Introduction
Immune surveillance by dendritic cells (DCs) is
important in the mediation of innate and acquired immunity. To
activate T lymphocytes following infection, DCs internalize and
eliminate invading microbes and exhibit pathogen-derived antigens
on their surface in the form of major histocompatibility complex
(MHC) molecules (1,2). Salmonella is a facultative
intracellular pathogen and the successful engagement of T
lymphocytes is required for the initiation of protective immunity
(3). The activity of
pathogen-specific T cells significantly reduces the levels of
infection and colonization by virulent bacteria, via direct
elimination of bacteria or by enhancing the innate immune response.
However, certain microbial pathogens have evolved molecular
mechanisms aimed at interfering with DC activity in order to evade
the specific adaptive immune response. These mechanisms are termed
‘immune evasion’ and significantly influence pathogen virulence
(4,5).
The action of multiple pathogenic factors of
Salmonella, encoded by the chromosome and the plasmid, is
required during infection (6). The
significance of large plasmids in determining the virulence of
Salmonella has been the subject of numerous studies
(7–9). In a previous study by our group, the
98.6 mDa, 150 kb Salmonella enterica serotype Typhi
(S. Typhi) plasmid, designated as pRST98,
was isolated during a survey of multidrug-resistant S. Typhi
strains, and is known to mediate bacterial multidrug resistance to
chloramphenicol, streptomycin, trimethoprin, sulphonamide,
gentamicin, neomycin, kanamycin, cephalosporin, ampicillin,
carbencillin and tetracycline. Patients infected with S.
Typhi carrying pRST98 exhibited more severe symptoms and
complications with high rates of mortality (10). However, only one plasmid existed in
the isolates. It was therefore hypothesized that pRST98
may be a mosaic-like structure responsible for not only drug
resistance, but also increased bacterial virulence.
On the plasmids of all pathogenic Salmonella
spp., except S. Typhi, there is a highly conserved 8 kb
region which encodes virulence phenotypes, and this gene is
designated as the Salmonella plasmid virulence (spv) gene
(11). The virulence genes on
pRST98 were previously detected by spv-specific
polymerase chain reaction and DNA analysis, and the results
indicated that pRST98 shared 99.8% homology with spv
(10). This study was the first,
to the best of our knowledge, to reveal that spv homologous genes
existed on the pRST98 plasmid. These early studies
demonstrated that pRST98 enhanced the virulence of host
bacteria by increasing survival and mediating macrophage apoptosis
via the mitochondrial pathway (12). However, the phenotype remained of
interest, since the mechanism of pRST98-increased
bacterial virulence has remained to be elucidated.
The virulence of Salmonella is frequently
serovar-specific, for example, S. Typhi causes typhoid fever
in humans, but no disease is associated with S. Typhi
infection of mice. In the present study, in order to determine
whether pRST98 was involved in the specific immune
response in vivo, pRST98 was transferred into
S. Typhimurium χ3337 (a virulence plasmid-cured strain) and
the conjugative strain χ3337/pRST98 was used in a murine
salmonellosis model. The effect of pRST98 on important
functions of murine DCs, including maturation as well as survival
and cytokine production, was evaluated, as well as its effects on T
cells. The results of the present study may aid the elucidation of
the mechanism underlying S. Typhi immune evasion.
Materials and methods
Salmonella strains and growth
conditions
In the present study, S. Typhimurium SR11
χ3337 (a nalidixic acid-resistant, virulence plasmid-cured
derivative of χ3306) was used as a negative control. This strain
was provided by Dr Roy Curtiss III (Arizona State University,
Phoenix, AZ, USA). The multidrug-resistant S. Typhi strains
harboring pRST98 were obtained from patients’ blood
during a typhoid fever outbreak in Suzhou, China between 1987 and
1992. Plasmid-free Escherichia coli K12W1485 Rifr F-Lac(+)
(E. coli K12W1485) with a rifampicin resistance gene on the
chromosome and S. Typhimurium SR11 χ3337 were used as
recipients to create the conjugant χ3337/pRST98. E.
coli V517 (54.4, 7.3, 5.6, 5.2, 4.0, 3.0, 2.7 and 2.1 Kb) and
Shigella flexneri 24570 (159.6, 4.0 and 3.0 Kb) harboring
standard plasmids were used as size markers. All strains were grown
to mid-logarithmic phase at 37°C in Luria-Bertani (LB) broth
(Shanghai Kemin Biotechnology Co. Ltd., Shanghai, China) and
quantified spectrophotometrically by determining optical density
(OD) at 600 nm. Strains were subsequently centrifuged at 2,300 xg
for 5 min and resuspended in RPMI-1640 medium (Gibco-BRL) without
antibiotics prior to their addition to cells.
Conjugal transfer of pRST98
and electrophoretic analysis
The conjugal test was performed as follows:
pRST98 was transferred from the clinical isolated
multi-drug-resistant S. Typhi to a plasmid-free laboratory
strain of E. coli K12W1485, and Shigella and
Salmonella (SS) agar plates containing rifampicin (100
μg/ml) and chloramphenicol (20 μg/ml) (Beijing Biodee Biotechnology
Co., Ltd., Beijing, China) were used. E. coli is able to
ferment lactose, so that it can therefore be easily identified on
an SS agar plate. pRST98 was transferred from E.
coli K12W1485 to S. Typhimurium χ3337. E. coli
K12W1485 receiving pRST98 (conjugant
pRST98/E. coli K12W1485) was used as the donor,
while S. Typhimurium χ3337 was the recipient. The two
strains of bacteria were grown for 16 h at 37°C in LB broth and
mixed well by transferring 0.1 ml of each into 3 ml fresh LB broth
for 4 h at 37°C. The mixture was centrifuged at 2,300 xg for 5 min
and resuspended in normal saline. 0.1 ml of the suspension was
transferred to a casein hydrolysate agar plate and grown for 16 h
at 37°C. The lawn was collected and the serial dilution tube test
was conducted. 0.1 ml of the suspension was transferred to an SS
agar plate containing rifampicin (100 μg/ml), chloramphenicol (20
μg/ml) and ampicillin (25 μg/ml) (Beijing Biodee Biotechnology Co.,
Ltd., Beijing, China). The colonies producing hydrogen sulfide were
selected to be cultured a second time on the same selective agar,
reactivated on LB agar plates and labeled as conjugant
χ3337/pRST98. Plasmid DNA extraction and electrophoretic
analysis were applied using routine methods (13).
Mice
BALB/c mice were purchased from the Experimental
Animal Center (Chinese Academy of Science, Shanghai Laboratory
Animal Center, Shanghai, China). Mice were housed in a
pathogen-free animal facility and maintained under standard
environmental conditions (room temperature 24±1°C; relative air
humidity 50±2%; a 12-h light/dark cycle; fed standard rodent diet
and water ad libitum). Mice were used aged 8–10 weeks
(weight 30–40g) and animal experiments were performed following
protocols approved by the institutional animal care and use
committee of Bengbu Medical College.
Antibodies
Antibodies were obtained from eBioscience, Inc. (San
Diego, CA, USA) unless otherwise stated. The
fluorochrome-conjugated anti-mouse monoclonal antibodies (mAbs)
used in the present study were as follows: anti-CD11c fluorescein
isothiocyanate (FITC; N418), anti-MHC Class II(I-A) phycoerythrin
(PE)-Cy5 (NIMR-4), anti-CD40 (PE; 1C10), anti-CD80 PE (16-10A1),
anti-CD86 PE (PO3.1), anti-CD62L FITC (MEL-14), anti-CD44 PE (IM7),
anti-CD3e peridinin chlorophyll (PerCP)-Cy5.5 (145-2C11), anti-CD8a
allophycocyanin (APC) (53-6.7), anti-interleukin (IL)-12 PE (C
17.8), anti-interferon (IFN)-γ PE (XMG1.2), anti-tumor necrosis
factor (TNF)-α PE (TN3-19) and all isotype-matched control mAbs
were from Caltag Laboratories (Invitrogen Life Technologies,
Carlsbad, CA, USA).
