Introduction
Coxsackie virus B3 (CVB3) infection is considered to
be a predominant cause of human myocarditis, and infection may
eventually result in heart failure (1). CVB3-induced myocarditis is mainly
caused by inflammatory infiltration of myocardial tissue, and
remodeling of cardiac structure and function (2). Although the virus directly attacks
myocardial cells, activation of the immune response and a
subsequent inflammatory cascade is key to the pathogenesis of
CVB3-mediated viral myocarditis (VM). In a previous model of CVB3
infection, nuclear factor (NF)-κB was shown to be vital in
regulating host inflammatory defenses (3). NF-κB mediates the expression of
various pro-inflammatory cytokines, such as tumor necrosis factor
(TNF)-α, interleukin (IL)-1β and IL-6, and thus may aggravate
myocardial injury (4). Notably,
NF-κB inhibitors, including pyrrolidine dithiocarbamate and Bay
11-7082 were demonstrated to significantly inhibit the myocardial
tissue expression of pro-inflammatory factors, slowing myocarditis
pathogenesis (5). Despite
extensive investigation into VM prevention and treatment, there
remains no definitive therapy for this disease.
Hydrogen sulfide (H2S) is generated in
mammalian tissues mainly by two types of
pyridoxal-5′-phosphate-dependent enzymes, cystathionine β-synthase
and cystathionine γ-lyase (6).
H2S has long been recognized as a toxic gas with a
pungent odor, which pollutes the environment and endangers human
health. However, a previous study has shown that H2S may
become the third endogenous gaseous signaling transmitter
identified, following nitric oxide (NO) and carbon monoxide, due to
newly-detected physiological and pathophysiological functions
(7). It has been demonstrated that
H2S exerts protective effects against myocardial
ischemia (8); however, great
controversy remains regarding the exact role of H2S in
inflammation (9). Furthermore,
considerable evidence suggests that H2S also exerts an
anti-apoptotic effect (10,11).
In local and systemic inflammation experimental models,
H2S was shown to have a pro-inflammatory role (12). Conversely, accumulating evidence
suggests that H2S exerts an anti-inflammatory effect in
various models (13,14). In endotoxic shock rat models,
H2S inhibited lipopolysaccharide (LPS)-mediated
inflammatory cytokine production and reduced plasma C-reactive
protein (15). In LPS-irritated
human cartilage cells, the H2S release agent GYY4137
significantly repressed cyclooxygenase 2, inducible NO synthase and
TNF-α expression, by restraining the activation of NF-κB (16). To confirm the potent
anti-inflammatory mechanism of H2S in CVB3-infected
cardiomyocytes, the present study investigated whether
H2S, administered via GYY4137 treatment, exerts
anti-inflammatory effects in CVB3-infected cardiomyocytes, and
whether this occurs through the inhibition of pro-inflammatory
processes, such as the downregulation of IL-6, IL-1β and TNF-α.
Mitogen-activated protein kinases (MAPKs) are
considered to constitute an intracellular signal transduction
pathway important in inflammatory reactions induced by viral
infections and oxidative stress, and are involved in regulating the
expression levels of various inflammatory factors and cell adhesion
factors (17). In cell ligation
and puncture-induced models of sepsis, H2S was
demonstrated to regulate the systemic inflammatory response via the
MAPK signaling pathway (18). In a
mouse model of VM, the MAPK signaling pathway (ERK1/2, p38 and
JNK1/2) was shown to be activated and involved in the inflammatory
reaction, upregulating inflammatory factor expression (19). In the present study, the underlying
mechanism and intracellular signaling pathways affected by
H2S in CVB3-infected cardiomyocytes were
investigated.
Materials and methods
Animals
Sprague Dawley rats, aged 1–2-days, were purchased
from the Center of Experimental Animals of Tongji Medical College,
Huazhong University of Science and Technology, Wuhan, China. All
animals used in this study were cared for in accordance with the
Guide for the Care and Use of Laboratory Animals published by the
United States National Institute of Health (NIH publication no.
85-23, revised 1996) and all procedures were approved by the
committee of Experimental Animals of Tongji Medical College,
Huazhong University of Science and Technology.
Reagents
Dulbecco’s modified Eagle’s medium (DMEM)/F12,
Trypsin 0.25% EDTA and fetal bovine serum (FBS) were obtained from
Gibco-BRL (Carlsbad, CA, USA). Collagenase B was purchased from
Roche Diagnostics GmbH (Mannheim, Germany). GYY4137 was bought from
Cayman Chemical Company (Ann Arbor, MI, USA). The IL-6, IL-1β and
TNF-α ELISA kits were purchased from USCN Life Science, Inc.
