Introduction
Cyclin-dependent kinases (CDKs) strictly orchestrate
the cell cycle machinery through the binding to their specific
cyclin counterparts at cell cycle checkpoints. The CDK-cyclin
complex formation ensures the appropriate and accurate transition
between cell cycle phases, which is essential for cell
proliferation (1–3). Cancer cells frequently demonstrate
de-regulated CDK activity that causes uncontrolled cell
proliferation, which is a hallmark of malignant cell turnover.
Therefore, recent approaches targeting CDKs with specific small
molecule inhibitors have a crucial role both in developmental
biology and cancer therapy. Roscovitine (CYC202 or seliciclib), a
purine derivative, is a novel promising candidate for CDK
inhibition and is able to induce apoptosis in a variety of cancer
cell types; including lung, prostate, breast and colon cancers as
well as multiple myeloma (4–5).
Roscovitine mainly acts as a competitive inhibitor of CDKs while
showing a broad affinity for specific CDK ends, and blocks the
ATP-binding catalytic site of kinases (1,3).
Recent studies demonstrated that roscovitine induces cell cycle
arrest at the G1/S or G2/M phase transition in MCF-7 breast
(1), lung cancer (2), HeLa cervical (3) and HCT116 colon cancer (4) cells. In addition, roscovitine
treatment may synergize the anti-tumor effects of other
chemotherapeutics by inducing further apoptotic cell death
(5).
Although previous studies have established the
apoptotic potential of roscovitine in HeLa cells (3), the autophagic regulation following
roscovitine treatment has not been fully elucidated. Autophagy is a
regulated process of degradation and recycling of cellular
components in vacuoles inside the cell, allowing organelle and
energy turnover. Autophagy has a crucial role in tissue dynamics,
homeostasis, development and disorders. Autophagy is responsible
for the degradation of cytoplasmic macromolecular and protein
structures and damaged or aged organelles in the cells. The most
significant marker of autophagy is the appearance of double
membrane-enclosed vesicles, which engulf portions of cytoplasm
and/or organelles in the cytoplasm (6–8). Due
to different stress factors, including starvation, autophagy can
result in cellular adaptation as an alternative to cell death or
survival. Although autophagy has been considered to be a stress
adaptation mechanism in response to certain conditions, including
starvation, apoptosis and autophagy is now considered to have a
specific function. In variable cellular settings and in the
presence of different external factors, autophagy is regarded as an
alternative cell death pathway. Depending on the type of the
stimulus, the two types of cell death may be triggered at the same
time, corresponding to each other by common upstream signals, or
occasionally the cell switches in between these death pathways in a
successive manner. Thus, the clarification of the joint molecular
targets in both apoptosis and autophagy regulation processes is
critical to evaluate the therapeutic potential of novel
chemotherapeutic drugs. One of the links between apoptosis and
autophagy is B-cell lymphoma 2 (Bcl-2), which is an anti-apoptotic
member of the Bcl-2 family. Bcl-2 exerts a dual role to inhibit
apoptosis and eliminate pro-apoptotic Bcl-2 family members or
autophagy by binding Beclin-1, which has a similar structure to
that of pro-apoptotic Bcl-2 proteins. Beclin-1 is a Bcl-2 homology
(BH) 3 only protein which binds to anti-apoptotic Bcl-2 family
members; myeloid cell leukemia-1 (Mcl-1), Bcl-2 or Bcl-2 extra
large (Bcl-xL) (9,10).
It has been suggested that anti-apoptotic Bcl-2 family members
exert a dual role in inhibiting both apoptotic and autophagic
pathways in the cell. Beclin-1 is an initiator factor for autophagy
and recruits Vps34 and Vps15 to form pre-autophagosome structures
(11). Therefore, the expression
profile of Beclin-1 akin to Bcl-2 may be a crucial switch molecule
between apoptosis and autophagy (12).
The present study aimed to examine the potential
therapeutic role of roscovitine in HeLa cervical cancer cells by
investigating apoptotic and autophagic molecular signaling
molecules in a time-dependent manner.
Materials and methods
Drugs, chemicals and antibodies
Roscovitine (seliciclib, 2-(1-
ethyl-2-hydroxy-ethylamino)-6-benzylamino-9-isopropylpurine) was
purchased from Sigma-Aldrich (St Louis, MO, USA) and dissolved in
dimethyl sulfoxide (DMSO) to establish a 10 mM stock solution, and
the aliquots were maintained at −20°C. Propidium iodide (PI), DAPI,
3,3′-dihexyloxacarbocyanine iodide (DiOC6) and acridine orange (AO)
were purchased from Applichem (Darmstadt, Germany).
