Introduction
At present, surgical resection is the primary
treatment method for gastric cancer. However, for gastric cancer in
the advanced or metastasized stages, surgery may not be an option
and therefore, other therapies, including adjuvant therapy, salvage
chemotherapy and cytotoxic treatment, are used (1); however, the effect of these therapies
is often limited.
Trastuzumab, in combination with chemotherapy, has
been reported to have a significant impact on the treatment of
advanced human epidermal growth factor receptor 2
(HER2)-overexpressing gastric cancer. The results of this study
demonstrated an increase in the long-term survival of patients,
therefore suggesting its potential as a targeted therapy for
gastric cancer (2). The
identification of effective drug targets for novel therapies is an
increasingly important field of drug research.
Epidermal growth factor receptor (EGFR), a receptor
tyrosine kinase, is an important transmembrane receptor, with its
protein-tyrosine kinase activity residing in the intracellular
domain. Activation of EGFR via growth factor ligand-binding
triggers an intracellular signal transduction pathway, which
further initiates intracellular responses by regulating downstream
molecules. EGFR was reported to be highly associated with the
incidence and development of gastric cancer(3). Furthermore, it was reported that
patients with EGFR-overexpression had worse prognoses compared with
those of EGFR-negative patients (4).
c-Met is a high-affinity hepatocyte growth factor
(HGF) receptor, which also possesses tyrosine kinase activity.
Studies have revealed that c-Met was frequently overexpressed in
46.1–77.3% of patients with gastric cancer (5–7); in
addition, increased c-Met expression was reported to be highly
associated with gastric cancer staging and poor prognosis as well
as tumor cell migration, invasion and metastasis (8). Studies have shown that the HGF
fragment NK4 acted as a HGF antagonist, improving the sensitivity
of gastric cancer cells to the orally active EGFR tyrosine kinase
inhibitor (EGFR-TKI), gefitinib (9). Phase II clinical studies of
metastatic gastric and gastroesophageal junction adenocarcinoma
have shown that following gefitinib administration to 75 late-stage
patients, one patient showed a partial response (PR) and in 13
patients increase disease control was achieved (10). Another phase II clinical study
examined the effect of an EGFR-TKI, erlotinib, on gastrointestinal
and gastric adenocarcinoma. Out of 43 patients with
gastroeosophageal junction adenocarcinoma, one demonstrated a
complete response (CR) and four showed a PR; however, no
significant results were observed in any of the 25 gastric
adenocarcinoma patients (11).
These clinical studies provided evidence for the
minimal sensitivity of gastric cancers to EGFR-TKI. Therefore, it
has been hypothesized that this may be due to drug resistance;
however, the mechanism of sensitivity of certain gastroesophageal
junction carcinoma to EGFR-TK1 remains to be elucidated.
Numerous tyrosine kinase receptors are located on
the surface of tumor cells, and activation of these receptors
triggers signal transduction networks, which are able to crosstalk
with each other (12,13). In theory, these pathways may have a
synergistic role in cancer signaling and therefore, targeting these
pathways may be an effective novel strategy for disease management.
EGFR mutations in non-small-cell lung cancer (NSCLC) increased the
sensitivity of cells to EGFR-TKI treatment (14); in addition, the continuous
overexpression of c-Met was reported to be functionally relevant to
EGFR-TKI resistance (15–17). Non-mutated EGFR and c-Met have been
shown to be overexpressed in the gastric cancer cell line MKN-45
(18,19). The aim of the present study was to
investigate whether altering c-Met gene expression by using small
interfering RNAs (siRNAs) affected the sensitivity and resistance
of MKN-45 cells to gefitinib.
Materials and methods
Cell line and culture
The human gastric cancer cell line MKN-45 was
purchased from American Type Culture Collection (Manassas, VA,
USA). MKN-45 was grown and passaged routinely at 37°C in a
humidified 5% CO2 atmosphere in high-glucose Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA, USA)
containing 10% fetal bovine serum (FBS; Gibco-BRL).
siRNA transfection
Three pairs of siRNAs for c-Met and one pair of
control siRNAs were designed and synthesized (Shanghai GeneChem
Co., Ltd., Shanghai, China). siRNA sequences were as follows:
c-Met-siRNA1 sense, 5′-GUGCCACUAACUACAUUUATT-3′ and anti-sense,
5′-UAAAUGUAGUUAGUGGCACTT-3′; c-Met-siRNA2 sense,
5′-GUCCCGAGAAUGGUCAUAATT-3′ and anti-sense,
5′-UUAUGACCAUUCUCGGGACTT-3′; c-Met-siRNA3 sense,
5′-GCCUGAAUGAUGACAUUCUTT-3′ and anti-sense,
5′-AGAAUGUCAUCAUUCAGGCTT-3′; control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′. The transfection efficiency was
analyzed using fluorescence microscopy (Axioskop4O; Carl Zeiss AG,
Jena, Germany) using the methods described previously (20).