Cell culture and infection
For the generation of bone marrow-derived DCs, bone
marrow was flushed from the tibias and femurs of 8 to 10-week-old
mice. Following red cell lysis with lysing solution (BD
Biosciences) and washing, the progenitor cells were cultured in
six-well plates at 1×106/well in RPMI containing 10%
fetal calf serum supplemented with 10 ng/ml recombinant mouse
granulocyte-macrophage colony-stimulating factor (GM-CSF) and 10
ng/ml IL-4 (Biomics Biotechnology Co., Ltd., Nantong, China). These
cells were cultured for six days at 37°C in 5% CO2, and
fresh medium and cytokines were added on days two and four. On day
six, the DCs were collected for use in the infection
experiments.
For infection of DCs, 1.5×105 cells
(invasion assays), 4×105 cells (apoptosis assays) or
106 cells (co-stimulatory molecule expression analysis)
were seeded in 24- or six-well plates in medium without antibiotics
and infected with the corresponding S. Typhimurium strains
at a multiplicity of infection (MOI) of 20 bacteria per DC.
Bacteria were centrifuged onto DC at 400 xg for 5 min. Following 1
h of infection, 100 μg/ml amikacin (AMK; Beijing Biodee
Biotechnology Co.) was added to exterminate extracellular bacteria.
Then, the cells were washed and further incubated with medium
containing 10 μg/ml AMK to prevent extracellular growth of bacteria
released from the infected DC for an additional, indicated length
of time.
Bacterial invasion and DC viability
assays
S. Typhimurium strains were grown to the
mid-log growth phase, at which the bacteria are most invasive. DCs
were infected in triplicate as aforementioned. To evaluate
bacterial infectivity, AMK-treated DCs were permeabilized for 30
min with 0.1% Triton X-100 (Shanghai Fushen Biotechnology Co.,
Ltd., Shanghai, China) in phosphate-buffered saline (PBS; Hefei
Bomei Biotechnology Co., Ltd., Hefei, China). Lysates of 2 h were
plated on agar plates for overnight incubation at 37°C. At the same
time-point, DC viability was analyzed using a FACSCalibur flow
cytometer (BD Biosciences), using staining of fragmented DNA with
propidium iodide and labeling of cells with fluorescently tagged
Annexin V (BD Biosciences), which binds to phosphatidylserine
present in the outer membrane of cells during early-stage
apoptosis.
Detection of cell-surface markers
The infected cells were harvested at 24 h
post-infection and incubated with mouse anti-FcRII/III mAb for 10
min at 4°C. Fluorochrome-conjugated antibodies were added to cells,
which were incubated on ice for 30 min and then washed twice with
wash buffer. Cells were kept on ice and analyzed by flow cytometry
and CellQuest™ version 3.3 software (BD Biosciences, Franklin
Lakes, NJ, USA) and WinMDI 2.8 (winmdi.software.informer.com/2.8/).
Bacterial infection of mice
S. Typhimurium strains were grown overnight
in LB medium, harvested in the logarithmic phase (OD600=0.8–1.0),
washed once in PBS and resuspended in PBS with 15% glycerol (Hefei
Bomei Biotechnology Co.). The cultures were stored in aliquots at
−80°C. The bacterial titers were determined by plating serial
dilutions on LB agar plates. For infection, aliquots were thawed
and appropriately diluted in PBS. In experiments in which DC and
T-cell activation were analyzed, mice were infected intravenously
with 500 organisms and sacrificed by cervical dislocation on the
fifth or tenth day following infection. In experiments in which the
intracellular cytokines of splenic DCs were studied, mice were
administered a single intravenous injection of 107
bacteria and sacrificed 24 h subsequently. Total splenocytes were
subjected to flow-cytometric analysis.
Flow-cytometric analysis of spleen
cells
Single-cell suspensions were obtained by passing
spleens through stainless steel meshes followed by erythrocyte
lysis. Cells (106) were incubated with anti-FcRII/III
mAb to block unspecific antibody binding. Following 10 min of
incubation, cells were stained with FITC-conjugated mouse
anti-CD11c, PE-conjugated mouse anti-CD40, PE-conjugated mouse
anti-CD80, PE-conjugated mouse anti-CD86, PerCP-Cy5.5-conjugated
mouse anti-CD3e, APC-conjugated mouse anti-CD8a, FITC-conjugated
mouse anti-CD62L and PE-conjugated mouse anti-CD44. Following 30
min on ice, cells were washed and analyzed.
Intracellular cytokine expression was analyzed in a
splenocyte preparation that had been incubated for 5 h in the
presence of GolgiPlug™ (brefeldin A solution, 10 μg/ml; BD
Biosciences). Cells were then washed with PBS supplemented with
0.1% bovine serum albumin (Beijing Leigen Biotechnology Co., Ltd.,
Beijing, China) and were surface stained for CD11c to identify
splenic DCs and for co-stimulatory molecules. Cells were
subsequently fixed, permeabilized and stained with anti-IL-12 PE,
anti-IFN-γ PE or anti-TNF-α PE using the cytofix/cytoperm kit
(eBioscience, Inc.) according to the manufacturer’s instructions.
Flow cytometry was performed on splenocytes from individual mice
stimulated separately.
Assessment of bacterial burden
Single-cell suspensions were obtained from the
spleens of infected mice in RPMI-1640. An aliquot of the suspension
was lysed and evaluated to determine the number of viable bacteria.
Colony-forming units (CFU) were determined by plating serial
dilutions of tissue homogenates on LB agar plates.
Statistical analysis
Values are presented as the mean ± standard
deviation. Data obtained from independent experiments were analyzed
using Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference between values. The
statistical software package SPSS 15.0 (SPSS, Inc., Chicago, IL,
USA) was used for all data analyses.