Monodansylacadaverine (MDC) was purchased from Sigma-Aldrich. The
monoclonal anti-rabbit antibodies against Bcl-2-associated X (Bax;
1:1,000), Bcl-2 (1:1,000), Bcl-xL (1:1,000),
P53-upregulated modulator of apoptosis (Puma; 1:1,000), Beclin-1
(1:1,000), microtubule-associated light chain 3B (LC3B; 1:1,000),
autophagy protein (Atg)5 (1:1,000), Atg12 (1:1,000), p62 (1:1,000),
β-actin (1:1000) and horseradish peroxidase (HRP)-conjugated
secondary anti-rabbit (1:5,000) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Cell culture
HeLa (CCL-2) cervical adenocarcinoma cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA). The cells were maintained in high-glucose Dulbecco’s modified
Eagle’s medium (DMEM; PAN Biotech, Aidenbach, Germany) with 10%
fetal calf serum (FCS; PAN Biotech) and 100 U/100 mg
ml−1 penicillin/streptomycin (PAN Biotech) at 37°C in a
humidified 5% CO2 incubator (Hera Cell 150i; Thermo
Fisher Scientific, San Jose, CA, USA). Serum starvation of cells
was performed via incubation of cells with DMEM and 0.5% FCS for 2
h prior to staining with MDC.
MTT cell viability assay
To determine the cytotoxicity of roscovitine
treatment, an MTT cell viability assay [Sigma; 50 mg/ml in
phosphate-buffered saline (PBS)] was used. For this purpose,
1×104 cells were seeded in each well of a 96-well plate
and treated with roscovitine for 24 h. MTT was added and the cells
were incubated at 37°C for 4 h. Following removal of the MTT
reagent including the media, 100 μl DMSO was added to each well and
cells were incubated for 5 min in the dark. Absorbance was
determined at 570 nm (A570) using a microplate reader (Model 680;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Survival assay (trypan blue dye exclusion
assay)
A total of 5×104 cells per well were
seeded into six-well plates and treated with roscovitine for 24, 48
and 72 h. The cells were then counted by staining with 0.4% (w/v)
trypan blue dye. Viable and dead cells were counted under light
microscopy (IX71; Olympus Corporation, Tokyo, Japan). The number of
viable cells (y-axis) was plotted against the time (x-axis).
Apoptotic cell death ELISA assay
The ability of roscovitine to induce apoptosis was
determined by the Cell Death Detection ELISA PLUS kit (Roche,
Indianapolis, IN, USA). The cell lysates were placed in a
streptavidin-coated microplate. A mixture of anti-histone-biotin
and anti-DNA-peroxisidase (POD) was incubated for 2 h at 15–25°C.
Following removal of unbound antibodies by washing, POD was
determined colorometrically at 405 nm with
2,2′-azino-di-(3-ethylbenzthiazoline sulfonate (6) diammonium salt (ABTS), which was used
as a substrate.
Fluorescence microscopy
The HeLa cells were seeded at a density of
5×104 per well in 12-well plates and treated with 20 μM
roscovitine for 24 and 48 h, respectively. The cells were stained
with different fluorescent dyes which are summarized in Table I. Following the indicated
incubation period of the cells at 37°C, the dye including the media
was removed, 1X PBS was added and cells were visualized by
fluorescent microscopy according to their specific excitation and
emission wavelengths at the indicated time-points.
 | Table IFluorescence dyes to detect apoptosis
and autophagy. |
Table I
Fluorescence dyes to detect apoptosis
and autophagy.
| Dye | Concentration | Incubation time
(min) | Excitation (nm) | Emission (nm) |
|---|
| PI | 5 mg/ml | 30 | 536 | 617 |
| DiOC6 | 4 nM | 15 | 488 | 525 |
| DAPI | 5 mg/ml | 10 | 350 | 570 |
| AO | 5 mg/ml | 10 | 460 | 650 |
| MDC | 10 mg/ml | 30 | 510 | 595 |
Immunoblot analysis
The cells were lysed on ice using ProteoJET
mammalian cell lysis reagent (Fermentas, Glen Burnie, MD, USA)
containing protease inhibitor (Roche, Mannheim, Germany). The cell
lysates were centrifuged at 16,100 × g for 20 min at 4°C and the
protein concentration was measured by a Bradford Assay (Bio-Rad
Laboratories, Inc.). Following separation of denatured proteins
according to size by SDS-PAGE, the proteins were transferred onto a
polyvinylidene fluoride membrane (Thermo Scientific) where they
were labeled using antibodies specific to the target protein. The
gels obtained by SDS-PAGE were placed in the Trans-Blot Turbo
transfer system (Bio-Rad Laboratories) and protein transfer was
conducted at 25 V for 5 min. The membrane was blocked in 5% bovine
serum albumin/TBS containing 0.1% Tween-20 (TBST) for 2 h at room
temperature, and then incubated with primary antibodies for each
target; Bax, Puma, Bcl-2, Beclin-1, LC3B, Atg5, Atg12, p62 and
β-actin overnight at 4°C and further incubated with anti-rabbit
immunoglobulin G-HRP secondary antibody overnight at 4°C. Following
washing with TBST, the proteins were detected using Super Signal
West FemtoLuminol/Enhancer Solution (Thermo Scientific) and exposed
to Hyperfilm-enhanced chemiluminescence (Amersham Pharmacia
Biotech, Piscataway, NJ, USA). Densitometric analysis of
immunoblots was performed using Image J (National Institutes of
Health, Bethesda, MD, USA) and GraphPad Prism 6 (GraphPad Software,
Inc., La Jolla, CA, USA) software; and all of the proteins were
quantified relative to the loading control.
