Introduction
Osteosarcoma is the most common primary malignant
bone tumor. A critical issue in the clinical therapy of
osteosarcoma is the development of treatment strategies that kill
cancer cells without harming normal healthy cells. (1). Cisplatin
(cis-diamminedichloroplatinum II) is one of the most
effective and widely used chemotherapeutic agents to treat solid
tumors (2). Cisplatin causes DNA
damage, which may lead to cell apoptosis; however, cisplatin
treatment is often ineffective due to acquired drug resistance
(3). Ion channels contribute to
massive ion fluxes across plasma membranes and have important roles
in diverse cell processes in healthy and disease states, including
excitability, contraction, cell cycle regulation and metabolism,
(4). Previous studies have
suggested that chloride channels are associated with cell volume
regulation, proliferation, cell cycle control and migration, as
well as apoptosis (5–7). Apoptotic volume decrease (AVD)
usually indicates cell apoptosis and is activated by ionic efflux,
particularly of chloride ions, through volume regulatory anion
channels (8). Ion channels have
previously been proposed as potential targets for cancer therapy. A
previous study suggested that treatment with a specific inhibitor
of the cystic fibrosis transmembrane conductance regulator may be a
novel therapeutic approach in the prevention of cisplatin-induced
nephrotoxicity, without affecting the antitumor efficacy of
cisplatin (9). Furthermore,
impaired activity of volume-sensitive, outwardly rectifying (VSOR)
chloride channels has been shown to contribute to the acquisition
of cisplatin resistance in A549/CDDP cells (10). Conversely, Su et al
(11) demonstrated that
suppression of chloride channel 3 resulted in the inhibition of Akt
and autophagy, which may enhance the therapeutic benefit of
cisplatin in U251 human glioma cells. The present study aimed to
investigate the role of chloride channels in cisplatin-induced
apoptosis of MG-63 cells.
Materials and methods
Materials
All of the chemicals used in the present study were
purchased from Sigma-Aldrich (St. Louis, MO, USA). The isotonic
bath solution contained (in mM): 70 NaCl, 0.5 MgCl2, 2
CaCl2, 10 HEPES and 140 D-mannitol. The isosmotic
solution was produced by replacing 70 mM NaCl with equimolar NaI,
NaBr or sodium gluconate. The pipette solution consisted of (in
mM): 70 N-methyl-D-glucamine chloride, 1.2 MgCl2, 10
HEPES, 1 EGTA, 140 D-mannitol and 2 ATP. Osmolarity of the
solutions was detected using an automatic cryoscopic osmometer
(Osmomat 030; Gonotec, Berlin, Germany). The pH of all bath and
pipette solutions was adjusted to 7.4 and 7.25, respectively. The
chloride channel blocker, 5-nitro-2-(3-phenylpropylamino)-benzoate
(NPPB; 100 μmol/l; Sigma-Aldrich), was dissolved in dimethyl
sulfoxide (DMSO; 100 mM; Sigma-Aldrich), and the other chloride
channel blocker tamoxifen (20 μmol/l; Sigma-Aldrich) was dissolved
in methanol anhydrous. NPPB and tamoxifen were diluted to final
concentrations using isotonic solutions.
Cell culture
The MG-63 human osteosarcoma cells (American Type
Culture Collection, Manassas, VA, USA; no. CRL-1427) were cultured
in Dulbecco’s modified Eagle’s medium (DMEM; Gibco Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal calf
serum (FCS), 100 IU/ml penicillin and 100 μg/ml streptomycin
(Sigma-Aldrich) in a humidified chamber containing 5%
CO2 and 95% O2, at 37°C. The cells were
collected at the logarithmic growth phase, resuspended, plated on
coverslips and incubated for 1 h prior to further analysis.
Chloride current recordings
Following stabilization of the background chloride
current in isotonic solution, the bath solution was changed to
isotonic solution containing 2 μg/ml cisplatin (CDDP) for 30–50
min. Once the cisplatin activated currents had reached their
maximum, the bath solution was changed to cisplatin solution
containing 100 μmol/l NPPB or 20 μmol/l tamoxifen for ~30min.