Reagents
LipofectamineTM 2000 transfection kits
were purchased from Invitrogen Life Technologies (Carlsbad, CA,
USA). Polyclonal goat anti-human-c-Met, -PI3K, -phosphorylated
(p)-PI3K, -AKT and -p-AKT (1:300) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) was
purchased from Proteintech (Chicago, IL, USA). Primers for
polymerase chain reaction (PCR) and the reverse-transcription PCR
(RT-PCR) kit were obtained from Takara Bio, Inc. (Dalian,
China).
Transfection
siRNA was transfected with the
LipofectamineTM 2000 kit according to the manufacturer’s
instructions. In brief, cells were seeded into six-well plates
until they reached 70–90% confluence. Cells were divided into five
groups: Control cells; cells transfected with Lipofectamine only;
cells transfected with 200 pmol siRNA1, siRNA2 or siRNA3; and cells
transfected with control siRNA. A total of 5 μl
LipofectamineTM 2000 was added to 250 μl serum-free
medium and mixed for 5 min at room temperature. An appropriate
amount of siRNA (final concentration of 200 pmol) was then added
and incubated for 20 min. The mixture was then added to
phosphate-buffered saline (PBS)-washed cells and incubated at 37°C
for 4 h. 10% FBS/DMEM medium was then added to achieve a final
volume of 2 ml.
Western blot analysis
Following transfection, cells were lysed for 48 h
and then separated using 8% SDS-PAGE. Prior to incubation, the
membrane was blocked with primary antibodies.
Rabbit-anti-human-c-Met, -PI3K, -p-PI3K, -AKT, -p-AKT or β-actin
(internal control) antibodies (Bejing Biosynthesis Biotechnology
Co., Ltd., Beijing, China) were incubated with the membranes for 2
h at room temperature, washed using 1X Tris-buffered saline with
Tween 20 and incubated with a HRP-labeled secondary antibody
(goat-anti-rabbit IgG; 1:500; Proteintech) for 90 min. An enhanced
chemiluminescence kit from Perkin-Elmer (Waltham, MA, USA) was used
to detect the signal Western blot analyses were quantified by
densitometry and analyzed using the Quantity One image analysis
system (Bio-Rad, Hercules, CA, USA).
RT-PCR
Total RNA was isolated using TRIzol®
(Invitrogen Life Technologies) 48 h following transfection. RNA
purity was measured using a spectrometer, and 2 μg RNA was
reverse-transcribed in a 20-μl reaction system. The specific
primers used were as follows: c-Met forward,
5′-CCTCACCATAGCTAATCTTGGGACA-3′ and reverse,
5′-CACAATCACTTCTGGAGACACTGGA-3′; PI3K forward
5′-AGGCTGTGATTGGGCGTA-3′ and reverse, 5′-AAGCAACCTCAAAGGGAAA-3′;
AKT forward, 5′-ATGGCACCTTCATTGGCTAC-3′ and reverse,
5′-CAGTCTGGATGGCGGTTG-3′. The housekeeping gene GAPDH was used as
the internal control (forward, 5′-CAAGGTCATCCATGACAACTTTG-3′ and
reverse, 5′-GTCCACCACCCTGTTGCTGTAG-3′). The cycling conditions were
95°C for 30 sec, 40 cycles of 95°C for 5 sec, and then 60°C for 30
sec.
MTT assay
In brief, cells were seeded into 96-well plates at a
density of 6,000 cells/well 24 h following transfection. A series
of concentrations of gefitinib were then added and incubated for 48
h. MTT was added with the final concentration of 5 mg/ml for 4 h.
Medium was replaced with 150 μl dimethyl sulfoxide and incubated
for 10 min. Optical density was measured at 490 nm using a Wellscan
MK3 ELISA reader (Labsystems, Dragon, Finland) in order to
determine the IC50 of gefitinib. IgIC50
served as a standard control, IgIC50 = Xm − I [P − (3 −
Pm − Pn) / 4] m where Xm is the numerical value of the maximum
designed concentration; I is the numerical value of the maximum
dose/adjacent doses, is the sum of positive reaction rates, Pm is
the maximum positive reaction rate and Pn is the smallest positive
reaction rate.
Fluorescence-activated cell sorting
(FACS)
Cells were dissociated into a single-cell suspension
and the apoptotic rate was assayed using flow cytometry
(Becton-Dickinson FACSCalibur flow cytometer; BD Biosciences,
Franklin Lakes, NJ, USA with Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) double staining according to the
manufacturer’s instructions (Hong Kong Jiamei Century
Biotechnology, Ltd., Hong Kong, Japan).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Differences between groups were assessed by one-way
analysis of variance using SPSS 19.0 statistical software package
(International Business Machines Corp., Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant difference
between values.