Determination of mitochondrial membrane
potential assay
The HeLa cells (1×105) were seeded in
12-well plates, treated with the indicated concentration of
roscovitne and allowed to attach overnight. The cells were washed
once with 1X PBS, and stained with 0.4 mM DiOC6
(Molecular Probes Life Technologies, Carlsbad, CA, USA) fluorescent
probe. Mitochondrial membrane potential (MMP) loss was measured
using a Fluoroskan Ascent fluorometer (excitation/emission = 488
nm/525 nm; Thermo Labsystems, Milford, MA, USA).
Statistical analysis
All of the experiments were statistically analyzed
using GraphPad Prism 6 software. Error bars in the graphs were
generated using ± standard deviation values. A P<0.05 was
considered to indicate a statistically significant difference.
Results
Roscovitine decreases cell viability and
induces apoptosis in a dose-dependent manner
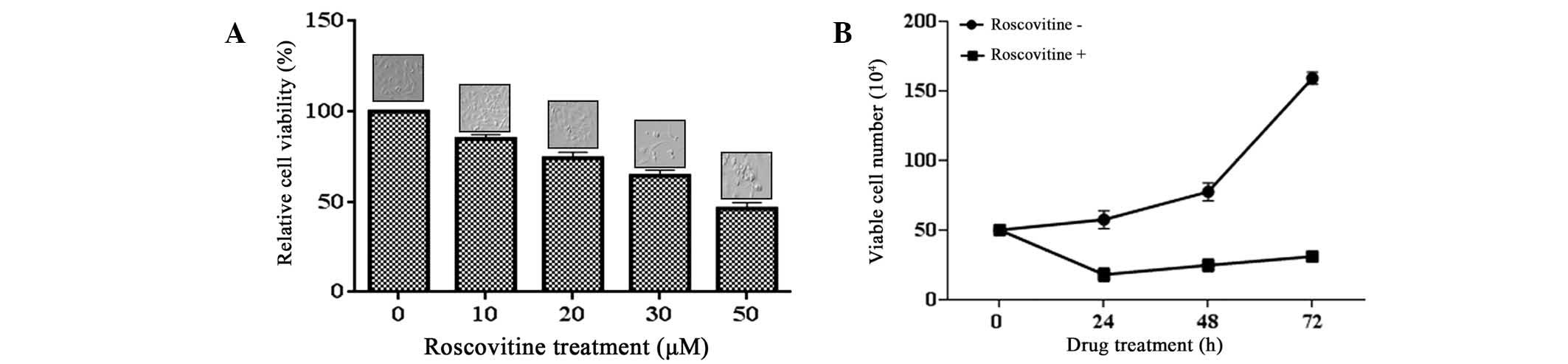
The effect of roscovitine on the viability of HeLa
cells was investigated by an MTT assay. Exposure of the cells to
roscovitine (0–50 μM) for 24 h decreased the cell viability in a
dose-dependent manner (P<0.0001, each compared with the
untreated control). A moderate cytotoxic effect was triggered
following 20 μM roscovitine treatment, which caused 25% cell
viability loss compared with the untreated control samples. The
highest concentration of 50 μM roscovitine reduced the cell
viability ratio by 50% within 24 h (Fig. 1A).
To confirm the cytotoxic effect of roscovitine on
cell proliferation in HeLa cells, a trypan blue exclusion assay was
also performed (Fig. 1B).
Exponentially growing HeLa cells were treated with roscovitine (20
μM) for different time periods within 72 h. The doubling time of
HeLa cells was determined to be 23 h, which was in accordance with
the information provided by the supplier. Treatment of the cells
with roscovitine suppressed the cell viability following 24 h.
However, the cells gained proliferation capacity following
long-term exposure to roscovitine for 48 and 72 h. Although
roscovitine diminished the cell proliferation within 24 h, it was
concluded that longer treatment with roscovitine may lead to the
accumulation of resistance factors in HeLa cells (P<0.0001).
Following exposure of HeLa cells to roscovitine for
24 and 48 h, the cells were stained with PI and DAPI to identify
the cell death mechanism (Fig.