(Wuhan, China). The Cell Counting kit-8 (CCK-8) was obtained from
Dojindo Molecular Technologies (Kunamoto, Japan).
5-bromo-2′-deoxyuridine (BrdU) was purchased from Sigma-Aldrich
(St. Louis, MO, USA). The lactate dehydrogenase (LDH) and creatine
kinase-MB (CK-MB) ELISA kits were bought from Nanjing Jiancheng
Biochemical Reagent Co., Ltd (Nanjing, China). The primary
antibodies for the NF-κB and MAPK signaling pathways were from Cell
Signaling Technology, Inc. (Danvers, MA, USA) and horse radish
peroxidase (HRP)-conjugated secondary antibodies were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
CVB3 virus
A Nancy variant of CVB3, which was provided by the
State Key Laboratory of Virology (Wuhan, China), was propagated in
HeLa cell monolayers and stored at −80°C until use. The HeLa cells
were provided by Prof. ZeHua Wang (Department of Obstetrics and
Gynecology, Union Hospital, Tongji Medical College, Huazhong
University of Science and Technology, Wuhan, China.). The HeLa
cells were grown in DMEM, supplemented with 10% FBS. After reaching
≥90% confluence, CVB3 was added to the cells for 2 h, the virus was
then released from the cells by three freeze-thaw cycles. The
samples were centrifuged at 5,000 × g at 4°C for 10 min, and the
supernatants were collected. The viral titer was determined to be
10−4.5 in terms of the viral cytopathic effects, and was
expressed as the tissue culture infective dose
(TCID50).
Primary culture of neonatal rat
cardiomyocytes
The 1–2-days old Sprague Dawley rats were sacrificed
by ether inhalation and the hearts were harvested. The primary
cardiomyocytes were isolated from the ventricles, as described
previously (20), with certain
modifications. Briefly, the hearts were lightly minced and digested
with 0.05% trypsin and 0.025% collagenase B at 37°C for 5 min.
Subsequent to digestion five times, the supernatants were filtered,
centrifuged for 10 min (120 × g, 4°C) and resuspended in the
complete medium, which consisted of DMEM/F12 containing 10% FBS,
100 U/ml penicillin and 100 ug/ml streptomycin. The resuspended
cells were then plated in a petri dish in a humidified incubator
(5% CO2, 37°C) for 1.5 h to reduce the numbers of
endothelial cells and cardiac fibroblasts. Any non-adherent cells
were collected, filtered once, resuspended in the complete medium
and seeded in 6-well plates. After 48 h, myocyte morphology was
observed and the cells started beating spontaneously. By
observation of the cell morphology and beat frequency, >90%
cells were identified as cardiomyocytes. The cells were incubated
for three days prior to further experiments.
Cell treatment
Prior to each experiment, the cardiomyocytes were
serum-starved for 12 h. The cells were then treated with different
concentrations of GYY4137 (10, 50 and 100 μM) for different time
periods prior to the addition of 100 TCID50 CVB3 for 2 h
under serum-free conditions.
Cell viability assay
The CCK-8 assay was used to detect cell activity.
The cardiomyocytes were seeded at a density of 1×104
cells/well in a 96-well plate in order to observe the potential
cytotoxicity. The cells were serum-starved for 12 h and then
incubated with GYY4137 (10–100 μM) for a further 24 h. Following
the indicated treatments, 10 μl CCK-8 solution was added to each
well of the plate and the cells were incubated for an additional 4
h at 37°C. The absorbance at 450 nm was measured with a microplate
reader (Bio-Rad, Hercules, CA, USA). The mean optical density (OD)
of six wells in the indicated groups were used to calculate the
percentage cell viability using the following calculation:
Percentage cell viability = (ODtreatment
group/ODcontrol group) ×100%. The experiment was
repeated three times.
LDH and CK-MB assays
The cells were pre-incubated with 100
TCID50 CVB3 for 2 h and then different concentrations of
GYY4137 (10, 50 and 100 μM) for another 72 h. The supernatant media
from the myocardial cell cultures were collected from all groups
and stored at −80°C. LDH and CK-MB isoenzyme activity in all media
samples was measured according to the manufacturer’s instructions
of the respective ELISA kits. Each measurement was repeated three
times. Enzyme activity was indicated by the enzyme unit (U), which
signified the quantity of a particular enzyme.