Results
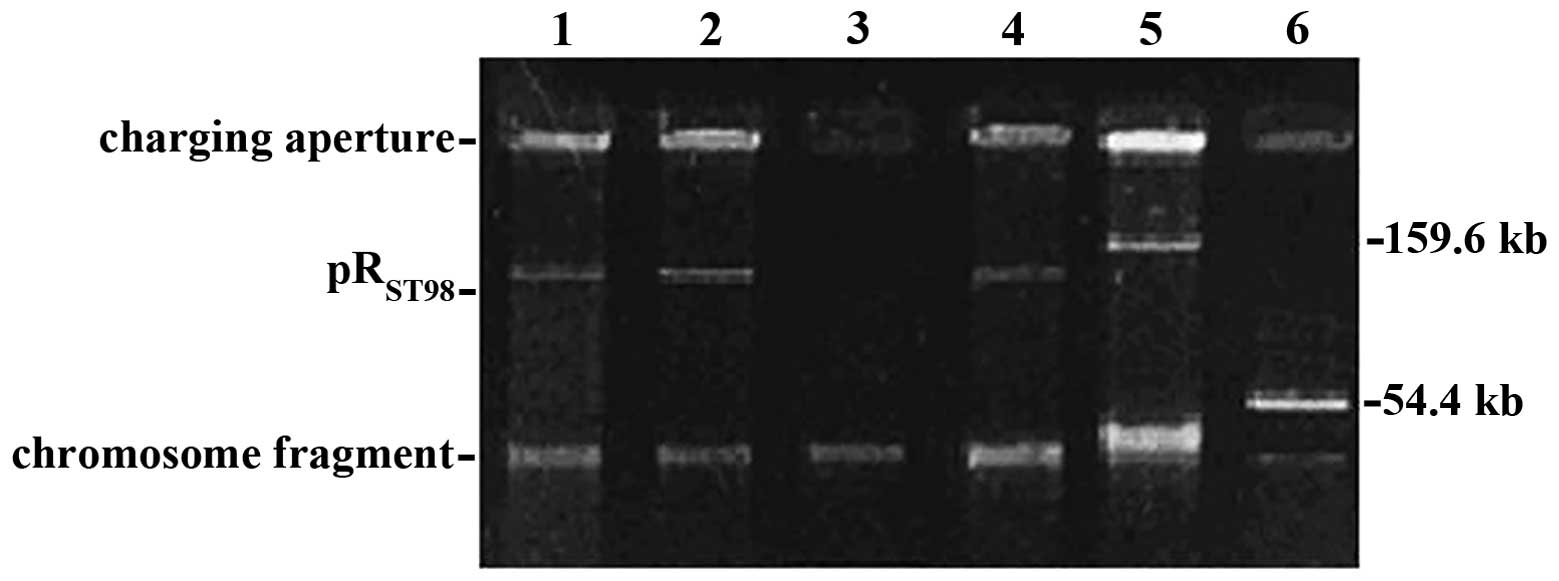
Conjugal transfer of plasmid
pRST98
S. Typhi causes typhoid fever in humans only;
therefore, S. Typhimurium was employed to infect mice in
order to study the pathogenesis of pRST98 in
vivo. pRST98 was initially transferred from the
multi-drug-resistant S. Typhi to E. coli K12W1485 and
then transferred from E. coli K12W1485 to S.
Typhimurium χ3337 to create the conjugant χ3337/pRST98.
Electrophoretic analysis indicated that an additional 150-Kb
pRST98 plasmid was found in χ3337/pRST98,
compared to the recipient χ3337, which confirmed that
pRST98 was transferred into χ3337 (Fig. 1).
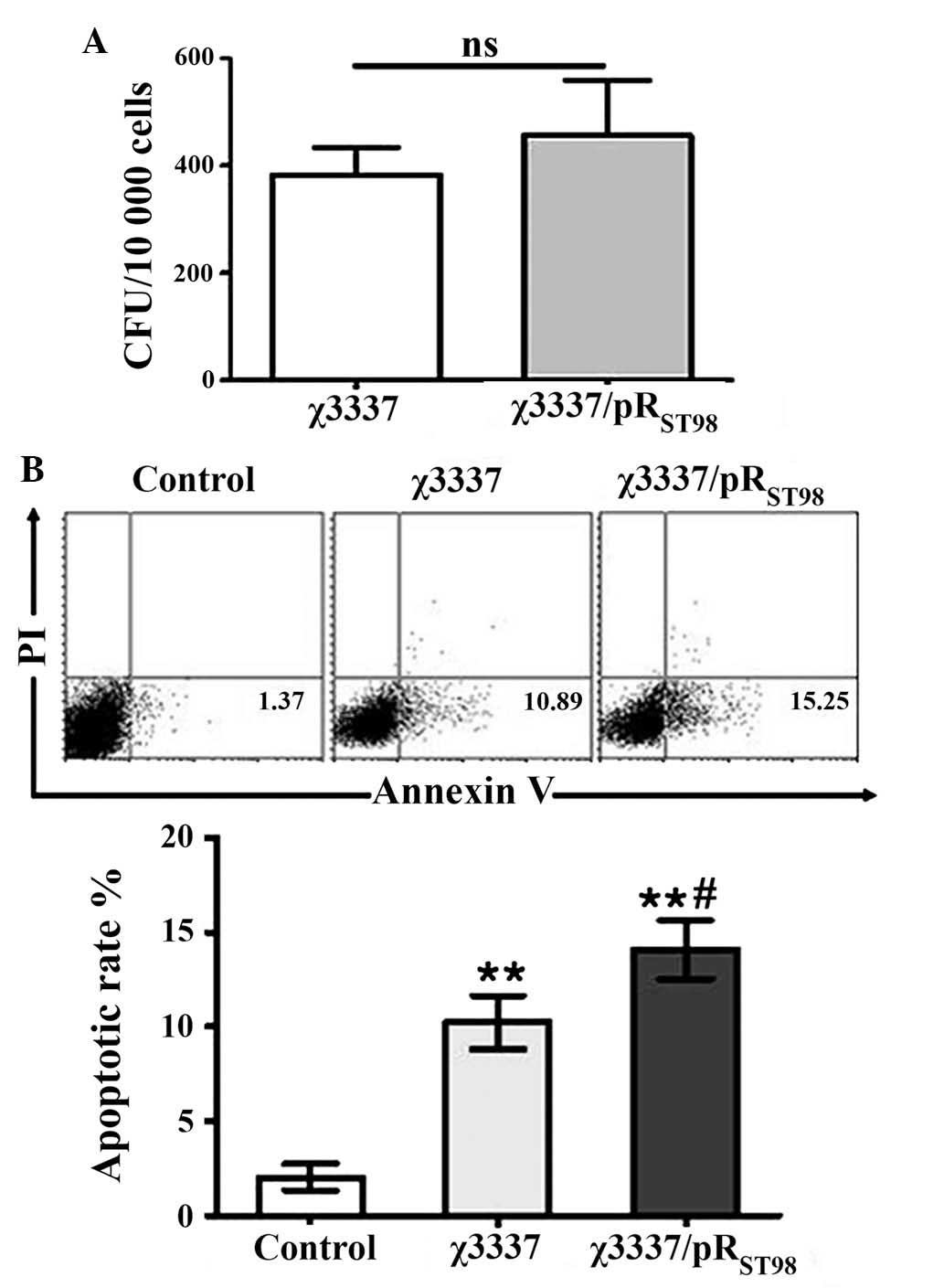
pRST98 does not influence
bacterial invasion
To date the role of pRST98 in bacterial
invasion has remained elusive. The invasion ability of S.
Typhimurium strains with or without pRST98 was compared.
The results demonstrated that S. Typhimurium χ3337 and
conjugant χ3337/pRST98 were equally invasive (Fig. 2A). This therefore indicated that
the expression of pRST98 did not increase the ability of
the bacteria to invade DCs.
DC viability is decreased following
infection with pRST98 S. Typhimurium
A previous study demonstrated that the infection of
macrophages or DCs with S. Typhimurium expressing the type
III secretion system resulted in early apoptosis (14). To assess whether pRST98
of S. Typhimurium induced mortality of murine DCs and
whether this may be involved in mediating the ability of DCs to
stimulate T cells, an apoptosis assay was performed using S.
Typhimurium-infected murine DCs. As illustrated in Fig. 2B, S. Typhimurium induced
apoptosis in murine DCs. Two hours after infection, S.
Typhimurium χ3337/pRST98-infected DCs exhibited a higher
rate of apoptosis than that of χ3337-infected cells. Therefore,
pRST98 may be an important factor involved in
Salmonella-induced cell death.