2A). The percentage of PI-positive cells, which are regarded as
dead cells, was increased following roscovitine treatment of HeLa
cells (red stain) compared with the untreated cells in a
time-dependent manner. In addition, DAPI staining was performed to
identify apoptotic cells which exhibited fragmented DNA and nuclear
condensation. The number of apoptotic cells (bright blue stain) was
increased following 24 and 48 h roscovitine treatment compared with
the untreated control cells. According to the fluorescence images,
the ratio of nuclear condensation was increased following 48 h of
roscovitine treatment.
In order to investigate the cytotoxic effect of
roscovitine on HeLa cells, a Cell Death ELISA assay was performed,
which demonstrated the cleavage of DNA due to drug treatment
(Fig. 2B). Exposure of cells to
lower concentrations of roscovitine (10 and 15 μM) induced
apoptotic cell death by 1.65 and 1.85-fold compared with untreated
control samples, respectively. The appropriate concentration of
roscovitine, 20 μM, increased apoptosis by 2-fold compared with the
untreated control cells (P<0.0001, each compared with the
untreated control). Therefore, it was concluded that roscovitine
treatment for 24 h decreased the cell viability and induced
apoptosis in a dose-dependent manner.
Roscovitine induces apoptosis and
autophagy through modulating levels of Bcl-2 family members
Since Bcl-2 family members have a decisive role in
apoptotic cell death mechanisms, the effect of roscovitine on the
expression levels of pro- and anti-apoptotic Bcl-2 family member
proteins in HeLa cells was determined. As revealed in Fig. 3A, roscovitine treatment decreased
the Bcl-2 and Bcl-xL expression levels time-dependently,
whereas the activation of pro-apoptotic Bcl-2 family members, Puma
and Bax, was observed as an early response. Although roscovitine
treatment for 24 h downregulated Bcl-xL expression
levels, no significant alteration was observed for Bcl-2 at the
same time-point. It was concluded that roscovitine is an
apoptosis-inducing agent via modulating the ratio between anti and
pro-apoptotic Bcl-2 family members in HeLa cells.
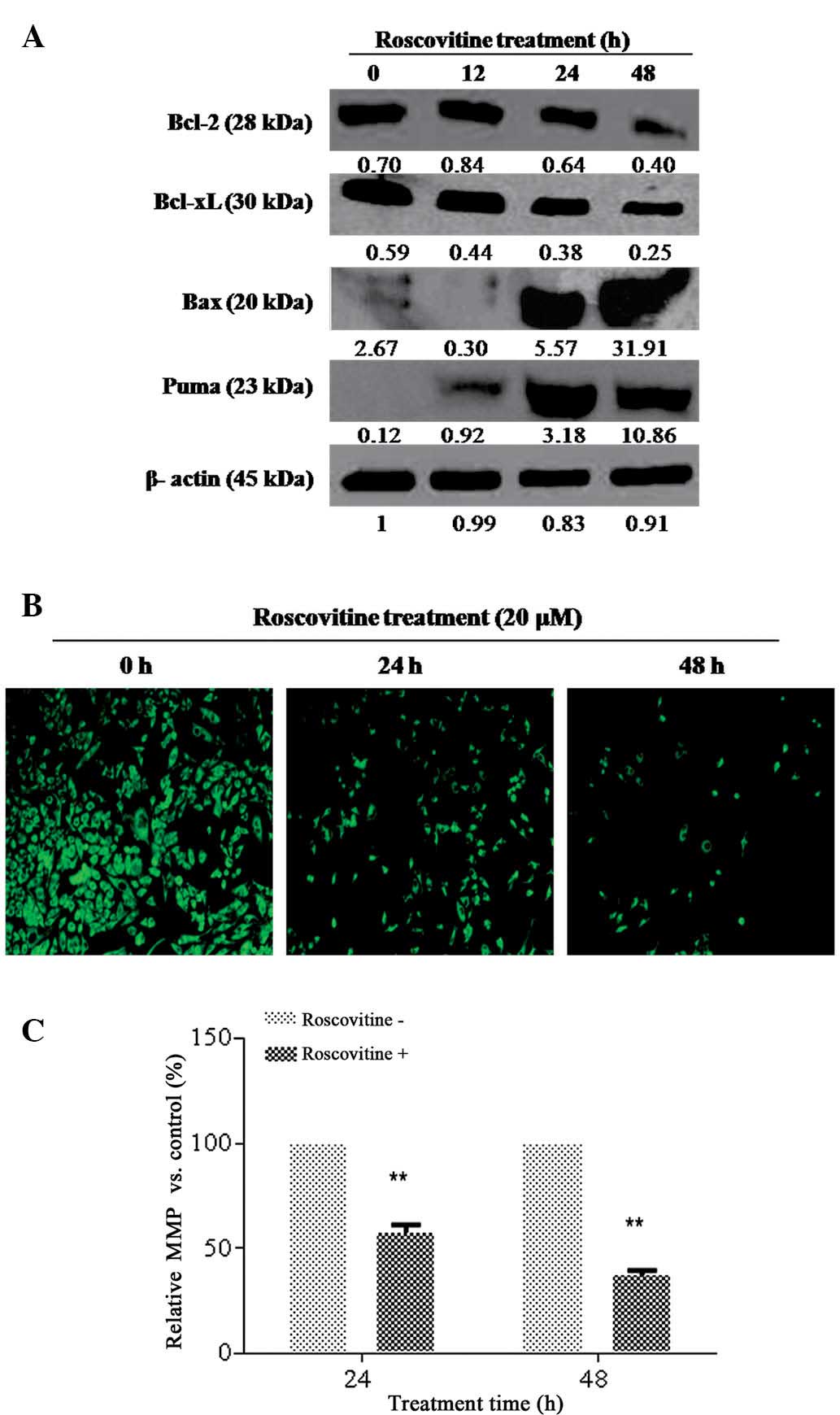 | Figure 3Roscovitine induced
mitochondria-mediated apoptosis. (A) HeLa cells were seeded at a
density of 1×106 in 60-mm plates and treated with
roscovitine (20 μM) for 12, 24 and 48 h. A total of 20 μg of
protein lysate was purified by 12% SDS-PAGE. β-actin was used as a
loading control. (B) The loss of MMP was determined by fluoresence
microscopy (magnification, ×200). (C) The MMP was measured by a
fluorometer (P<0.0001, vs the untreated control group;
excitation, 485 nm; emission, 538 nm). MMP, mitochondrial membrane
potential; Bcl-2, B-cell lymphoma 2; Bcl-xL, Bcl-2 extra large;
Bax, Bcl-2-associated X; PUMA, P35-upregulated modulator of
apoptosis. |
In order to examine the potential role of
roscovitine on mitochondria-mediated apoptosis, DiOC6 staining was
performed (Fig. 3B and C). The
number of viable cells (green stained), which have normal