Whole-cell Cl− currents were recorded using the
patch-clamp technique with 5–10 MΩ pipette resistance and an EPC-9
patch clamp amplifier (HEKA Electronik, Lambrecht/Pfalz, Germany).
Whole-cell currents of individual cells were maintained at a
constant voltage, then amplified and filtered at 2.9 kHz. The
Cl− equilibrium potential was set to 0 mV, then stepped
to ±40 and ±80 mV for 200 ms repeatedly (12), with a 4 sec interval between pulses
in voltage clamp mode, at 20–24°C. The currents were measured 10
msec after the onset of voltage steps. The background current was
normalized in isotonic solution. The percentage of inhibition of
the chloride channel blockers was calculated using the following
equation: Inhibition
(%)=[(CCDDP-CIso)−(CBlocker-CISO)]/(CCDDP-CIso)
× 100, where CIso is the background current under
isotonic conditions; CCDDP is the maximal stable current
following exposure to cisplatin; and CBlocker is the
current recorded following treatment with the chloride channel
inhibitors.
Measurements of cell volume
Cells in the control group were incubated under
isotonic conditions for 360 min. Cells in the treatment groups were
incubated under isotonic conditions for 10 mins, then administered
2 μg/ml cisplatin alone or in combination with 20 μmol/l tamoxifen,
and then incubated under isotonic conditions for a further 350
mins. Cells in the same field of view were imaged using an inverted
phase contrast microscope (DMI6000 B; Leica Microsystems GmbH,
Wetzlar, Germany) at 20–24°C, then analyzed using Scion software
(Scion Corporation, Torrance, CA, USA) at the following time
points: 0, 5, 10, 15, 20, 30, 40, 60, 80, 100, 140, 180, 220, 260,
300 and 360 min. The cell volume (V) was calculated from measured
cell diameters (d) using the following equation: V=4/3π
(d/2)3.
Proliferation and apoptosis analysis
An MTT assay was used to detect the rate of cell
proliferation. Cells were cultured in control medium (DMEM
supplemented with FCS and antibiotics) or medium containing
cisplatin (2 μg/ml) alone or in combination with 100 μmol/l NPPB or
20 μmol/l tamoxifen for 72 h. Cell suspensions containing
2.5×107 cells/l were plated in 96-well culture plates,
at a volume of 100 μl per well. The cells were incubated in normal
media or media containing 2 μg/ml cisplatin and/or chloride channel
blockers (NPPB; 100 μmol/l). MTT solution (10 μl/well;
Sigma-Aldrich) was added to the cells, which were then incubated
for 4 h prior to detection. The MTT solution was removed and
replaced with DMSO, in order to dissolve the formazan crystals, for
15–30 min. The absorbance was then measured at a wavelength of 570
nm, using a Safire2 microplate reader equipped with the
Magellan (5.0) reader software (Tecan Group, Ltd, Männedorf,
Switzerland).
The rate of apoptosis of the cells was measured by
flow cytometry. The cells were cultured for 72 h,
(1–2×105 cells/sample), then collected and analyzed.
Briefly, the cells were washed three times with phosphate-buffered
saline (PBS), fixed in chilled 70% ethanol at −20°C for 30 min,
washed a further two times with PBS and incubated with RNase A (50
μg/ml in PBS) at 37°C for 30 min. The cells were then stained with
propidium iodide (50 μg/ml; Beyotime, Shanghai, China) for 15 min
and analyzed by flow cytometry (FACS101; BD Biosciences, Franklin
Lakes, NJ, USA). The percentage of apoptotic cells was quantified
using DNA histograms (FlowJo 7.6.1 software; FlowJo, LLC, Ashland,
OR, USA).
Statistical analysis
Data are expressed as the mean ± standard error.