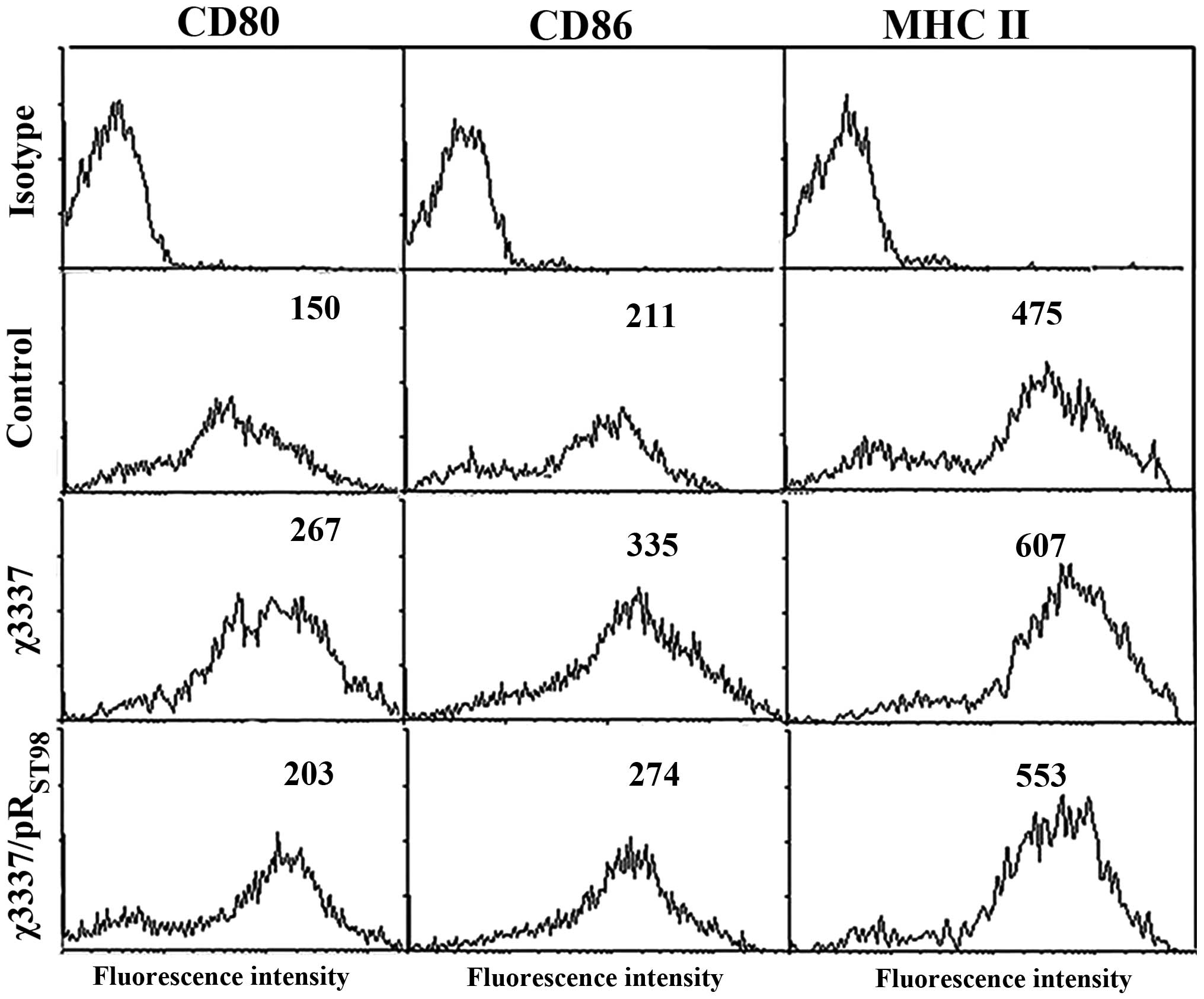
S. Typhimurium induces DC
maturation
To study the effects of Salmonella
pRST98 on DC maturation, cells were infected with the
corresponding bacterial strains, and the expression of
co-stimulatory molecules involved in T-cell activation was measured
by flow cytometric analysis following 24 h of further incubation to
facilitate the biosynthesis of novel surface molecules.
Unstimulated DCs are physiologically immature; however, following
infection with S. Typhimurium χ3337 or
χ3337/pRST98, DCs exhibited a mature phenotype,
manifested as an increase in the expression of co-stimulatory
molecules. Phenotypic changes following the infection of DCs with
corresponding strains are indicated in Fig. 3. Compared to S. Typhimurium
χ3337/pRST98- and χ3337-infected DCs exhibited increased
expression levels of all three surface molecules (CD80, CD86 and
MHC II), indicated by increased fluorescence intensity.
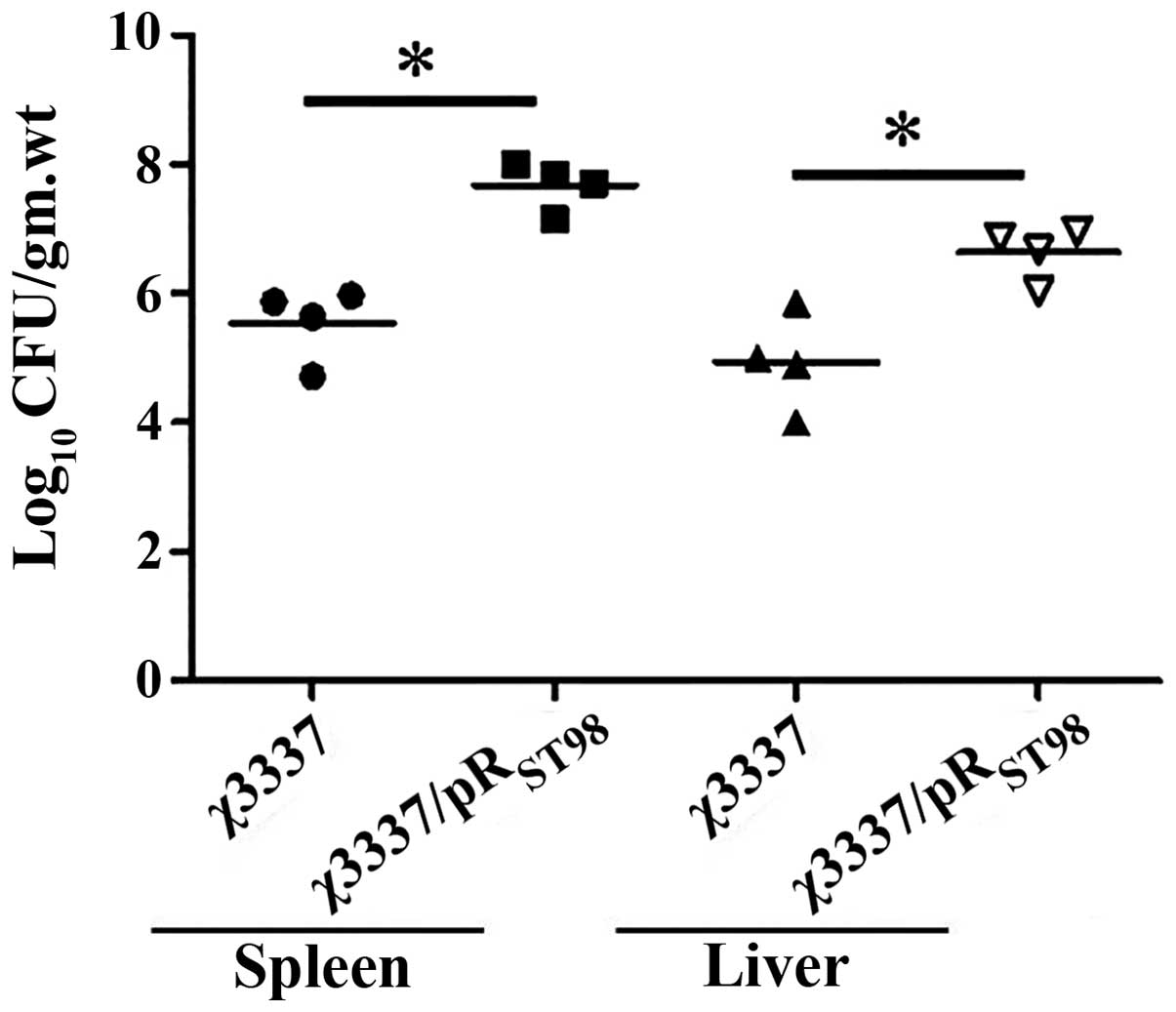
pRST98 enhances bacterial
burden following S. Typhimurium infection
BALB/c mice were intravenously infected with 500
bacteria of corresponding S. Typhimurium strains. Infection
resulted in massive expansion of bacteria in the spleen and liver.