Cytokine assays
The cardiomyocytes were cultured in 96-well plates
(1×104 cells/ml) and preincubated with 100
TCID50 CVB3 for 2 h, followed by incubation with
different concentrations of GYY4137 (10, 50 and 100 μM) for another
24 h. The cell culture supernatants were used to measure the IL-6,
IL-1β and TNF-α levels using ELISA kits, following the
manufacturer’s instructions.
Electrophoretic mobility shift assay
(EMSA)
The NF-κB DNA-banding activity was detected by EMSA
using a commercial kit (Thermo Fisher Scientific, Rockford, IL,
USA). The cardiomyocytes were plated in 100 mm dishes
(1×106 cells per dish) and grown in complete DMEM/F12
until confluent. The cells were preincubated with 100
TCID50 CVB3 for 2 h and then incubated with different
concentrations of GYY4137 (10, 50 and 100 μM) for another 12 h.
Subsequent to washing three times with cold phosphate-buffered
saline (PBS; pH 7.4), the cells were then scraped and centrifuged
at 15,000 × g for 5 min, to collect the pellets. Nuclear proteins
were extracted using the NE-PER® Nuclear Extraction
Reagent (Thermo Fisher Scientific) according to the manufacturer’s
instructions. Biotin-labeled NF-κB specific oligonucleotides, with
the following sequence: 5′-AGTTGAGGGGACTTTCCCAGGC-′3, were prepared
as labeled probes, according to the manufacturer’s instructions
(Promega Corporation, Madison, WI, USA). Biotin end-labeled
double-stranded DNA and the nuclear extracts were incubated at room
temperature for 20 min, and then 10 μl protein-DNA complex was
subjected to 6.5% PAGE at 100 V for 1 h at 4°C and transferred to a
nylon membrane. Following ultra-violet light cross-linking,
HRP-labeled streptavidin incubation with chemiluminescent substrate
(North2South® Chemiluminescent Hybridization and
Detection kit; Thermo Fisher Scientific, Waltham, MA, USA) was used
to detect the biotin end-labeled DNA. The nylon membrane was
subjected to X-ray film exposure and the results obtained were
analyzed using AlphaEase software (Alpha Innotech, Santa Cruz, CA,
USA).
Western blot analysis of protein
expression levels
Subsequent to infection with CVB3 for 2 h, the cells
were treated with GYY4137 at concentrations of 10, 50 and 100 μM
for different time periods (6 h for detection of the MAPK signaling
pathway, 24 h for the NF-κB signaling pathway). Following the
indicated treatments, the cells were harvested and lysed with cell
lysis buffer (Thermo Fisher Scientific), and centrifuged at 10,000
× g for 10 min. The supernatant served to represent total protein.
Nuclear and membranous proteins were extracted using the NE-PER
extraction kit, according to the manufacturer’s instructions. The
protein concentrations were determined using a bicinchoninic acid
protein assay kit (Beyotime, Biotech., Jiangsu, China). The protein
samples (40 μg) were separated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes. The membranes were blocked
with 5% non-fat milk in Tris-buffered saline containing 0.05%
Tween-20 (TBST) at room temperature for 2 h, washed and incubated
at 4°C overnight with various primary antibodies (phosphor-ERK1/2,
1:500; ERK1/2, 1:1,000; p38, 1:1,000; JNK, 1:1,000; IκBα, NF-κB
p65, Histone3 and β-actin, 1:1,000). Following washing with TBST,
the membranes were incubated with the HRP-conjugated secondary
antibodies (goat anti-rabbit IgG, rabbit anti-goat IgG, rabbit
anti-rat IgG and rabbit anti-mouse IgG; all 1:3,000). Subsequent to
washing the membranes, an Enhanced Chemiluminescence kit (Thermo
Fisher Scientific) was used to detect the immunoreactivity, then
the membranes were exposed to X-ray film. The density of the
signals was quantified using AlphaEase FC software (Genetic
Technologies, Inc., Miami, Florida, USA).