Results
Transfection efficiency
Fluorescent-labeled negative control siRNAs were
transfected into MKN-45 cells in order to monitor siRNA uptake. Six
hours post-transfection, transfection efficiency was analyzed using
fluorescence microscopy. As shown in Fig. 1, 80% transfection was achieved
using an siRNA: LipofectamineTM 2000 ratio of 40 pmol:1
μl, which was adopted throughout the study.
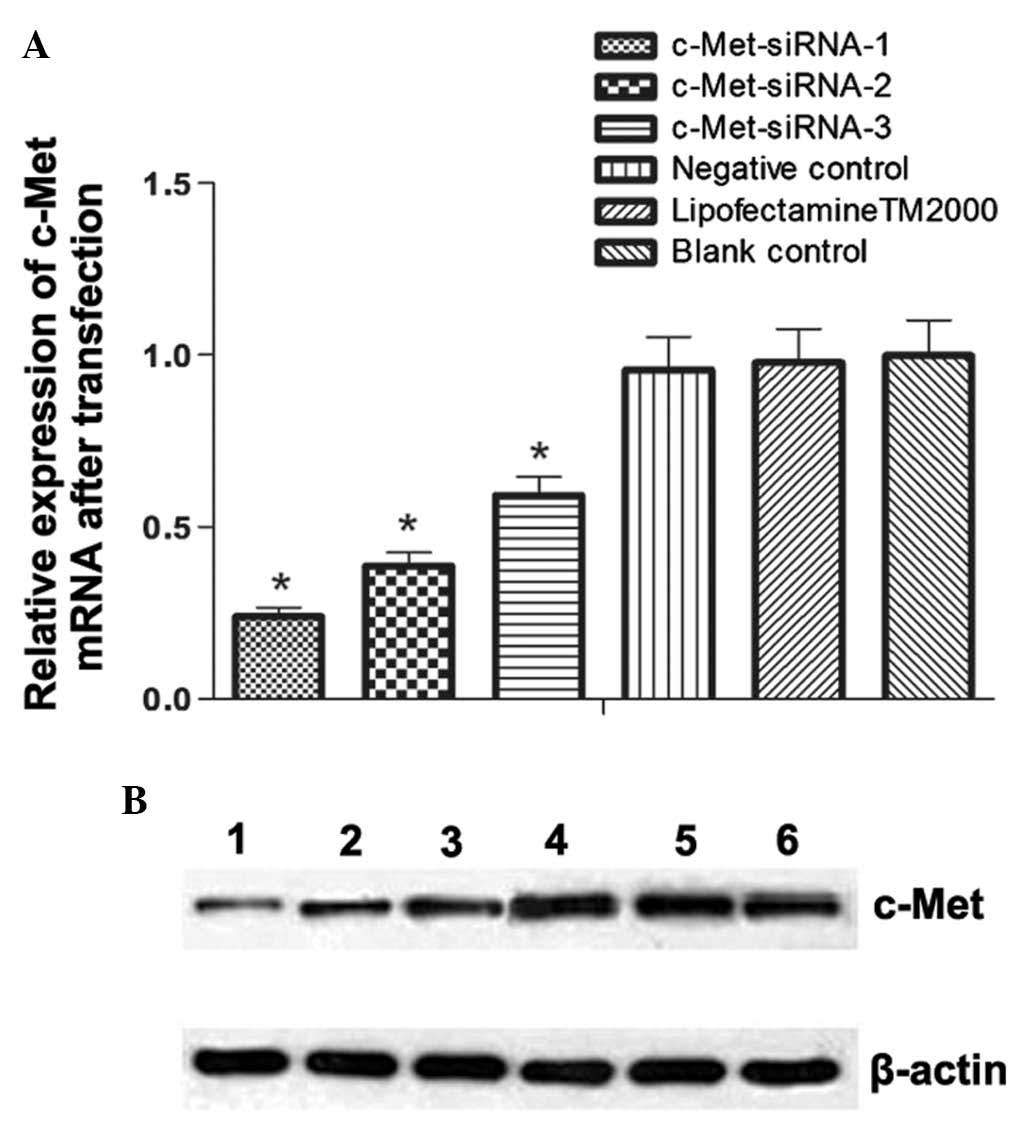
c-Met mRNA levels following
transfection
The expression of c-Met was calculated by
normalizing values relative to GAPDH. The results demonstrated that
all c-Met siRNA constructs significantly downregulated c-Met
expression (P<0.05); however, siRNA-c-Met-1 had the most obvious
effect (Fig. 2A).
c-Met protein expression following
transfection
c-Met protein expression was normalized to β-actin
and compared following transfection. The relative expression levels
of c-Met in siRNA groups 1, 2 and 3 were 0.258±0.021, 0.379±0.018
and 0.485±0.040, respectively; each siRNA group showed
significantly decreased c-Met protein expression compared to that
of the control (P<0.05). The strongest suppression of c-Met
expression was observed following transfection of c-Met-siRNA-1
(Fig. 2B). Accordingly,
c-Met-siRNA-1 was used in the subsequent functional experiment.
