH, glutathione; LDH, lactate dehydrogenase; CMF,
5-chloromethylfluorescein; Z-VAD,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; Z-DEVD,
benzyloxycarbonyl-Asp-Glu-Thr-Asp-fluoromethylketone; Z-IETD,
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone; Z-LEHD,
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone; FITC,
fluorescein isothiocyanate. |
In association with the levels of
O2•− and GSH, Z-VAD and Z-DEVD significantly
reduced levels of O2•− (DHE) in HeLa cells
treated with Au and prevented the depletion of GSH in those cells
(Fig. 3C and D). While Z-IETD
prevented depletion of GSH in Au-treated HeLa cells, it did not
affect levels of O2•− in those cells
(Fig 3C and D). Z-LEHD did not
result in any alterations in the levels of
O2•− or GSH (Fig. 3C and D).
Effects of NAC and BSO on cell growth,
cell death, O2•− and GSH levels in Au-treated
HeLa cells
The effects of NAC and BSO on cell growth, cell
death and levels of ROS and GSH were then assessed in 2 μM
Au-treated HeLa cells at 24 h. As shown in Fig. 4A, NAC significantly recovered the
inhibition of cell growth induced by Au. NAC also prevented
apoptotic cell death and release of LDH in Au-treated HeLa cells
(Fig. 4B and C). However, as an
inhibitor of GSH synthesis, BSO enhanced the inhibition of cell
growth, apoptosis and the release of LDH induced by Au in HeLa
cells (Fig. 4A–C). In the
assessment of whether NAC and BSO affect the levels of
O2•− and GSH in Au-treated HeLa cells, NAC
attenuated the levels of O2•− and the
depletion of GSH in these cells (Fig.
4D and E). By contrast, BSO significantly enhanced the levels
of O2•− and the depletion of GSH induced by
Au in HeLa cells (Fig. 4D and
E).
 | Figure 4Effects of NAC and BSO on cell
growth, cell death, O2•− and GSH levels in
Au-treated HeLa cells. Exponentially growing cells were treated
with 2 μM Au and/or 2 mM NAC or 10 μM BSO for 24 h. (A) Graph of
cellular growth changes. (B) Annexin V-FITC and PI cells were
measured with a FACstar flow cytometer. The numbers in each figure
indicate the percentages of annexin V-FITC positive cells
regardless of PI negative and positive cells. (C) Graph showing the
release of LDH. (D and E) Graphs showing levels of DHE
(O2•−) and the percentages of (−) CMF
(GSH-depleted) cells in the HeLa cells. *P<0.05,
compared with the control group. #P<0.05, compared
with the cells treated with Au only. Au, Auranofin; NAC, N-acetyl
cysteine; BSO, L-buthionine sulfoximine; PI, propidium iodide; LDH,
lactate dehydrogenase; DHE, dihydroethidium; CMF,
5-chloromethylfluorescein; GSH, glutathione; FITC, fluorescein
isothiocyanate. |
Discussion
In the present study, the anti-cancer effects of Au
on HeLa cervical cancer cells was investigated in association with
the levels of ROS and GSH. Au significantly and efficiently
decreased the growth of HeLa cells in a dose and time-dependent
manner. When the cell cycle distributions were examined in
Au-treated HeLa cells, Au did not induce any specific phase arrest
of the cell cycle at 24 h (data not shown) and did not increase the
number of sub-G1 cells in the HeLa cells. However, Au induced cell
death via apoptosis, which was accompanied by the cleavage of PARP.
This agent also led to loss of MMP in HeLa cells. It has been
suggested that a high ratio of Bax to Bcl-2 can cause the collapse
of MMP (29). Similarly, apoptosis
and loss of MMP caused by Au were accompanied by the downregulation
of Bcl-2 and the upregulation of Bax. Furthermore, this drug also
induced necrosis, which was supported by the release of LDH.
Therefore, these results suggested that Au induced apoptotic and
necrotic cell death in HeLa cells.