Immunofluorescence analysis
The cardiomyocytes were grown on glass slides in
6-well plates. Following incubation with CVB3 for 2 h, the cells
were treated with 50 μM GYY4137 for a further 24 h. The cells were
washed three times and fixed with 4% paraformaldehyde for 15 min,
and then incubated with an immune blocking solution for 1 h to
reduce non-specific binding. Subsequently, the cells were washed
three times and incubated at 4°C overnight with the primary NF-κB
p65 antibody. Following washing, the cells were incubated with
Cy3-conjugated secondary antibody (1:200 dilution) at room
temperature for 1 h, then the nuclei were stained with DAPI for 5
min prior to observation. Images were captured at excitation
wavelengths of 350 nm (for DAPI) and 540 nm (for Cy3). The p65
protein and DAPI-stained nuclei exhibited red and blue
fluorescence, respectively, which was visualized by laser confocal
microscopy (A1Si; Nikon Corporation, Tokyo, Japan). Image-Pro Plus
software (Media Cybernetics, Georgia, MD, USA) was used to analyze
the results.
Statistics
All experiments were repeated at least three times
with equivalent results. The data are expressed as the means ±
standard error. One-way analysis of variance was used to analyze
the differences between groups. The data was analyzed using SPSS
18.0 software (SPSS, Inc., Chicago, IL, USA). A P<0.05 was
considered to indicate a statistically significant difference.
Results
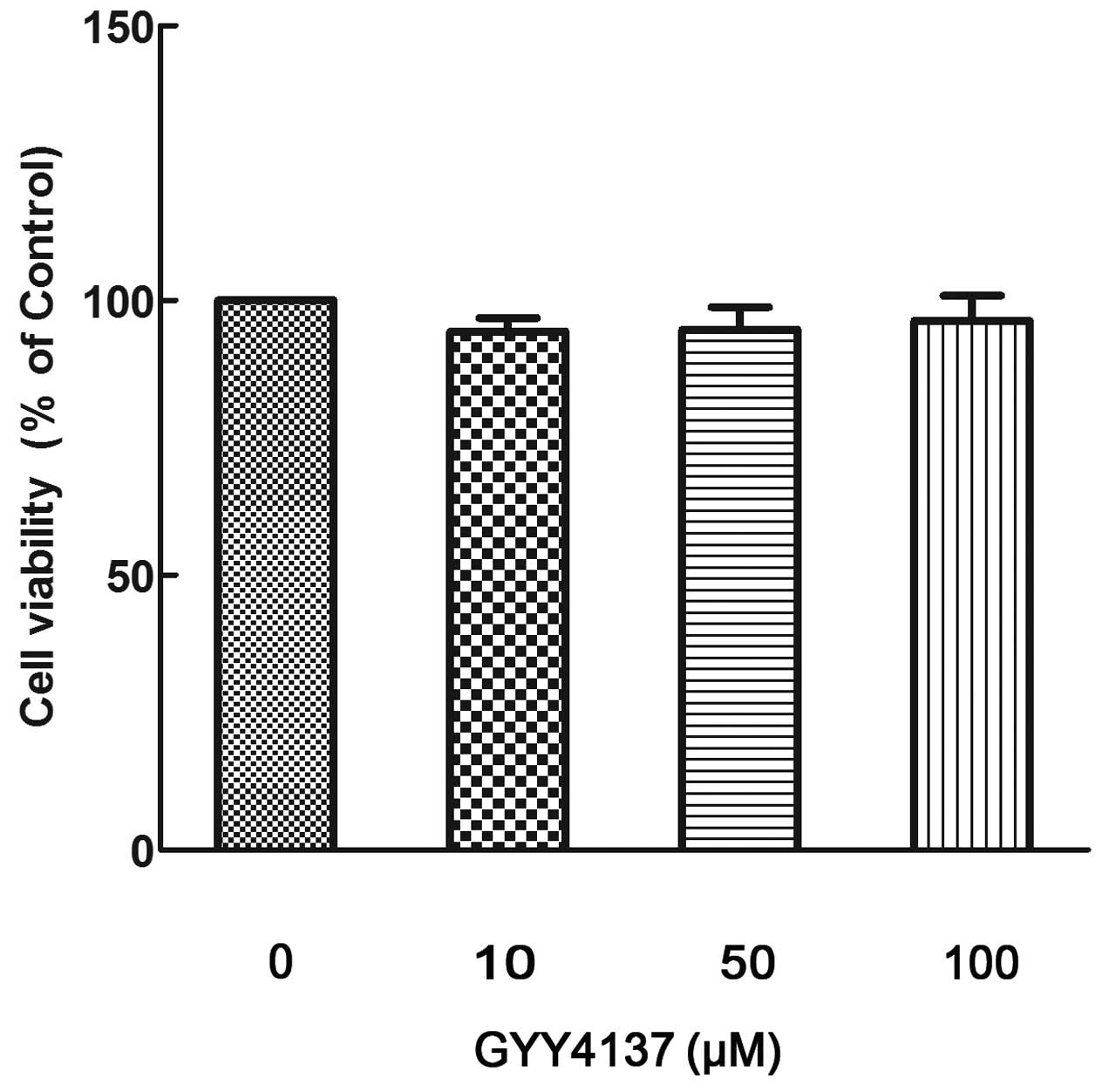
GYY4137 is not cytotoxic to
cardiomyocytes
Following exposure of the cardiomyocytes to
different GYY4137 concentrations (10, 50 and 100 μM) for 24 h, no
significant differences in cell viability were detected (Fig. 1). These data indicate that
different GYY4137 concentrations exerted no cytotoxic effect in
cardiomyocytes; therefore, the GYY4137 concentrations used in
subsequent experiments did not exceed 100 μM.