Apoptosis of MKN-45 cells prior to and
following c-Met gene silencing
Annexin V-FITC/PI double staining and FACS analysis
was used to evaluate the apoptotic rate of MKN-45 cells (Fig. 3). Early apoptotic cells are
Annexin-positive and PI-negative, and are therefore represented in
the lower-right quadrant of the photomicrographs; Annexin- and PI-
positive cells in the upper-right quadrant are late apoptotic or
necrotic cells. The total apoptotic rate was obtained by
calculating the sum of these two quadrants. The apoptotic rate of
MKN-45 cells (Table I) following
c-Met-siRNA transfection was significantly higher than that in
control siRNA-transfected or LipofectamineTM 2000
only-transfected cells (35.43±4.6% vs. 7.02±2.24 and 11.82±2.30%,
respectively; P<0.05); this therefore indicated that c-Met was
involved in MKN-45 apoptosis.
 | Figure 3Fluorescene-activated cell sorting and
Annexin V-FITC/PI double-labeled staining was used to determine the
apoptotic rate of MKN-45 cells. Sorting of MKN-45 cells following
transfection with (A) Normal siRNA control; (B)
LipofectamineTM 2000; and (C) c-Met-siRNA-1. Early
apoptotic cells are Annexin-positive and PI-negative, lower-right
quandrant of the photomicrographs; late apoptotic or necrotic cells
are Annexin- and PI- positive cells, upper-right quadrant. FITC,
fluorescein isothiocyanate; PI, propidium iodide; siRNA, small
interfering RNA; Quad, quadrant; UL, upper left; UR, upper right;
LL, lower left; LR, lower right. |
 | Table IApoptotic rates of MKN-45 cells
following transfection. |
Table I
Apoptotic rates of MKN-45 cells
following transfection.
| Group | Apoptotic rate
(%) | P-value |
|---|
| c-Met-siRNA-1 | 35.43±4.6 | <0.05 |
|
LipofectamineTM 2000 | 11.82±2.30 | >0.05 |
| Normal control | 7.02±2.24 | |
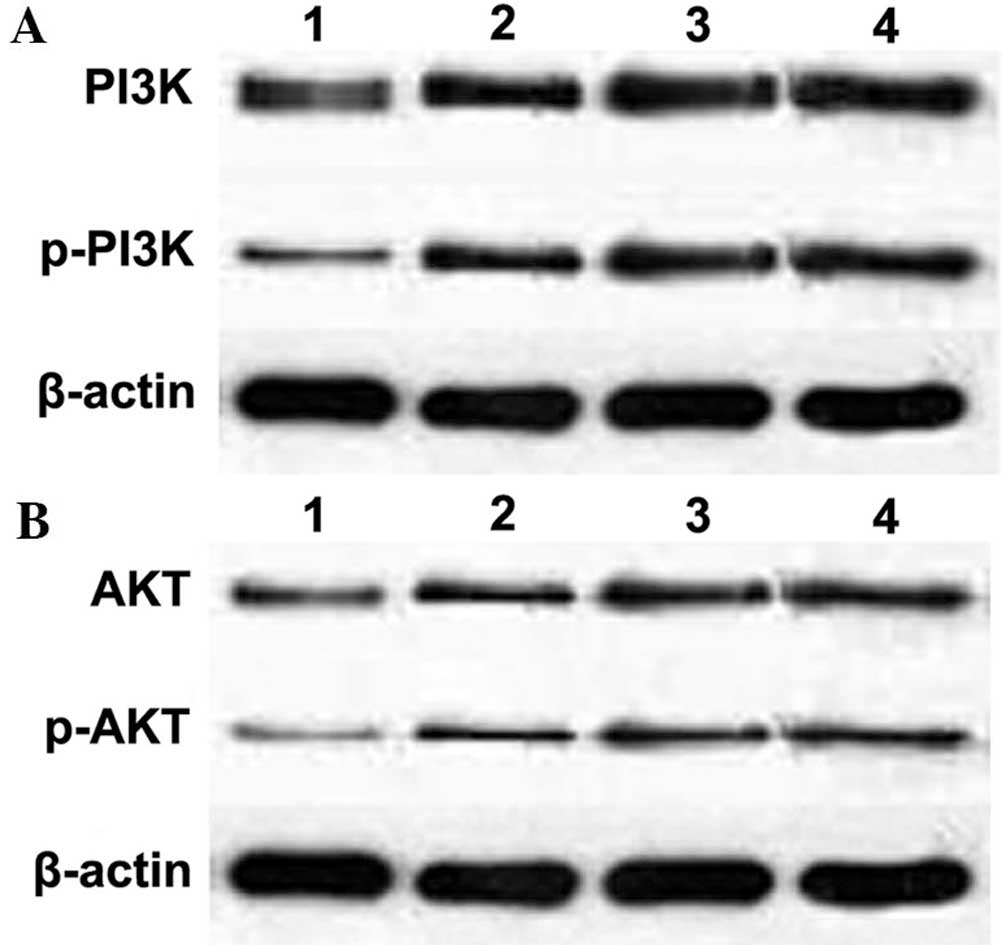
Impact of c-Met knockdown on PI3K and AKT
signaling
PI3K and AKT are important downstream genes of c-Met
(21). Following transfection, the
relative mRNA expression of PI3K and AKT was examined using
quantitative PCR. As shown in Table
II, expression levels of PI3k and AKT showed no significant
difference to those of the groups transfected with control siRNA
(P>0.05). In addition, protein expression levels of PI3K and AKT
were not altered by c-Met knockdown (P>0.05) (Table III; Fig. 4). By contrast, protein levels of
p-PI3K and p-AKT were significantly downregulated compared to those
of the group transfected with control siRNA (Table IV; Fig. 4). These results therefore indicated
that c-Met signaling was attenuated by the downregulation of c-Met
transcription.