In determining which caspases were involved in cell
death in Au-treated HeLa cells, Z-VAD significantly prevented
apoptosis and necrosis induced by Au. Z-DEVD and Z-IETD also
rescued certain cells from Au-induced apoptotic and necrotic cell
death. Although Z-LEHD did not significantly prevent apoptosis in
the Au-treated HeLa cells, it inhibited the necrosis induced by Au.
Therefore, apoptotic cell death caused by Au was mediated by the
extrinsic apoptotic pathway of caspase-8 and the intrinsic
apoptotic pathway of caspase-9. In addition, the activation of
various caspases was tightly involved in necrosis in the Au-treated
HeLa cells, which supported the hypothesis that caspase activation
contributes to necrotic cell death (30,31).
Au, as an inhibitor of TrxR, can affect the redox
status of cells. It is reported that Au generates ROS in solid
tumor and leukemia cells and induces apoptosis in these cells
(14,32). Similarly, levels of intracellular
O2•− were markedly increased in the
Au-treated HeLa cells. The pan-caspase inhibitor, Z-VAD, which
demonstrated anti-apoptotic effects, appeared to decrease the
levels of O2•− in the cells. These results
suggested that alterations in the levels of intracellular
O2•− were closely associated with apoptotic
and necrotic cell death caused by Au. Furthermore, NAC attenuated
the inhibition of cell growth and cell death in Au-treated HeLa
cells and significantly reduced levels of
O2•− in these cells. Taken together,
Au-induced HeLa cell death was mediated by oxidative stress, mainly
derived from O2•−.
The Trx system consists of Trx, TrxR and NADPH,
which are important for cellular redox balance (1). The present study detected changes in
the levels of Trx system-related proteins, revealing that Au
decreased the levels of TrxR1 and Trx2. It is possible that the
downregulation of Trx2 affected an increase in the level of
O2•− in Au-treated HeLa cells, which
consequently contributed to the induction of oxidative stress in
these cells. GSH is important in protecting against cell damage
caused by free radicals and toxins. It can remove
O2•− and reduce H2O2 to
H2O by providing electrons to GSH peroxidase (33). The intracellular GSH content has a
crucial effect on cell death (34,35).
Similarly, it was demonstrated that Au increased the number of
GSH-depleted cells in the HeLa cells. All the inhibitors assessed,
with the exception of Z-LEHD, attenuated the depletion of GSH in
the Au-treated HeLa cells and, as expected, NAC markedly prevented
the depletion. In addition, BSO significantly increased the
depletion of GSH in the Au-treated HeLa cells and simultaneously
intensified cell death in these cells. These results suggested that
inhibition of the Trx and GSH systems by Au was closely associated
with the death of HeLa cells and supports the hypothesis that the
inhibition of Trx and GSH systems potentiates the effect of
anti-cancer drugs (36,37).
In conclusion, Au induced the growth inhibition of
HeLa cervical cancer cells via apoptosis as well as necrosis, which
was accompanied by intracellular increases in the levels of ROS and
the depletion of GSH. The present study provides an important
insight into the anti-cancer effects of Au on HeLa cells with
respect to oxidative stress and GSH.
Acknowledgements
This study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Korean
government (MSIP; no. 2008-0062279) and supported by the Basic
Science Research Program through the NRF funded by the Ministry of
Education (no. 2013006279).