Cardiomyocyte damage-related enzyme
activity in the supernatant of myocardial cells
The levels of LDH and CK-MB in the supernatant media
are shown in Fig. 2. In the CVB3
infection group, the levels of LDH and CK-MB were significantly
increased (P<0.05 respectively) in comparison with those of the
control group. The GYY4137 treatment (at concentrations of 10, 50
and 100 μM) groups exhibited significantly reduced LDH and CK-MB
enzyme levels (P<0.05 respectively) in comparison with those of
the CBV3 infection group. These data indicate that GYY4137 protects
against damage to myocytes in viral infection.
GYY4137 inhibits CVB3-induced
cardiomyocyte inflammatory factor secretion
The cardiomyocytes were incubated with CVB3 (100
TCID50) with or without GYY4137 treatment (10, 50 and
100 μM). The levels of inflammatory cytokines in the cell culture
media were determined by ELISA assays. As shown in Fig. 3, the production of IL-6, IL-1β and
TNF-α was significantly increased following infection with CVB3, as
compared with the unstimulated control (P<0.05). Incubating the
cells with GYY4137 at 10, 50 and 100 μM concentrations
dose-dependently inhibited the production of IL-6, IL-1β and TNF-α,
respectively (all P<0.05). These results indicate that GYY4137
effectively inhibited the expression of inflammatory cytokines in
CVB3-infected myocardial cells.
GYY4137 inhibits NF-κB translocation to
the nucleus
To assess the potential effects on NF-κB
translocation, indirect immunofluorescence techniques were used to
detect the distribution of NF-κB in the myocardial cells. As shown
in Fig. 4, compared with normal
cells (shown as control), intracellular p65 was observed to
transfer from the cytoplasm to the nucleus in CVB3-infected cells.
Subsequent to incubation with 50 μM GYY4137, this nuclear
translocation was suppressed.
GYY4137 inhibits CVB3-mediated NF-κB DNA
binding activity
NF-κB is a key regulator of numerous genes in all
types of cells. The cardiomyocytes were pretreated with 10, 50 or
100 μM GYY4137 for 12 h prior to incubation with CVB3 for 2 h. The
inhibition of CVB3-induced NF-κB DNA binding activity in the
cardiomyocytes was detected by EMSA. As shown in Fig. 5, NF-κB DNA binding activity was
significantly increased in the CVB3 group, compared with the
control group (P<0.05); however treatment with different GYY4137
concentrations significantly reduced the CVB3-induced NF-κB DNA
binding activity in a dose-dependent manner (P<0.05). The
CVB3-induced DNA binding was significantly reduced by 100 μM
GYY4137 (P<0.05).
Effects of GYY4137 on inducing NF-κB and
IκBα degradation
To elucidate the possible mechanism of
GYY4137-mediated reduction in the secretion of inflammatory
cytokines, the NF-κB signaling pathway was evaluated and the effect
of GYY4137 treatment on IκBα degradation was examined. The data
showed that treatment with GYY4137 significantly prevented the IκBα
degradation induced by CVB3 (P<0.05; Fig. 6A). p65 was expressed mainly in the
cytoplasm prior to CVB3 infection (Fig. 6B), but incubation with CVB3
resulted in the accumulation of p65 in the nucleus (Fig. 6C). However, this nuclear
translocation of p65 was significantly suppressed by GYY4137 in a
concentration-dependent manner (P<0.05).