| Table IIPI3K/AKT mRNA levels following c-Met
knockdown. |
Table II
PI3K/AKT mRNA levels following c-Met
knockdown.
| Group | PI3K mRNA | P-value | AKT mRNA | P-value |
|---|
| c-Met-siRNA-1 | 0.450±0.017 | >0.05 | 0.215±0.018 | >0.05 |
| Negative control | 0.455±0.030 | >0.05 | 0.225±0.016 | >0.05 |
|
LipofectamineTM 2000 | 0.453±0.021 | >0.05 | 0.219±0.025 | >0.05 |
| Blank control | 0.465±0.025 | - | 0.229±0.024 | - |
| Table IIIPI3K/AKT protein levels following
c-Met knockdown. |
Table III
PI3K/AKT protein levels following
c-Met knockdown.
| Group | PI3K protein | P-value | AKT protein | P-value |
|---|
| c-Met-siRNA-1 | 0.466±0.050 | >0.05 | 0.200±0.030 | >0.05 |
| Negative
control | 0.475±0.020 | >0.05 | 0.218±0.050 | >0.05 |
|
LipofectamineTM 2000 | 0.470±0.030 | >0.05 | 0.215±0.010 | >0.05 |
| Blank control | 0.477±0.010 | - | 0.229±0.020 | - |
| Table IVp-PI3K and p-AKT levels following
c-Met knockdown. |
Table IV
p-PI3K and p-AKT levels following
c-Met knockdown.
| Group | p-PI3K protein | P-value | p-AKT protein | P-value |
|---|
| c-Met-siRNA-1 | 0.190±0.020 | <0.05 | 0.125±0.040 | <0.05 |
| Negative
control | 0.380±0.020 | >0.05 | 0.195±0.020 | >0.05 |
|
LipofectamineTM 2000 | 0.388±0.035 | >0.05 | 0.188±0.020 | >0.05 |
| Blank control | 0.395±0.030 | - | 0.198±0.030 | - |
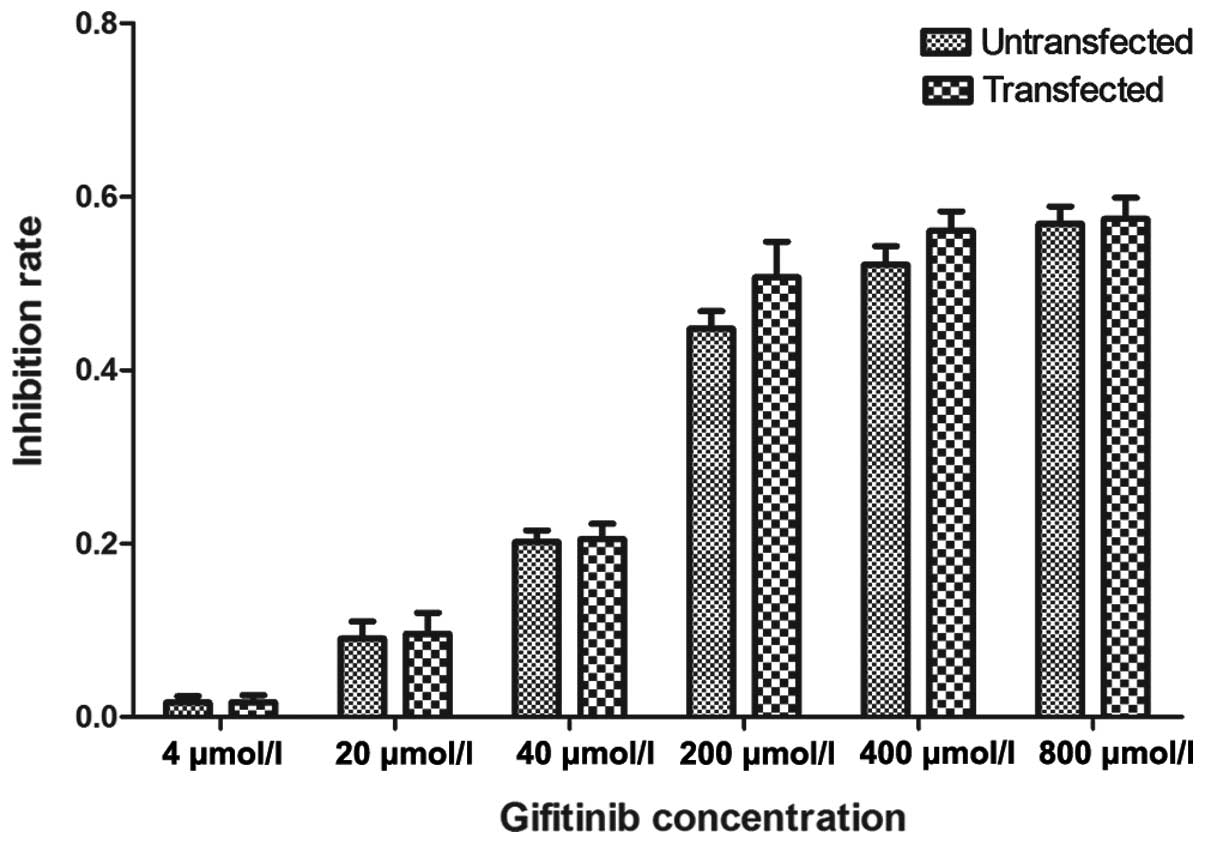
IC50 of gefitinib in MKN-45
cells
The IC50 values of gefitinib on MKN-45
cells were determined following transfection using an MTT assay
(Table V), with the
IgIC50 values in the un-transfected and transfected
cells being 2.595±0.010 and 2.566±0.206, respectively.
Un-transfected and transfected cells demonstrated comparable
responses to gefitinib (Fig. 5),
indicating that drug-sensitivity was independent of c-Met
expression. siRNA transfection did not affect the inhibition rate
of MKN-45 cells following treatment with different concentrations
of gefitinib.
| Table VIC50 of gefitinib in
MNK-45 cells. |
Table V
IC50 of gefitinib in
MNK-45 cells.
| Group | IC50
(μmol/l) |
IgIC50 | P-value |
|---|
| Untransfected | 393.650±8.594 | 2.595±0.010 | 0.136 |
| Transfected | 368.648±17.368 | 2.566±0.206 | |
Discussion
EGFR and c-Met are tyrosine kinase receptors that
share downstream signaling transduction pathways, including the