Abbreviations:
|
Au
|
auranofin
|
|
ROS
|
reactive oxygen species
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PI
|
propidium iodide
|
|
FITC
|
fluorescein isothiocyanate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-Asp-Glu-Thr-Asp-fluoromethylketone
|
|
Z-IETD-FMK
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
|
LDH
|
lactate dehydrogenase
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
Trx
|
thioredoxin
|
|
TrxR
|
thioredoxin reductase
|
References
|
1
|
Lu J and Holmgren A: Thioredoxin system in
cell death progression. Antioxid Redox Signal. 17:1738–1747. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collet JF and Messens J: Structure,
function, and mechanism of thioredoxin proteins. Antioxid Redox
Signal. 13:1205–1216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burke-Gaffney A, Callister ME and Nakamura
H: Thioredoxin: friend or foe in human disease? Trends Pharmacol
Sci. 26:398–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rundlöf AK and Arnér ES: Regulation of the
mammalian selenoprotein thioredoxin reductase 1 in relation to
cellular phenotype, growth, and signaling events. Antioxida Redox
Signal. 6:41–52. 2004. View Article : Google Scholar
|
|
5
|
Noike T, Miwa S, Soeda J, Kobayashi A and
Miyagawa S: Increased expression of thioredoxin-1, vascular
endothelial growth factor, and redox factor-1 is associated with
poor prognosis in patients with liver metastasis from colorectal
cancer. Hum Pathol. 39:201–208. 2008. View Article : Google Scholar
|
|
6
|
Karlenius TC, Shah F, Di Trapani G, Clarke
FM and Tonissen KF: Cycling hypoxia up-regulates thioredoxin levels
in human MDA-MB-231 breast cancer cells. Biochem Biophys Res
Commun. 419:350–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fernandes AP, Capitanio A, Selenius M,
Brodin O, Rundlöf AK and Björnstedt M: Expression profiles of
thioredoxin family proteins in human lung cancer tissue:
correlation with proliferation and differentiation. Histopathology.
55:313–320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poerschke RL and Moos PJ: Thioredoxin
reductase 1 knockdown enhances selenazolidine cytotoxicity in human
lung cancer cells via mitochondrial dysfunction. Biochem Pharmacol.
81:211–221. 2011. View Article : Google Scholar
|
|
9
|
Fu JN, Li J, Tan Q, et al: Thioredoxin
reductase inhibitor ethaselen increases the drug sensitivity of the
colon cancer cell line LoVo towards cisplatin via regulation of G1
phase and reversal of G2/M phase arrest. Invest New Drugs.
29:627–636. 2011. View Article : Google Scholar
|
|
10
|
Madeira JM, Gibson DL, Kean WF and
Klegeris A: The biological activity of auranofin: implications for
novel treatment of diseases. Inflammopharmacology. 20:297–306.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suarez-Almazor ME, Spooner CH, Belseck E
and Shea B: Auranofin versus placebo in rheumatoid arthritis.
Cochrane Database Syst Rev. CD0020482000.PubMed/NCBI
|
|
12
|
Han S, Kim K, Kim H, et al: Auranofin
inhibits overproduction of pro-inflammatory cytokines,
cyclooxygenase expression and PGE2 production in macrophages. Arch
Pharm Res. 31:67–74. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han S, Kim K, Song Y, et al: Auranofin, an
immunosuppressive drug, inhibits MHC class I and MHC class II
pathways of antigen presentation in dendritic cells. Arch Pharm
Res. 31:370–376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Liu Z, Li M, et al: Enhancement of
auranofin-induced apoptosis in MCF-7 human breast cells by
selenocystine, a synergistic inhibitor of thioredoxin reductase.
PloS One. 8:e539452013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakaya A, Sagawa M, Muto A, Uchida H,
Ikeda Y and Kizaki M: The gold compound auranofin induces apoptosis
of human multiple myeloma cells through both down-regulation of
STAT3 and inhibition of NF-kappaB activity. Leuk Res. 35:243–249.
2011. View Article : Google Scholar
|
|
16
|
Rigobello MP, Scutari G, Boscolo R and
Bindoli A: Induction of mitochondrial permeability transition by
auranofin, a gold(I)-phosphine derivative. Br J Pharmacol.
136:1162–1168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rigobello MP, Folda A, Baldoin MC, Scutari
G and Bindoli A: Effect of auranofin on the mitochondrial
generation of hydrogen peroxide. Role of thioredoxin reductase.