mitochondrial function, was gradually decreased in a time-dependent
manner. The loss in mitochondrial membrane potential (MMP) was also
confirmed by fluorometric analysis (excitation, 485 nm; emission,
538 nm).
To investigate the effect of roscovitine on
autophagic regulation mechanisms, the expression levels of the key
signaling molecules associated with autophagy were investigated
following roscovitine treatment in HeLa cells. As demonstrated in
Fig. 4A, Beclin-1 expression
levels were progressively upregulated with increasing exposure time
to the drug in HeLa cells. Treatment with roscovitine for 12 and 24
h upregulated the expression of two LC3 isoforms (LC3-I and
LC3-II); however, long-term treatment for 48 h with the drug
downregulated the LC3-II isoform in HeLa cells (Fig. 4A). By contrast, the expression
profiles of Atg5 and Atg12 were not altered following roscovitine
treatment. p62 is referred to as a significant biomarker for the
autophagic vacuole formation process. In the present study, it was
determined that roscovitine treatment at 48 h eliminated the p62
expression levels in HeLa cells. Therefore, it was concluded that
autophagic mechanisms are finalized later than roscovitine-induced
apoptosis.
To demonstrate the stages of roscovitine-induced
autophagy, cells were stained with MDC or AO (Fig. 4B). MDC staining was performed to
visualize acidic autophagic vesicles. As a positive control,
nutrient-deprived cells were stained with MDC, displaying bright
green dots. Although MDC-stained cells were identified following
12, 24 and 48 h of roscovitine treatment, maximum fluorescent
staining was observed after 12 h. The lysosomotrophic dye AO was
used to identify lysosomes. As a positive control,
nutrient-deprived cells were stained with AO, displaying bright red
dots. The accumulation of AO dye was increased following
roscovitine treatment in a time-dependent manner.
Discussion
Cervical cancer is the second most common cancer
type among females worldwide and the disease etiology is mostly
linked to infection with Human Papilloma Virus (HPV) (13,14).
Depending on the tumor size the preferred treatments for cervical
cancer are medical surgery, radiation therapy, chemotherapy or
their combination. However, the presence of an increasing number of
recurrent cases demonstrates an urgent requirement for improvement
of therapeutic application models. Although a number of the
classical chemotherapeutic agents, including cisplatin, mitomycin C
and bleomycin, are used as therapeutic agents against cervical
cancer, these drugs induce intrinsic resistance factors, which
renders the efficiency of therapy (15,16).
Therefore, the development of novel anti-tumor agents is critical
for the treatment of cervical cancer patients.
Since the loss of functional cell cycle control is a
common problem in numerous malignancies, CDK inhibitors are
considered to inhibit the uncontrolled proliferation in cancer
cells. Roscovitine was the first orally bioavailable CDK inhibitor
to enter clinical trials for breast, lung, B-cell lymphoma and
advanced solid tumors (17–19).