The two bacterial strains exhibited the same course. The number of
bacteria recovered from χ3337/pRST98-infected mice was
significantly higher than that of χ3337-infected mice ten days
post-infection (Fig. 4). As
expected, the χ3337/pRST98 strain was virulence-enhanced
compared to the χ3337 strain, and the titer in the spleen and liver
was 107 bacteria/organ.
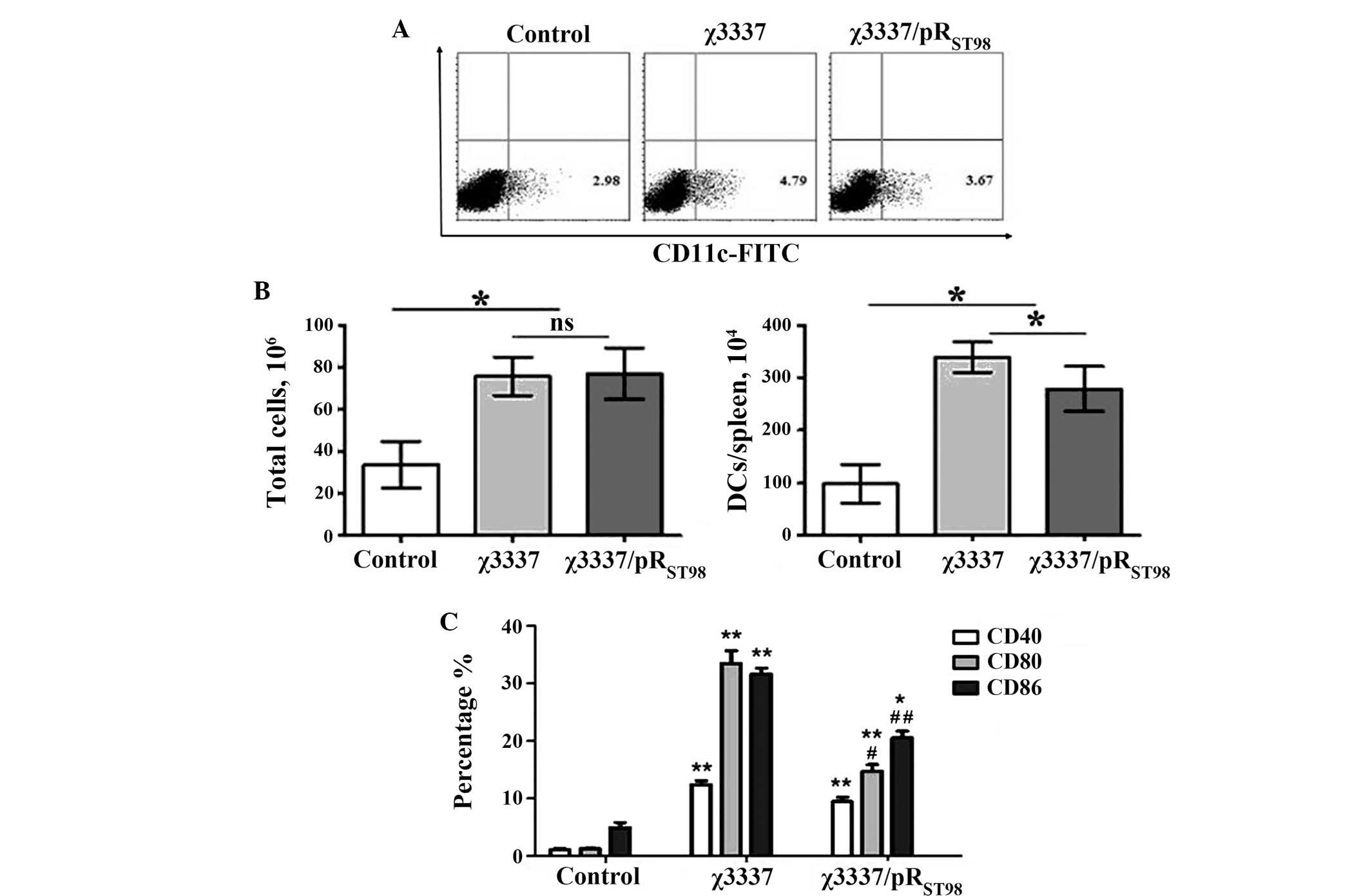
Suboptimal activation of DCs results from
infection with the conjugant, χ3337/pRST98
In vivo, Salmonella is usually
contained within splenic DCs, which are the most efficient
antigen-presenting cell (APC) types capable of stimulating naïve T
cells (15). The present study
therefore aimed to determine how the numbers and activation states
of splenic DCs were altered during infection with S.
Typhimurium. It was demonstrated that the overall number of splenic
DCs increased significantly five days following infection with
either strain, compared to that of mock-infected animals. A
significant difference in the number of DCs in the spleen was
observed between χ3337- and χ3337/pRST98-inoculated mice
(Fig. 5A and B). To determine
whether DC activation was distinct following wild-type χ3337 or the
conjugant χ3337/pRST98 infection, cell surface
expression of co-stimulatory molecules (CD40, CD80 and CD86) on
CD11c(+) splenic DCs, as well as their ability to secrete the
pro-inflammatory cytokines, was analyzed. Fig. 5C indicates that the extent of CD40,
CD80 and CD86 upregulation is distinct following χ3337 infection,
compared to that following χ3337/pRST98 immunization.
S. Typhimurium χ3337 infection induced greater
co-stimulatory molecule expression than χ3337/pRST98
immunization five days post-infection.