Effects of GYY4137 on MAP kinases in
CVB3-infected cardiomyocytes
The activation and phosphorylation of the MAPK
signaling pathway has been shown to exert a crucial role in
CVB3-induced NF-κB activation and the subsequent activation of
inflammatory mediators (21). To
investigate whether MAPK signaling pathway activation is involved
in the regulation of inflammatory response by GYY4137 in
CVB3-infected cardiomyocytes, MAPK phosphorylation (indicated by
the expression levels of phosphorylation (P)-p38, P-ERK1/2 and
P-JNK1/2) was analyzed by western blotting. As shown in Fig. 7, P-p38, P-ERK1/2 and P-JNK1/2
expression levels in cells were significantly increased after 2 h
treatment with CVB3 (P<0.05; Fig.
7), but GYY4137 treatment (10–100 μM) inhibited this
phosphorylation (P<0.05). These results suggest that GYY4137 may
reduce the expression of CVB3-induced inflammatory factors by
suppressing the phosphorylation of p38, ERK1/2 and JNK1/2 molecules
in the MAPK signaling pathway.
Discussion
VM is manifested by attacks on infection-induced
myocytes. Following viral infection, immune signaling pathways are
activated. The presence of Coxsackie virus in host cells and the
subsequent immune reaction may result in dilated cardiomyopathy and
heart failure (1). Increasing
evidence indicates that the myocardial cell inflammatory reaction
induced by CVB3 infection is crucial in VM development and
myocardial remodeling (2).
The NF-κB signaling pathway is a proinflammatory
pathway involved in the regulation of a number of inflammatory
responses. A previous study confirmed that the activation of NF-κB,
a nuclear transcription factor is important in the development of
CVB3-mediated myocarditis (21).
NF-κB as a central component of the inflammatory reaction regulates
the expression levels of various inflammatory factors (21). In CVB3-mediated myocarditis, NF-κB
is activated and the expression levels of downstream chemical
factors, cell adhesion factors and inflammatory factors, such as
IL-6, IL-1β and TNF-α, markedly increase, thereby further damaging
the heart (4). Inactivated NF-κB
reacts with IκB-α to form a compound that is distributed in the
cytoplasm. When an inflammatory factor, growth factor or chemokine,
is present that activates NF-κB, IκB-α is phosphorylated at Serine
32 and 36, and then degraded via the ubiquitin-protease pathway.
When NF-κB and IκB-α are depolymerized, the nuclear localization
sequences are exposed and the molecules are transported into the
nucleolus to combine with the relevant DNA gene sequences, induce
the transcription of target genes and accelerate the transcription
of NF-κB-dependent genes, including IL-6, IL-1β and TNF-α.
Therefore, following suppression of the NF-κB pathway, the heart is
prevented from undergoing an inflammatory response through the
downregulation of proinflammatory factor expression (25).
GYY4137 is a novel H2S-releasing agent
and has been found to exert anti-inflammatory and anti-apoptotic
actions, expand the coronary artery, improve myocardial oxygen
supply and protect the heart (22). The anti-inflammatory effect is
realized via inhibiting the expression of NF-κB-regulated
inflammatory genes and proinflammatory factors (23). In the present study, GYY4137
exerted a protective effect, significantly reducing the secretion
of IL-6, IL-1β and TNF-α proteins following 10–100 μM GYY4137
treatment in myocardial cells subsequent to CVB3 infection.
Enzymatic determination of myocardial injury revealed that the
levels of LDH and CK-MB were significantly reduced, which
alleviated the damage to myocardial cells. This result is
concurrent with that of a previous study that reported that
H2S pretreatment significantly reduced the inflammatory
reaction in a rat total hepatic ischemia and reperfusion model
(24).
To analyze the underlying mechanism of how GYY4137
protects CVB3-infected myocardial cells, the effects of GYY4137 on
the CVB3-activated NF-κB signaling pathway were investigated in the
present study. The data show that GYY4137 reduced CVB3-mediated
IκBα degradation in myocardial cells in a concentration-dependent
manner, and resulted in NF-κB and IκB-α depolymerization, thereby
resulting in translocation of NF-κB from the cytoplasm to the
nucleolus. To verify the activation of NF-κB, detection of this
change in cellular localisation is important. With the dislocation
and activation of NF-κB, the levels of CVB3-mediated inflammatory
factors were significantly increased. Whiteman et al
(26) found that GYY4137
restrained the expression of proinflammatory factors in macrophages
and upregulated IL-10 expression through negatively affecting NF-κB
nuclear translocation, thereby inducing an anti-inflammatory
reaction (26). To further confirm
whether GYY4137 induces an anti-inflammatory reaction through
restraining NF-κB DNA binding ability, the effects of GYY413 on
this binding ability were examined in the present study. EMSA
revealed that CVB3-mediated NF-κB DNA binding ability is inhibited
in a concentration-dependent manner by GYY4137, indicating that
GYY4137 restrains the expression of IL-6, IL-1β and TNF-α through
repressing NF-κB DNA binding ability. Therefore, in the
CVB3-infected myocardial cells, GYY4137 significantly supressed the
expression of these proinflammatory factors by inhibiting the NF-κB
signaling pathway.