Previous studies established that roscovitine predominantly
inhibits CDK2 and induces cell cycle arrest at G2/M phase. In the
present study, it was identified that roscovitine decreases cell
viability in a dose-dependent manner in HeLa cells. A moderately
cytotoxic concentration of the drug (20 μM) prevented cell
proliferation and induced apoptosis by 2-fold compared with the
untreated control cells within 24 h. As demonstrated in previous
studies, roscovitine induced apoptosis mainly through modulating
MMP and activating caspase-9, -3 and poly(adenosine
diphosphate-ribose) polymerase cleavage in different cancer cells
(4,18,20–28).
Similar to these observations, it was determined in the present
study that 20 μM roscovitine decreased the MMP in a time-dependent
manner in HeLa cells [MMP loss, 45% (24 h) and 65% (48 h)
vs. the control group, respectively]. Although roscovitine
(20 μM) upregulated Bax and Puma expression levels
time-dependently, anti-apoptotic Bcl-2 was downregulated following
long-term exposure of cells to the drug for 48 h. By contrast,
Bcl-xL was downregulated in a time-dependent manner
following roscovitine treatment. Therefore, it was concluded that
roscovitine exerts its apoptotic effect on HeLa cells by altering
the MMP and modulating Bcl-2 family members in HeLa cells. A
previous study revealed that roscovitine (20 μM) induced apoptosis
via reducing the cell proliferation caused by G2/M cell cycle
arrest in tamoxifen therapy-resistant breast cancer cell lines. In
addition, roscovitine successfully reduced breast tumor volume,
which was evaluated in an in vivo xenograft tumor model
(26). Although roscovitine mostly
activated caspases to induce the apoptotic cell death mechanism
(4), it was established that
roscovitine may successfully induce apoptosis in caspase-3 null
MCF-7 cells through upregulating the p53 signaling pathway
(29). In response to this
finding, the present study aimed to examine the potential role of
roscovitine on autophagy, which is also accepted as a
caspase-independent cell death type in HeLa cells. Previously, it
was demonstrated that roscovitine treatment enhanced
doxorubicin-induced apoptosis as well as autophagy in sarcoma cell
line models (30). In addition,
roscovitine downregulated mitogen-activated protein kinase 1/3,
which exhibits a high activity in mature oocytes of mammals, frogs
or invertebrates, such as starfish, to maintain cell cycle arrest.
By this way, mature oocytes underwent apoptosis if they were not
fertilized. Roscovitine increased calcium-dependent mitotic cell
entry and delayed cell death in sea urchins by downregulating
MAPK1/3. Based on these observations, it was suggested that
autophagy with a survival-promoting function may be activated when
cell cycle arrest was triggered by a specific CDK inhibitor
(31). The molecular targets of
roscovitine on the autophagic regulation processes have not been
fully elucidated yet. To examine its possible molecular targets,
the present study first conducted experiments to clarify the
potential effect of roscovitine on the key converging molecules
between apoptosis and autophagy. Beclin-1 is an important mediator
between cell death and survival mechanisms due to its binding
ability to anti-apoptotic Bcl-2 family members. Pro-apoptotic Bcl-2
family members have a crucial role in the dissociation of the
complex of Beclin-1 and Bcl-2. It was suggested that upregulation
of several pro-apoptotic Bcl-2 family members, including
BimEL, Bcl-2-interacting killer, Bcl-2 homology domain 3
interacting-domain death agonist, Bcl-2-associated death promoter,
Puma, but not Bax or Bak, competitively bind to hydrophobic grooves
on Bcl-2 and lead to autophagy by inducing the dissociation of
Beclin-1 (32). Beclin-1
homodimerization due to Bcl-2 interaction renders autophagic
regulation in the cells by preventing heterodimerization of
Beclin-1 with Atg14 or other complex proteins. In addition, the
interaction between Bcl-2 and Beclin-1 is regulated by c-Jun
N-terminal kinase and extracellular signal-regulated kinase (ERK)
led to phosphorylation of Bcl-2 or Beclin-1, respectively (33). Death-associated protein kinase is
one of the crucial targets of ERK, which consequently induces the
phosphorylation of Beclin-1 (34).