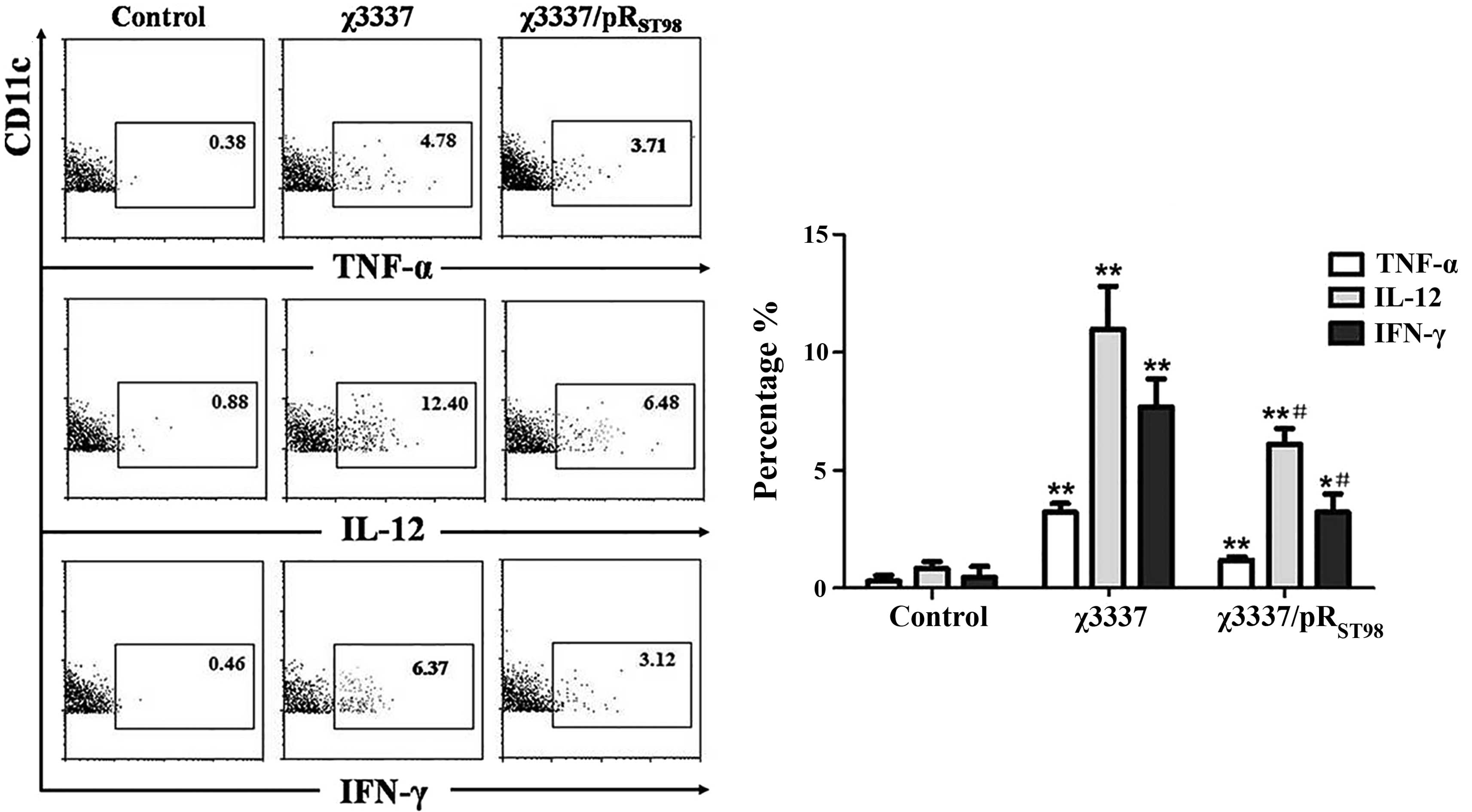
Furthermore, it was investigated whether the level
of T-cell function induced by splenic DCs was correlated with the
relative cytokine levels produced by these cells. As shown in
Fig. 6, CD11c(+) splenic DCs from
mice injected with S. Typhimurium produced IL-12, IFN-γ or
TNF-α 24 h following infection without in vitro
restimulation. The percentage of IL-12- or IFN-γ-producing splenic
DCs in the mice infected with χ3337/pRST98 was
significantly lower than that of those infected with χ3337. No
significant difference was detected in the percentage of splenic
TNF-α-producing DCs of mice infected with χ3337/pRST98
compared to that of χ3337-infected mice.
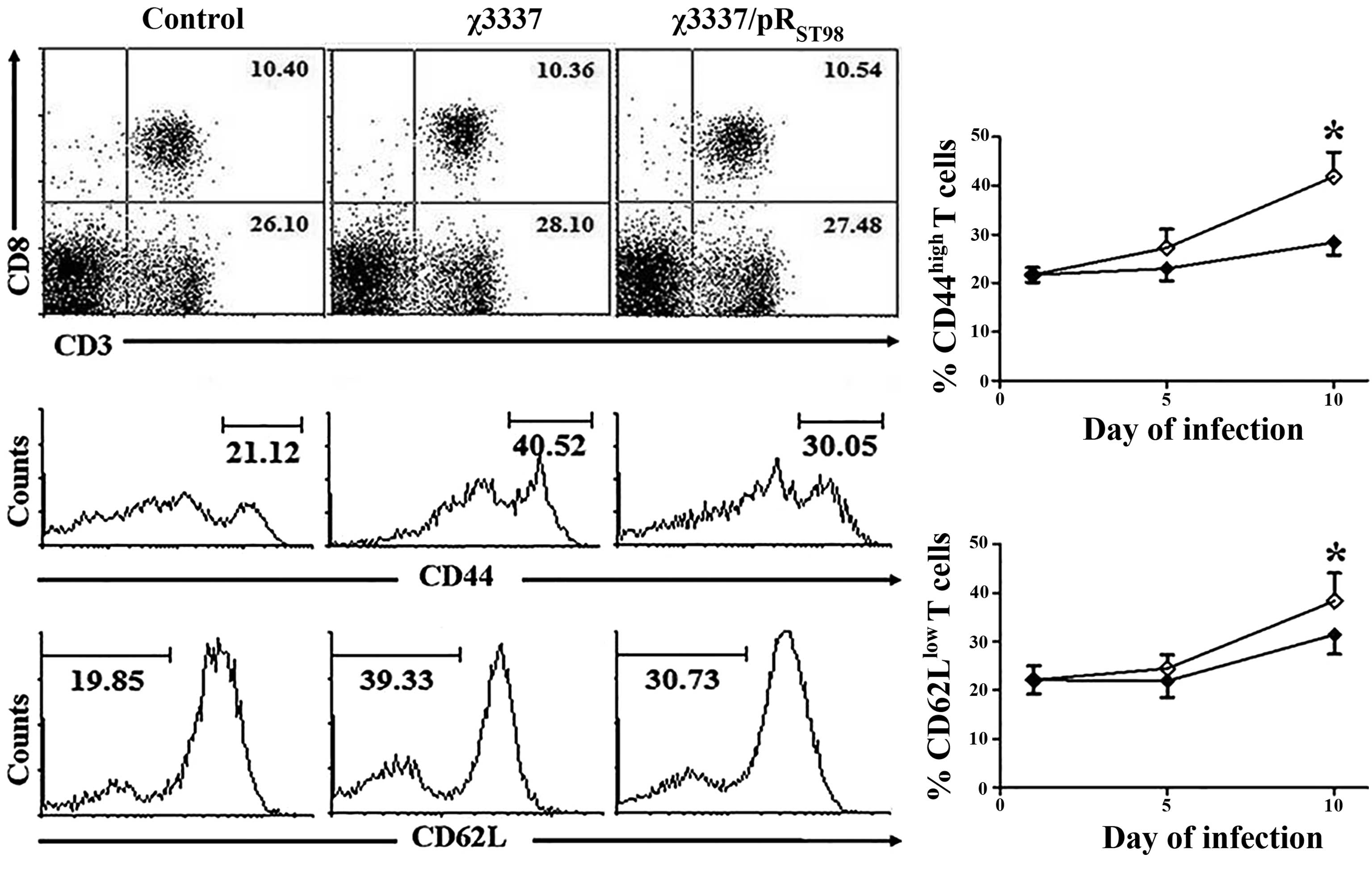
S. Typhimurium infection decreases T-cell
activation
S. Typhimurium infection induced massive
splenomegaly. On the tenth day post-infection, there was an almost
three-fold increase in spleen cellularity. Total splenocytes were
isolated from the naïve mice and mice infected with χ3337 or the
conjugant χ3337/pRST98 at the fifth and tenth day
post-infection and stained with specific antibodies. The effect of
pRST98 on the status of the CD4(+) and CD8(+)
T-lymphocyte populations was determined. The mice infected with
either strain of S. Typhimurium did not demonstrate a
significant change in T-cell population compared to that in the
control mice. T lymphocytes were subsequently further analyzed for
the expression of the immune activators CD44 and CD62L. In the
immune system, CD62L is expressed on naïve T cells, but its
expression declines upon activation. By contrast, the expression of
CD44 is upregulated following activation of naïve T lymphocytes
during their response against invading microbes (16). The mice were kept under
pathogen-free conditions; however, it was observed that in naïve
mice ~20% of splenic T cells exhibited a CD44(high) and CD62L(low)
(activated) phenotype. Following infection with S.