The MAPK intracellular signaling pathway is
important in inflammatory reactions induced by viral infection and
oxidative stress, and is involved in regulating the expression of
various inflammatory factors and cell adhesion factors (17). Therefore, to further analyze the
underlying molecular mechanism with regard to how GYY4137 inhibits
the secretion of proinflammatory factors, the effects of GYY4137 on
the MAPK signaling pathway were investigated. A previous study
observed that in in vivo and in vitro models of
inflammation, the MAPK signaling pathway is involved in the
regulation of NF-κB activation (27). Furthermore, in a mouse VM model,
ERK1/2, p38 and JNK1/2 were shown to be activated, and involved in
the inflammatory reaction and upregulated expression of
inflammatory factors (28,29). In accordance with this, in the
present study, 6 h after CVB3 infection, ERK1/2, p38 and JNK1/2
were significantly activated, as revealed by increased
phosphorylation of these molecules. Following intervention with
GYY4137, the levels of phosphorylated ERK1/2, p38 and JNK1/2 were
inhibited in a concentration-dependent manner. Therefore, GYY4137
is hypothesized to inhibit the activation of the MAPK signaling
pathway and thereby suppress the expression of inflammatory factors
in CVB3-infected rat myocardial cells.
In conclusion, in the myocardial cells of
CVB3-infected rats, GYY4137 downregulated the expression of
proinflammatory factors, reduced the levels of myocardial
injury-related enzymes (LDH and CK-MB) and alleviated damage to
myocardial cells. Thus, GYY4137 may provides a novel therapeutic
method in the treatment of VM. The anti-inflammatory effect was
achieved by inhibiting the NF-κB and MAPK signaling pathways.
However, whether the anti-inflammatory effect through inhibition of
the MAPK signaling pathway is achieved through regulation of NF-κB
activation requires further investigation.
Acknowledgements
The authors would like to thank Professor ZhanQiu
Yang (State Key Laboratory of Virology) for providing key reagents
and technical guidance, and the biotechnicians of the laboratories
of Pediatrics and Gastroenterology of Union Hospital (Wuhan, China)
for technical support.
References
|
1
|
Esfandiarei M and McManus BM: Molecular
biology and pathogenesis of viral myocarditis. Annu Rev Pathol.
3:127–155. 2008. View Article : Google Scholar
|
|
2
|
Yajima T and Knowlton KU: Viral
myocarditis: from the perspective of the virus. Circulation.
119:2615–2624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sobotta K, Wilsky S, Althof N, Wiesener N,
Wutzler P and Henke A: Inhibition of nuclear factor kappa B
activation reduces Coxsackievirus B3 replication in lymphoid cells.
Virus Res. 163:495–502. 2012. View Article : Google Scholar
|
|
4
|
Li K, Xu W, Guo Q, et al: Differential
macrophage polarization in male and female BALB/c mice infected
with coxsackievirus B3 defines susceptibility to viral myocarditis.
Circ Res. 105:353–364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gui J, Yue Y, Chen R, Xu W and Xiong S:
A20 (TNFAIP3) alleviates CVB3-induced myocarditis via inhibiting
NF-kappaB signaling. PLoS One. 7:e465152012. View Article : Google Scholar
|
|
6
|
Predmore BL, Lefer DJ and Gojon G:
Hydrogen sulfide in biochemistry and medicine. Antioxid Redox
Signal. 17:119–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lowicka E and Beltowski J: Hydrogen
sulfide (H2S) - the third gas of interest for
pharmacologists. Pharmacol Rep. 59:4–24. 2007.
|
|
8
|
Bian JS, Yong QC, Pan TT, et al: Role of
hydrogen sulfide in the cardioprotection caused by ischemic
preconditioning in the rat heart and cardiac myocytes. J Pharmacol
Exp Ther. 316:670–678. 2006. View Article : Google Scholar
|
|
9
|
Li L, Bhatia M and Moore PK: Hydrogen
sulphide - a novel mediator of inflammation? Curr Opin Pharmacol.