It was identified that, although HeLa cells were lacking basal
Beclin-1 expression, roscovitine treatment for 48 h upregulated the
expression levels of Beclin-1. Concomitantly, Bcl-2 downregulation
due to roscovitine treatment was marked. It was concluded that the
upregulation of pro-apoptotic Puma and Bax was noteworthy following
drug treatment for up to 48 h, which may dissociate the Beclin-1
and Bcl-2 complex and lead to heterodimerization of autophagy
complex proteins. Additionally, LC3, a cytosolic soluble protein,
was cleaved during autophagic induction and its involvement in the
autophagic vacuole membrane formation was determined. The 18 and 16
kDa forms of LC3 were called LC3-I and LC3-II, respectively. Amino
acid starvation-induced increases in the levels of LC3-II were
found to be associated with membrane compartments, whereas LC3-I
was localized in the cytoplasm (35). Previous findings stated that
roscovitine may synergize both apoptosis and autophagy by enhancing
the DNA-damaging effect of doxorubicin in different sarcoma cell
lines (30). The number of green
fluorescence protein-tagged LC3 puncta was increased due to
prolonged cell cycle arrest at G2/M phase triggered by roscovitine
treatment in the presence of doxorubicin, a DNA damage-inducing
agent. AO staining, which was concentrated in acidic vacuoles, also
confirmed that roscovitine synergized the doxorubicin-induced
autophagosome formation. In addition to Beclin-1 upregulation, LC3
cleavage was increased following roscovitine treatment of HeLa
cells for 12 h. This suggested that when pro-apoptotic Bcl-2 family
members were upregulated in a parallel manner, the
autophagy-inducing signaling molecules were also upregulated. p62
has also been demonstrated to be an autophagic marker, which is
depleted when autophagosome complexes are engulfed with lysosomes.
Exposure of HeLa cells to roscovitine for 48 h eliminated the p62
expression levels. Therefore, lysosomes induced the degradation of
p62 and LC3-II. To confirm lysosome-mediated degradation of p62 and
LC3-II (16 kDa) in HeLa cells, AO staining was performed. The
perceptible acidic vacuoles were distinguished gradually in a
time-dependent manner following the exposure of cells to
roscovitine. By contrast, as an early response to roscovitine
treatment at 12 h, MDC staining indicated acidic vacuole formation
in HeLa cells. Therefore, it was concluded that roscovitine (20 μM)
induced both apoptosis and autophagy, with each process following
one another in a time-dependent manner.
Acknowledgements
The authors are grateful to the Istanbul Kultur
University Scientific Projects Support Center.
Abbreviations:
|
ABTS
|
2,2′-azino-di-3-ethylbenzthiazoline
sulfonate 6 diammonium salt
|
|
AO
|
acridine orange
|
|
DiOC6
|
3,3′-dihexyloxacarbocyanine iodide
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
DMSO
|
dimethylsulfoxide
|
|
MDC
|
monodansylcadaverine
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
|
POD
|
peroxidase
|
References
|
1
|
Wesierska-Gadek J, Gueorguieva M,
Wojciechowski J and Horky M: Cell cycle arrest induced in human
breast cancer cells by cyclin-dependent kinase inhibitors: a
comparison of the effects exerted by roscovitine and olomoucine.
Pol J Pharmacol. 56:635–641. 2004.PubMed/NCBI
|
|
2
|
Zhang T, Jiang T, Zhang F, et al:
Involvement of p21Waf1/Cip1 cleavage during roscovitine-induced
apoptosis in non-small cell lung cancer cells. Oncol Rep.
23:239–245. PubMed/NCBI
|
|
3
|
Wesierska-Gadek J, Wandl S, Kramer MP, et
al: roscovitine up-regulates p53 protein and induces apoptosis in
human HeLaS(3) cervix carcinoma cells. J Cell Biochem.
105:1161–1171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arisan ED, Coker A and Palavan-Ünsal N:
Polyamine depletion enhances the roscovitine-induced apoptosis
through the activation of mitochondria in HCT116 colon carcinoma
cells. Amino Acids. 42:655–665. 2012. View Article : Google Scholar
|
|
5
|
Appleyard MV, O’Neill MA, Murray KE, et
al: Seliciclib (CYC202, R-roscovitine) enhances the antitumor
effect of doxorubicin in vivo in a breast cancer xenograft model.
Int J Cancer. 124:465–472. 2009. View Article : Google Scholar
|
|
6
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gozuacik D and Kimchi A: Autophagy and
cell death. Curr Top Dev Biol. 78:217–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J: Beclin 1 bridges autophagy,
apoptosis and differentiation. Autophagy. 4:947–948. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ku B, Woo JS, Liang C, et al: An insight
into the mechanistic role of Beclin 1 and its inhibition by
prosurvival Bcl-2 family proteins. Autophagy. 4:519–520. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maiuri MC, Criollo A, Tasdemir E, et al:
BH3-only proteins and BH3 mimetics induce autophagy by
competitively disrupting the interaction between Beclin 1 and
Bcl-2/Bcl-X(L). Autophagy. 3:374–376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zalckvar E, Berissi H, Eisenstein M and
Kimchi A: Phosphorylation of Beclin 1 by DAP-kinase promotes
autophagy by weakening its interactions with Bcl-2 and Bcl-XL.