Typhimurium, expression of CD44 and CD62L on T cells was markedly
altered (Fig. 7). ~30–40% of T
cells acquired a CD44(high) and CD62L(low) phenotype at ten days
post-infection. Although no significant difference was observed in
the CD44(high) and CD62L(low) T-lymphocyte population in the
χ3337-infected mice at five days post-infection, there was a
<10% increase in this population at ten days post-infection
compared to that in the χ3337/pRST98-infected mice.
These results therefore suggested that pRST98 did not
affect the status of the T-lymphocyte population, but prevented
T-cell activation.
Discussion
The role of DCs in triggering anti-microbial
immunity, particularly in vivo, has been well defined.
S. Typhimurium is a bacterial pathogen that systemically
disseminates from the site of infection (17). Studies have demonstrated that
S. Typhimurium interferes with the activation of host
adaptive immunity at multiple levels (18–20).
Salmonella virulence factors perturb antigen processing and
presentation by DCs, as well as the activation of
Salmonella-specific T cells (21). These observations led to the
hypothesis that this feature of virulent Salmonella is
required to promote systemic dissemination in the host.
In previous studies by our group, it was
demonstrated that plasmid pRST98 enhanced the virulence
of host bacteria by increasing survival and mediating macrophage
apoptosis via the mitochondrial pathway (12). Consistent with these results,
pRST98 was found to influence multiple important
functions of murine DCs, including maturation, survival and
cytokine production. In addition, pRST98 was also
demonstrated to decrease T-cell activation. These results suggested
that by targeting the aforementioned functions of DCs,
pRST98 may at least partially abrogate the adaptive
immune defense mechanisms of the host, which are required for the
elimination of the pathogen from infected tissues. However, further
studies are required in order to determine the impact of
pRST98 on subsets of splenic DCs.
Previous studies by our group suggested that
pRST98 was not required for the invasion of
non-phagocytic cells, including HeLa and Hep-2 cells (unpublished
observations), but instead, its function was required to prevent
significant entrance of Salmonella to phagocytic DCs. In the
present study, pRST98 activity was demonstrated not to
differentially modulate the entry of Salmonella to DCs.
Sundquist and Wick (22) reported
that the death of DCs induced by Salmonella following
infection may have a negative impact on the initiation of
antibacterial immunity. However, besides macrophagy, DCs may also
undergo cell death following interaction with pathogenic
Salmonella, pRST98 may potentially be one of the
factors mediating the induction of DC death. Furthermore, it was
revealed that Salmonella stimulation of immature DCs
resulted in their maturation to APCs, characterized by enhanced
expression of cell surface antigens, including CD80, CD86 and MHC
II. This phenotypic alteration was similar to the response observed
when DCs were treated with lipopolysaccharides, a response which is
associated with increased levels of co-stimulatory surface
molecules (23). Furthermore, the
conjugant χ3337/pRST98-treated DCs exhibited lower
levels of activation and maturation than those of χ3337-treated
DCs. These results provided evidence that pRST98, one of
the pathogenic factors of Salmonella, impaired the functions
of DCs, predominantly via the induction of apoptosis and concurrent
suppression of maturation.
Following investigation of the virulence of
pRST98 in DCs as well as macrophage cell lines, the
virulence of the conjugant strain was evaluated in a murine model.
To analyze the bacterial colonization in various organs, groups of
BALB/c mice were intravenously infected with 500 CFU χ3337 or the
conjugant χ3337/pRST98. The organ load of the
χ3337-infected mice was significantly lower than that of the
χ3337/pRST98-infected mice in spleen and liver on the
tenth day post-infection. This suggested that pRST98 was
essential for conferring Salmonella virulence. Furthermore,
recent studies have shown that following intravenous infection of
S. Typhimurium, >50% of bacteria that reach the spleen
are located inside DCs (15).
Therefore, the virulence capacity of S. Typhimurium to
spread systemically into the host may be directly correlated with
the bacterium’s ability to avoid antigen presentation by DCs,
without the activation of specific T-cell immunity. Quantitative
changes in DCs were also apparent in response to Salmonella
infection and significantly increased DC numbers were observed five
days after infection. This increase may be due to DC migration to
the spleen during infection, which occurs as a result of the
functions of the spleen in initiating specific immunity and as a
site of Salmonella replication (15). A marked maturation response of the
splenic DCs was observed following infection and supported the
hypothesis that pRST98 hindered DC maturation without
influencing the total splenic DC number during infection.
Local inflammatory responses by innate cells within
infected tissues may also influence antigen presentation and T-cell
activation (17). The capacity of
DCs to produce TNF-α, IL-12 and IFN-γ during Salmonella
infection was therefore examined. An increase in DCs producing
TNF-α was detected among the splenic DCs of
Salmonella-infected mice. However, the absolute number of
splenic DCs producing TNF-α during the first day of infection was
relatively low (~104). The relative lack of DCs
producing TNF-α suggested that DCs may not make a significant
contribution to the control of initial bacterial replication.
However, TNF-α produced by DCs may induce local effects during
Salmonella infection in order to coordinate the maturation
and migration of DCs. In this way, DC-derived TNF-α may facilitate
a link between the innate and adaptive immune responses.
The results of the present study also revealed
pRST98-associated changes in DC cytokine secretion
during Salmonella infection. In vivo production of
IL-12 by DCs in response to intravenous administration of microbial
stimuli is rapid and transient (24–26).
Following 24 h of S. Typhimurium infection, a portion of DCs
produced IL-12 without restimulation in vitro, which
indicated that these DCs were likely involved in IL-12 production
in vivo. These results were consistent with studies in which
IL-12 production of liver CD11c(+) cells was analyzed by flow
cytometry following stimulation with S. Typhimurium
(27). IL-12 is an essential
cytokine, which may provide protective immunity against
Salmonella, as it is a key cytokine produced by APC to
stimulate a Th1-directed response (28,29).
Salmonella-specific T cells developing under the influence
of IL-12 produce cytokines, including IFN-γ, that enhance the
bactericidal capacity of phagocytes and facilitate microbial
elimination (30,31). These observations suggested that
pRST98 interference with DC function may prevent the
activation of T-cell-mediated immunity against antigens derived
from this pathogen.
The activation and differentiation status of T
cells during the course of infection was subsequently evaluated. T
cells had acquired an activated phenotype after one week of
infection. An almost 10% decrease in the CD44(high) and CD62L(low)
T-lymphocyte population was observed in the χ3337/pRST98
infected mice ten days post-infection when compared to that in the
χ3337 infected mice. It was therefore suggested that underlying the
ability of pRST98 to impair the functions of DCs, is the
inhibition of protective T-cell responses in vivo. However,
the genetic basis of pRST98 modulation of DCs and T-cell
activation remains to be elucidated.
In conclusion, the characterization of the chimeric
plasmid pRST98, isolated from S. Typhi, may be
potentially important for elucidating the virulence mechanism of
this pathogen. The results of the present study indicated that
pRST98 was a potent pathogenic factor of
Salmonella and prevented DC-mediated activation of T cells,
predominantly by inhibiting DC maturation. These results also
potentially have significance in the treatment and prevention of
typhoid fever.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (no. 30972768), the Natural Science
Foundation of Jiangsu province (no. BK2011286), the Special
Research Fund for the Doctoral Program of High Education (no.