mitogen-activated protein kinase and PI3K/AKT pathways (22). Gefitinib has been reported to
inhibit the intracellular tyrosine kinase domain of EGFR and be
effective in the treatment of NSCLC, which harbors mutations in
EGFR at exons 19 and 21. Gastric cancers highly express EGFR;
however, studies have shown that gastric cancers exhibited minimal
sensitivity or were unresponsive to gefitinib. Certain studies have
indicated that high c-Met expression may be responsible for
acquired resistance to EGFR-TKI in lung cancer (23–26)
and c-Met amplification may activate the receptor tyrosine-protein
kinase, human epidermal growth factor receptor 3 (ErbB3;
HER3)-dependent PI3K/AKT signaling and therefore result in
gefitinib resistance (27).
A previous study (8) reported that the HGF inhibitor NK4
enhanced the sensitivity of peritoneally spread gastric cancer to
gefitinib in vivo (8). HGF
is a ligand for the c-Met receptor, therefore suggesting that
abnormal c-Met expression may alter the sensitivity of gefitinib to
EGFR-TKI. The aim of the present study was to explore whether
downregulation of c-Met enhanced the sensitivity of gastric cancer
to gefitinib.
Numerous studies have demonstrated that blocking
c-Met signaling inhibited cell proliferation and invasion as well
as induced apoptosis (28–31). In addition, the use of siRNAs to
knockdown c-Met in gastric cancer cells was reported to trigger
apoptosis (31). The results of
the present study revealed that the apoptotic rate of MKN-45 cells
transfected with c-Met siRNA was significantly higher than that of
the control groups; this therefore confirmed that c-Met mediated
apoptosis in gastric cancer.
In the present study, three siRNA oligos for c-Met
were transfected into MKN-45 cells, which resulted in high rates of
c-Met inhibition (P<0.05), indicating successful c-Met
downregulation. c-Met-siRNA-1 showed the most effective results and
therefore was used for the subsequent experiments.
Previous studies have shown that the IC50
value of gefitinib on gastric cancer cells was 400 μmol/l;
therefore, throughout the present study, this value was used as a
guideline to test the inhibition rate of different doses of
gefitinib on MKN-45 cells (18).