Autophagy. 5:720–722. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anorlu RI: What is the significance of the
HPV epidemic? Can J Urol. 15:3860–3865. 2008.PubMed/NCBI
|
|
14
|
Weinstein LC, Buchanan EM, Hillson C and
Chambers CV: Screening and prevention: cervical cancer. Prim Care.
36:559–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh KC, Agarwal A, Agarwal S, et al:
‘Quick course’ neoadjuvant chemotherapy with cisplatin, bleomycin
and vincristine in advanced cervical cancer. Gynecol Obstet Invest.
58:109–113. 2004. View Article : Google Scholar
|
|
16
|
van Luijk IF, Coens C, van der Burg ME, et
al: Phase II study of bleomycin, vindesine, mitomycin C and
cisplatin (BEMP) in recurrent or disseminated squamous cell
carcinoma of the uterine cervix. Ann Oncol. 18:275–281. 2007.
View Article : Google Scholar
|
|
17
|
Benson C, White J, De Bono J, et al: A
phase I trial of the selective oral cyclin-dependent kinase
inhibitor seliciclib (CYC202; R-roscovitine), administered twice
daily for 7 days every 21 days. Br J Cancer. 96:29–37. 2007.
View Article : Google Scholar
|
|
18
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guzi T: CYC-202 Cyclacel. Curr Opin
Investig Drugs. 5:1311–1318. 2004.
|
|
20
|
Coley HM, Shotton CF and Thomas H:
Seliciclib (CYC202; r-roscovitine) in combination with cytotoxic
agents in human uterine sarcoma cell lines. Anticancer Res.
27:273–278. 2007.PubMed/NCBI
|
|
21
|
Dey A, Wong ET, Cheok CF, Tergaonkar V and
Lane DP: R-roscovitine simultaneously targets both the p53 and
NF-kappaB pathways and causes potentiation of apoptosis:
implications in cancer therapy. Cell Death Differ. 15:263–273.
2008. View Article : Google Scholar
|
|
22
|
Duffin R, Leitch AE, Sheldrake TA, et al:
The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by
down-regulation of Mcl-1. FEBS Lett. 583:2540–2546. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goodyear S and Sharma MC: roscovitine
regulates invasive breast cancer cell (MDA-MB231) proliferation and
survival through cell cycle regulatory protein cdk5. Exp Mol
Pathol. 82:25–32. 2007. View Article : Google Scholar
|
|
24
|
Maurer M, Komina O and Wesierska-Gadek J:
roscovitine differentially affects asynchronously growing and
synchronized human MCF-7 breast cancer cells. Ann NY Acad Sci.
1171:250–256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mitchell C, Park MA, Zhang G, et al:
Extrinsic pathway- and cathepsin-dependent induction of
mitochondrial dysfunction are essential for synergistic
flavopiridol and vorinostat lethality in breast cancer cells. Mol
Cancer Ther. 6:3101–3112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nair BC, Vallabhaneni S, Tekmal RR and
Vadlamudi RK: roscovitine confers tumor suppressive effect on
therapy-resistant breast tumor cells. Breast Cancer Res.
13:R802011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ribas J, Boix J and Meijer L:
(R)-roscovitine (CYC202, Seliciclib) sensitizes SH-SY5Y
neuroblastoma cells to nutlin-3-induced apoptosis. Exp Cell Res.
312:2394–2400. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zolnierczyk JD, Bloński JZ, Robak T,
Kiliańska ZM and Wesierska-Gadek J: roscovitine triggers apoptosis
in B-cell chronic lymphocytic leukemia cells with similar
efficiency as combinations of conventional purine analogs with
cyclophosphamide. Ann NY Acad Sci. 1171:124–131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wesierska-Gadek J, Gueorguieva M and Horky
M: roscovitine-induced up-regulation of p53AIP1 protein precedes
the onset of apoptosis in human MCF-7 breast cancer cells. Mol
Cancer Ther. 4:113–124. 2005.PubMed/NCBI
|
|
30
|
Lambert LA, Qiao N, Hunt KK, et al:
Autophagy: a novel mechanism of synergistic cytotoxicity between
doxorubicin and roscovitine in a sarcoma model. Cancer Res.
68:7966–7974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Houel-Renault L, Philippe L, Piquemal M
and Ciapa B: Autophagy is used as a survival program in
unfertilized sea urchin eggs that are destined to die by apoptosis
after inactivation of MAPK1/3 (ERK2/1). Autophagy. 9:1527–1539.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sinha S and Levine B: The autophagy
effector Beclin 1: a novel BH3-only protein. Oncogene. 27(Suppl 1):
S137–S148. 2008. View Article : Google Scholar
|
|
33
|
Wei Y, Sinha S and Levine B: Dual role of
JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis
regulation. Autophagy. 4:949–951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zalckvar E, Berissi H, Mizrachy L, et al:
DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1
promotes dissociation of beclin 1 from Bcl-XL and induction of
autophagy. EMBO Rep. 10:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. Embo J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|