6:125–129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rinaldi L, Gobbi G, Pambianco M, Micheloni
C, Mirandola P and Vitale M: Hydrogen sulfide prevents apoptosis of
human PMN via inhibition of p38 and caspase 3. Lab Invest.
86:391–397. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu LF, Lu M, Wu ZY, Wong PT and Bian JS:
Hydrogen sulfide inhibits rotenone-induced apoptosis via
preservation of mitochondrial function. Mol Pharmacol. 75:27–34.
2009. View Article : Google Scholar
|
|
12
|
Li L, Bhatia M, Zhu YZ, et al: Hydrogen
sulfide is a novel mediator of lipopolysaccharide-induced
inflammation in the mouse. FASEB J. 19:1196–1198. 2005.PubMed/NCBI
|
|
13
|
Li T, Zhao B, Wang C, et al: Regulatory
effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the
plasma and pulmonary tissue of rats with acute lung injury. Exp
Biol Med (Maywood). 233:1081–1087. 2008. View Article : Google Scholar
|
|
14
|
Tamizhselvi R, Sun J, Koh YH and Bhatia M:
Effect of hydrogen sulfide on the phosphatidylinositol
3-kinase-protein kinase B pathway and on caerulein-induced cytokine
production in isolated mouse pancreatic acinar cells. J Pharmacol
Exp Ther. 329:1166–1177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Salto-Tellez M, Tan CH, Whiteman M
and Moore PK: GYY4137, a novel hydrogen sulfide-releasing molecule,
protects against endotoxic shock in the rat. Free Radic Biol Med.
47:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Fox B, Keeble J, et al: The complex
effects of the slow-releasing hydrogen sulfide donor GYY4137 in a
model of acute joint inflammation and in human cartilage cells. J
Cell Mol Med. 17:365–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hommes DW, Peppelenbosch MP and van
Deventer SJ: Mitogen activated protein (MAP) kinase signal
transduction pathways and novel anti-inflammatory targets. Gut.
52:144–151. 2003. View Article : Google Scholar
|
|
18
|
Zhang H, Moochhala SM and Bhatia M:
Endogenous hydrogen sulfide regulates inflammatory response by
activating the ERK pathway in polymicrobial sepsis. J Immunol.
181:4320–4331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Si X, Luo H, Morgan A, et al:
Stress-activated protein kinases are involved in coxsackievirus B3
viral progeny release. J Virol. 79:13875–13881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Lin G, Wan W, et al: Resveratrol,
a polyphenol phytoalexin, protects cardiomyocytes against
anoxia/reoxygenation injury via the TLR4/NF-kappaB signaling
pathway. Int J Mol Med. 29:557–563. 2012.PubMed/NCBI
|
|
21
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szabó C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang C, Yang Z, Zhang M, et al: Hydrogen
sulfide protects against chemical hypoxia-induced cytotoxicity and
inflammation in HaCaT cells through inhibition of
ROS/NF-kappaB/COX-2 pathway. PLoS One. 6:e219712011. View Article : Google Scholar
|
|
24
|
Chen Y, Liu Z and Xie X: Hydrogen sulphide
attenuates renal and cardiac injury after total hepatic ischemia
and reperfusion. J Surg Res. 164:e305–e313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Whiteman M, Li L, Rose P, Tan CH,
Parkinson DB and Moore PK: The effect of hydrogen sulfide donors on
lipopolysaccharide-induced formation of inflammatory mediators in
macrophages. Antioxid Redox Signal. 12:1147–1154. 2010. View Article : Google Scholar :
|
|
27
|
Chen TH, Kao YC, Chen BC, Chen CH, Chan P
and Lee HM: Dipyridamole activation of mitogen-activated protein
kinase phosphatase-1 mediates inhibition of
lipopolysaccharide-induced cyclooxygenase-2 expression in RAW 264.7
cells. Eur J Pharmacol. 541:138–146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuen S, Smith J, Caruso L, Balan M and
Opavsky MA: The coxsackie-adenovirus receptor induces an
inflammatory cardiomyopathy independent of viral infection. J Mol
Cell Cardiol. 50:826–840. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marchant D, Dou Y, Luo H, et al: Bosentan
enhances viral load via endothelin-1 receptor type-A-mediated p38
mitogen-activated protein kinase activation while improving cardiac
function during coxsackievirus-induced myocarditis. Circ Res.
104:813–821. 2009. View Article : Google Scholar : PubMed/NCBI
|