Free Rad Res. 39:687–695. 2005. View Article : Google Scholar
|
|
18
|
Haverkos H, Rohrer M and Pickworth W: The
cause of invasive cervical cancer could be multifactorial. Biomed
Pharmacother. 54:54–59. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Marco F, Bucaj E, Foppoli C, et al:
Oxidative stress in HPV-driven viral carcinogenesis: redox
proteomics analysis of HPV-16 dysplastic and neoplastic tissues.
PloS One. 7:e343662012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim NH, Park HJ, Oh MK and Kim IS:
Antiproliferative effect of gold(I) compound auranofin through
inhibition of STAT3 and telomerase activity in MDA-MB 231 human
breast cancer cells. BMB Rep. 46:59–64. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marzano C, Gandin V, Folda A, Scutari G,
Bindoli A and Rigobello MP: Inhibition of thioredoxin reductase by
auranofin induces apoptosis in cisplatin-resistant human ovarian
cancer cells. Free Rad Biol Med. 42:872–881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rigobello MP, Gandin V, Folda A, et al:
Treatment of human cancer cells with selenite or tellurite in
combination with auranofin enhances cell death due to redox shift.
Free Rad Biol Med. 47:710–721. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of carbonyl cyanide p-(trifluoromethoxy)
phenylhydrazone on the growth inhibition in human pulmonary
adenocarcinoma Calu-6 cells. Toxicology. 265:101–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
25
|
You BR and Park WH: Zebularine inhibits
the growth of HeLa cervical cancer cells via cell cycle arrest and
caspase-dependent apoptosis. Mol Biol Rep. 39:9723–9731. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2 (*−) generator induces apoptosis via the
depletion of intracellular GSH contents in Calu-6 cells. Lung
Cancer. 63:201–209. 2009. View Article : Google Scholar
|
|
27
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
You BR and Park WH: Suberoyl bishydroxamic
acid-induced apoptosis in HeLa cells via ROS-independent,
GSH-dependent manner. Mol Biol Rep. 40:3807–3816. 2013. View Article : Google Scholar
|
|
29
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niquet J, Allen SG, Baldwin RA and
Wasterlain CG: Evidence of caspase-3 activation in hyposmotic
stress-induced necrosis. Neurosci Letters. 356:225–227. 2004.
View Article : Google Scholar
|
|
31
|
Honda A, Abe S, Hiroki E, et al:
Activation of caspase 3, 9, 12, and Bax in masseter muscle of mdx
mice during necrosis. J Muscle Res Cell Motil. 28:243–247. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park SJ and Kim IS: The role of p38 MAPK
activation in auranofin-induced apoptosis of human promyelocytic
leukaemia HL-60 cells. Br J Pharmacol. 146:506–513. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hatem E, Berthonaud V, Dardalhon M, et al:
Glutathione is essential to preserve nuclear function and cell
survival under oxidative stress. Free Radic Biol Med. 67:103–114.
2014. View Article : Google Scholar
|
|
34
|
Ortega AL, Mena S and Estrela JM:
Glutathione in cancer cell death. Cancers. 3:1285–1310. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Franco R and Cidlowski JA: Glutathione
efflux and cell death. Antioxid Redox Signal. 7:1694–1713. 2012.
View Article : Google Scholar
|
|
36
|
Sobhakumari A, Love-Homan L, Fletcher EV,
et al: Susceptibility of human head and neck cancer cells to
combined inhibition of glutathione and thioredoxin metabolism. PloS
One. 7:e481752012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scarbrough PM, Mapuskar KA, Mattson DM,
Gius D, Watson WH and Spitz DR: Simultaneous inhibition of
glutathione- and thioredoxin-dependent metabolism is necessary to
potentiate 17AAG-induced cancer cell killing via oxidative stress.
Free Radic Biol Med. 52:436–443. 2012. View Article : Google Scholar
|