20103201110009), the Qinglan Project of Jiangsu Province (no.
SR13400211) the Provincial Project of Natural Science Research for
Colleges and Universities of Anhui Province of China (no.
KJ2013B142) and the Program for Scientific Innovation Research of
College Graduates in Jiangsu Province (no. CX10B_042Z).
References
|
1
|
Steinman RM: Dendritic cells and the
control of immunity: enhancing the efficiency of antigen
presentation. Mt Sinai J Med. 68:160–166. 2001.PubMed/NCBI
|
|
2
|
Walseng E, Bakke O and Roche PA: Major
histocompatibility complex class II-peptide complexes internalize
using a clathrin- and dynamin-independent endocytosis pathway. J
Biol Chem. 283:14717–14727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mittrücker HW and Kaufmann SH: Immune
response to infection with Salmonella typhimurium in mice. J Leukoc
Biol. 67:457–463. 2000.PubMed/NCBI
|
|
4
|
Bosio CM, Bielefeldt-Ohmann H and Belisle
JT: Active suppression of the pulmonary immune response by
Francisella tularensis Schu4. J Immunol. 178:4538–4547. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lecours MP, Gottschalk M, Houde M, Lemire
P, Fittipaldi N and Segura M: Critical role for Streptococcus suis
cell wall modifications and suilysin in resistance to
complement-dependent killing by dendritic cells. J Infect Dis.
204:919–929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Madajczak G and Binek M: Influence of spv
plasmid genes group in Salmonella Enteritidis virulence for
chickens. I. Occurrence of spv plasmid genes group in Salmonella
Enteritidis large virulence plasmid. Med Dosw Mikrobiol.
57:163–174. 2005.(In Polish).
|
|
7
|
García-Quintanilla M and Casadesús J:
Virulence plasmid interchange between strains ATCC 14028, LT2, and
SL1344 of Salmonella enterica serovar Typhimurium. Plasmid.
65:169–175. 2011. View Article : Google Scholar
|
|
8
|
García-Quintanilla M, Ramos-Morales F and
Casadesús J: Conjugal transfer of the Salmonella enterica virulence
plasmid in the mouse intestine. J Bacteriol. 190:1922–1927. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sajid SU and Schwarz S: Plasmid
fingerprinting and virulence gene detection among indigenous
strains of Salmonella enterica serovar enteritidis. J Ayub Med Coll
Abbottabad. 21:83–86. 2009.PubMed/NCBI
|
|
10
|
Huang R, Wu S, Zhang X and Zhang Y:
Molecular analysis and identification of virulence gene on pR(ST98)
from multi-drug resistant Salmonella Typhi. Cell Mol Immunol.
2:136–140. 2005.PubMed/NCBI
|
|
11
|
Kurita A, Gotoh H, Eguchi M, et al:
Intracellular expression of the Salmonella plasmid virulence
protein, SpvB, causes apoptotic cell death in eukaryotic cells.
Microb Pathog. 35:43–48. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu S, Li Y, Xu Y, Li Q, Chu Y, Huang R and
Qin Z: A Salmonella enterica serovar Typhi plasmid induces rapid
and massive apoptosis in infected macrophages. Cell Mol Immunol.
7:271–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takahashi S and Nagano Y: Rapid procedure
for isolation of plasmid DNA and application to epidemiological
analysis. J Clin Microbiol. 20:608–613. 1984.PubMed/NCBI
|
|
14
|
Cirillo DM, Valdivia RH, Monack DM and
Falkow S: Macrophage-dependent induction of the Salmonella
pathogenicity island 2 type III secretion system and its role in
intracellular survival. Mol Microbiol. 30:175–188. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yrlid U, Svensson M, Håkansson A, Chambers
BJ, Ljunggren HG and Wick MJ: In vivo activation of dendritic cells
and T cells during Salmonella enterica serovar Typhimurium
infection. Infect Immun. 69:5726–5735. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baaten BJ, Li CR and Bradley LM:
Multifaceted regulation of T cells by CD44. Commun Integr Biol.
3:508–512. 2010. View Article : Google Scholar
|
|
17
|
Hurley D, McCusker MP, Fanning S and
Martins M: Salmonella-host interactions - modulation of the host
innate immune system. Front Immunol. 5:4812014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alaniz RC, Cummings LA, Bergman MA,
Rassoulian-Barrett SL and Cookson BT: Salmonella Typhimurium
coordinately regulates FliC location and reduces dendritic cell
activation and antigen presentation to CD4+ T cells. J Immunol.
177:3983–3993. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Halici S, Zenk SF, Jantsch J and Hensel M:
Functional analysis of the Salmonella pathogenicity island
2-mediated inhibition of antigen presentation in dendritic cells.
Infect Immun. 76:4924–4933. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Srinivasan A, Nanton M, Griffin A and
McSorley SJ: Culling of activated CD4 T cells during typhoid is
driven by Salmonella virulence genes. J Immunol. 182:7838–7845.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bueno SM, Riedel CA, Carreño LJ and
Kalergis AM: Virulence mechanisms displayed by Salmonella to impair
dendritic cell function. Curr Med Chem. 17:1156–1166. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sundquist M and Wick MJ: Salmonella
induces death of CD8alpha(+) dendritic cells but not CD11c(int)
CD11b(+) inflammatory cells in vivo via MyD88 and TNFR1. J Leukoc
Biol. 85:225–234. 2009. View Article : Google Scholar
|
|
23
|
Jiang HR, Muckersie E, Robertson M, Xu H,
Liversidge J and Forrester JV: Secretion of interleukin-10 or
interleukin-12 by LPS-activated dendritic cells is critically
dependent on time of stimulus relative to initiation of purified DC
culture. J Leukoc Biol. 72:978–985. 2002.PubMed/NCBI
|
|
24
|
Huang LY, Reis e Sousa C, Itoh Y, Inman J
and Scott DE: IL-12 induction by a Th1-inducing adjuvant in vivo:
dendritic cell subsets and regulation by IL-10. J Immunol.
167:1423–1430. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiao X, Lo-Man R, Guermonprez P, et al:
Dendritic cells are host cells for mycobacteria in vivo that
trigger innate and acquired immunity. J Immunol. 168:1294–1301.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schulz O, Edwards AD, Schito M, Aliberti
J, Manickasingham S, Sher A and Reis e Sousa C: CD40 triggering of
heterodimeric IL-12 p70 production by dendritic cells in vivo
requires a microbial priming signal. Immunity. 13:453–462. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Johansson C and Wick MJ: Liver dendritic
cells present bacterial antigens and produce cytokines upon
Salmonella encounter. J Immunol. 172:2496–2503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lehmann J, Springer S, Werner CE, et al:
Immunity induced with a Salmonella enterica serovar Enteritidis
live vaccine is regulated by Th1-cell-dependent cellular and
humoral effector mechanisms in susceptible BALB/c mice. Vaccine.
24:4779–4793. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoon WS, Choi HJ and Park YK: Salmonella
Typhimurium harboring plasmid expressing interleukin-12 induced
attenuation of infection and protective immune responses. J Gen
Appl Microbiol. 57:115–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
John B, Rajagopal D, Pashine A, Rath S,
George A and Bal V: Role of IL-12-independent and IL-12-dependent
pathways in regulating generation of the IFN-gamma component of T
cell responses to Salmonella Typhimurium. J Immunol. 169:2545–2552.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
MacLennan C, Fieschi C, Lammas DA, et al:
Interleukin (IL)-12 and IL-23 are key cytokines for immunity
against Salmonella in humans. J Infect Dis. 190:1755–1757. 2004.
View Article : Google Scholar : PubMed/NCBI
|