The results revealed that the IC50 values of gefitinib
were unchanged, despite c-Met knockdown. It was speculated that
MKN-45 cell resistance to gefitinib was not dependent on c-Met
expression and the molecular mechanism of EGFR-TKI-resistance in
MKN-45 cells remains to be elucidated. In c-Met-addicted gastric
cancer, the inhibition or gene silencing of c-Met may be an
effective approach for reversing drug resistance. However, EGFR and
HER activation were reported to induce drug resistance (32), suggesting that there is complex
cross-talk between EGFR and c-Met (33).
Phase II clinical studies on NSCLC patients
demonstrated that the combination of the c-Met inhibitor tivantinib
(ARQ197) and erlotinib did not increase the overall survival rate;
however, in patients with KRAS mutations, tivantinib was shown to
increase the beneficial effect of erlotinib (34). In the present study, the
hypothesized increase of gefitinib sensitivity in c-Met-silenced
MKN-45 cells was not observed. In addition, mRNA and protein levels
of the downstream targets of c-Met, PI3K and AKT were not
significantly altered following c-Met knockdown; by contrast, siRNA
transfection was shown to attenuate p-PI3K and p-AKT levels, which
may explain the unaltered sensitivity to gefitinib in MKN-45 cells.
However, the precise molecular mechanism of gastric cancer
insensitivity to gefitinib remains to be elucidated.
In NSCLC, c-Met was markedly associated with
high-grade amplification that conferred acquired resistance to
EGFR-TKIs in EGFR-mutant cancers. In gastric cancer, EGFR and HER3
activation was reported to result in acquired resistance to c-Met
inhibitors. The results of the present study indicated that c-Met
downregulation promoted MKN-45 cell apoptosis; however, it did not
increase the sensitivity of gastric cancer cells to gefitinib. In
conclusion, these results suggested that c-Met expression was not
associated with or had little effect on the resistance of gastric
cancer cells to gefitinib. Further studies are required in order to
determine whether KRAS gene mutations or other signaling molecules
downstream of EGFR and c-Met are involved in mediating gefitinib
resistance.
References
|
1
|
Lordick F and Siewert JR: Recent advances
in multimodal treatment for gastric cancer: a review. Gastric
Cancer. 8:78–85. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bang YJ, Van Cutsem E, Feyereislova A, et
al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): a phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Liu H, Zhu R, Hinterdorfer P,
Zhang B and Tang J: Single molecular dissection of the ligand
binding property of epidermal growth factor receptor. Analyst.
138:5325–5331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liakakos T, Xeropotamos N, Ziogas D and
Roukos D: EGFR as a prognostic marker for gastric cancer. World J
Surg. 32:1225–1226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Janjigian YY, Tang LH, Coit DG, et al: MET
expression and amplification in patients with localized gastric
cancer. Cancer Epidemiol Biomarkers Prev. 20:1021–1027. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang TJ, Wang JY, Lin SR, Lian ST and
Hsieh JS: Overexpression of the c-met protooncogene in human
gastric carcinoma - correlation to clinical features. Acta Oncol.
40:638–643. 2001. View Article : Google Scholar
|
|
7
|
Drebber U, Baldus SE, Nolden B, et al: The
overexpression of c-met as a prognostic indicator for gastric
carcinoma compared to p53 and p21 nuclear accumulation. Oncol Rep.
19:1477–1483. 2008.PubMed/NCBI
|
|
8
|
Amemiya H, Menolascino F and Peña A: Role
of the expression of c-Met receptor in the progression of gastric
cancer. Invest Clin. 51:369–380. 2010.(In Spanish).
|
|
9
|
Namiki Y, Namiki T, Yoshida H, et al:
Preclinical study of a ‘tailor-made’ combination of NK4-expressing
gene therapy and gefitinib (ZD1839, Iressa) for disseminated
peritoneal scirrhous gastric cancer. Int J Cancer. 118:1545–1555.
2006. View Article : Google Scholar
|
|
10
|
Rojo F, Tabernero J, Albanell J, et al:
Pharmacodynamic studies of gefitinib in tumor biopsy specimens from
patients with advanced gastric carcinoma. J Clin Oncol.
24:4309–4316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dragovich T, McCoy S, Fenoglio-Preiser CM,
et al: Phase II trial of erlotinib in gastroesophageal junction and
gastric adenocarcinomas: SWOG 0127. J Clin Oncol. 24:4922–4927.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng Y, Asara JM and Tyner AL:
Protein-tyrosine kinase 6 promotes peripheral adhesion complex
formation and cell migration by phosphorylating p130 CRK-associated
substrate. J Biol Chem. 287:148–158. 2012. View Article : Google Scholar :
|
|
13
|
Chell V, Balmanno K, Little AS, et al:
Tumour cell responses to new fibroblast growth factor receptor
tyrosine kinase inhibitors and identification of a gatekeeper
mutation in FGFR3 as a mechanism of acquired resistance. Oncogene.
32:3059–3070. 2013. View Article : Google Scholar
|
|
14
|
Tian W, Chen J, He H and Deng Y: MicroRNAs
and drug resistance of breast cancer: basic evidence and clinical
applications. Clin Transl Oncol. 15:335–342. 2013. View Article : Google Scholar
|
|
15
|
Karamouzis MV, Konstantinopoulos PA and
Papavassiliou AG: Targeting MET as a strategy to overcome
crosstalk-related resistance to EGFR inhibitors. Lancet Oncol.
10:709–717. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Onitsuka T, Uramoto H, Nose N, et al:
Acquired resistance to gefitinib: the contribution of mechanisms
other than the T790M, MET, and HGF status. Lung Cancer. 68:198–203.
2010. View Article : Google Scholar
|
|
17
|
Bean J, Brennan C, Shih JY, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao WG, Ma T, Li JF, et al: Effect of
gefitinib on radiosensitivity of gastric cancer cell lines. Chinese
Journal of Cancer. 26:1330–1335. 2007.(In Chinese).
|
|
19
|
Fushida S, Yonemura Y, Urano T, et al:
Expression of hepatocyte growth factor(hgf) and C-met gene in human
gastric-cancer cell-lines. Int J Oncol. 3:1067–1070.
1993.PubMed/NCBI
|
|
20
|
Casagrande G, te Kronnie G and Basso G:
The effects of siRNA-mediated inhibition of E2A-PBX1 on EB-1 and
Wnt16b expression in the 697 pre-B leukemia cell line.
Haematologica. 91:765–771. 2006.PubMed/NCBI
|
|
21
|
Li Y, Huang X, Zhong W, Zhang J and Ma K:
Ganglioside GM3 promotes HGF-stimulated motility of murine hepatoma
cell through enhanced phosphorylation of cMet at specific tyrosine
sites and PI3K/Akt-mediated migration signaling. Mol Cell Biochem.
382:83–92. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kao J, Sikora AT and Fu S: Dual EGFR and
COX-2 inhibition as a novel approach to targeting head and neck
squamous cell carcinoma. Curr Cancer Drug Targets. 9:931–937. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen HJ, Mok TS, Chen ZH, et al:
Clinicopathologic and molecular features of epidermal growth factor
receptor T790M mutation and c-MET amplification in tyrosine kinase
inhibitor-resistant Chinese non-small cell lung cancer. Pathol
Oncol Res. 15:651–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rosenzweig SA: Acquired resistance to
drugs targeting receptor tyrosine kinases. Biochem Pharmacol.
83:1041–1048. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Agarwal S, Zerillo C, Kolmakova J, et al:
Association of constitutively activated hepatocyte growth factor
receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in
non-small-cell lung cancer cells. Br J Cancer. 100:941–949. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kosaka T, Yamaki E, Mogi A and Kuwano H:
Mechanisms of resistance to EGFR TKIs and development of a new
generation of drugs in non-small-cell lung cancer. J Biomed
Biotechnol. 2011:1652142011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Que W and Chen J: Knockdown of c-Met
inhibits cell proliferation and invasion and increases
chemosensitivity to doxorubicin in human multiple myeloma U266
cells in vitro. Mol Med Rep. 4:343–349. 2011.PubMed/NCBI
|
|
29
|
Wang ZX, Lu BB, Yang JS, Wang KM and De W:
Adenovirus-mediated siRNA targeting c-Met inhibits proliferation
and invasion of small-cell lung cancer (SCLC) cells. J Surg Res.
171:127–135. 2011. View Article : Google Scholar
|
|
30
|
Xie B, Xing R, Chen P, et al:
Down-regulation of c-Met expression inhibits human HCC cells growth
and invasion by RNA interference. J Surg Res. 162:231–238. 2010.
View Article : Google Scholar
|
|
31
|
Shinomiya N, Gao CF, Xie Q, et al: RNA
interference reveals that ligand-independent met activity is
required for tumor cell signaling and survival. Cancer Res.
64:7962–7970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Teng L and Lu J: cMET as a potential
therapeutic target in gastric cancer (Review). Int J Mol Med.
32:1247–1254. 2013.PubMed/NCBI
|
|
33
|
Wheeler DL, Huang S, Kruser TJ, et al:
Mechanisms of acquired resistance to cetuximab: role of HER (ErbB)
family members. Oncogene. 27:3944–3956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sequist LV, von Pawel J, Garmey EG, et al:
Randomized phase II study of erlotinib plus tivantinib versus
erlotinib plus placebo in previously treated non-small-cell lung
cancer. J Clin Oncol. 29:3307–3315. 2011. View Article : Google Scholar : PubMed/NCBI
|