Significant differences were determined by analysis of variance
using SPSS version 13.0 software (SPSS Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
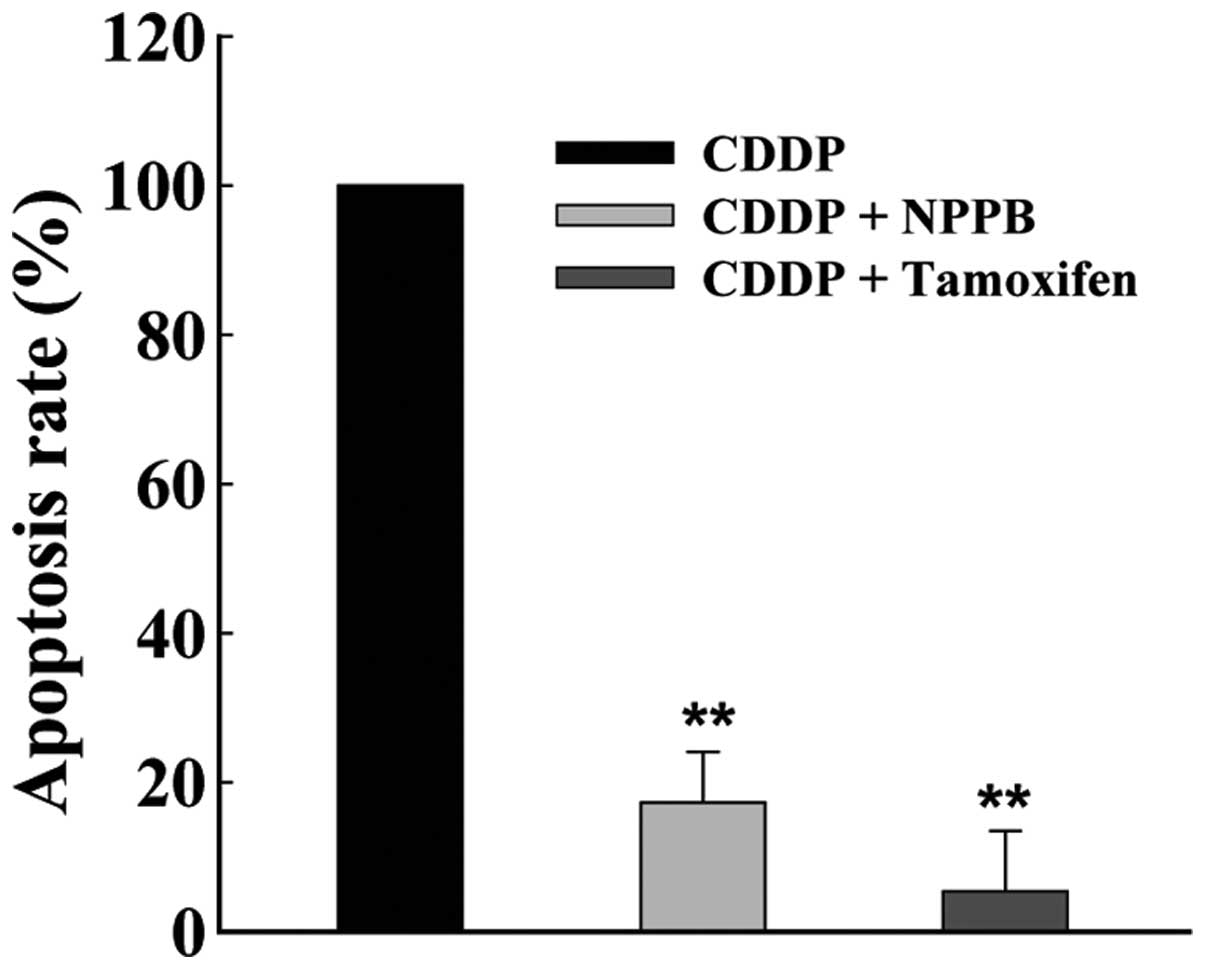
Cisplatin-induced apoptosis can be
suppressed by chloride channel blockers
To determine the effects of chloride channels on
apoptosis, MG-63 cells were cultured with cisplatin (2 μg/ml),
either alone, or in combination with chloride channel blockers NPPB
(100 μmol/l) and/or tamoxifen (20 μmol/l) for 72 h. The chloride
channel blockers significantly suppressed the rate of
cisplatin-induced apoptosis, as determined by flow cytometry. The
rate of apoptosis inhibition was 84.7±15.8 and 94.4±18.1%, in
response to treatment with NPPB and tamoxifen, respectively (n=6,
P<0.01, Fig. 1).
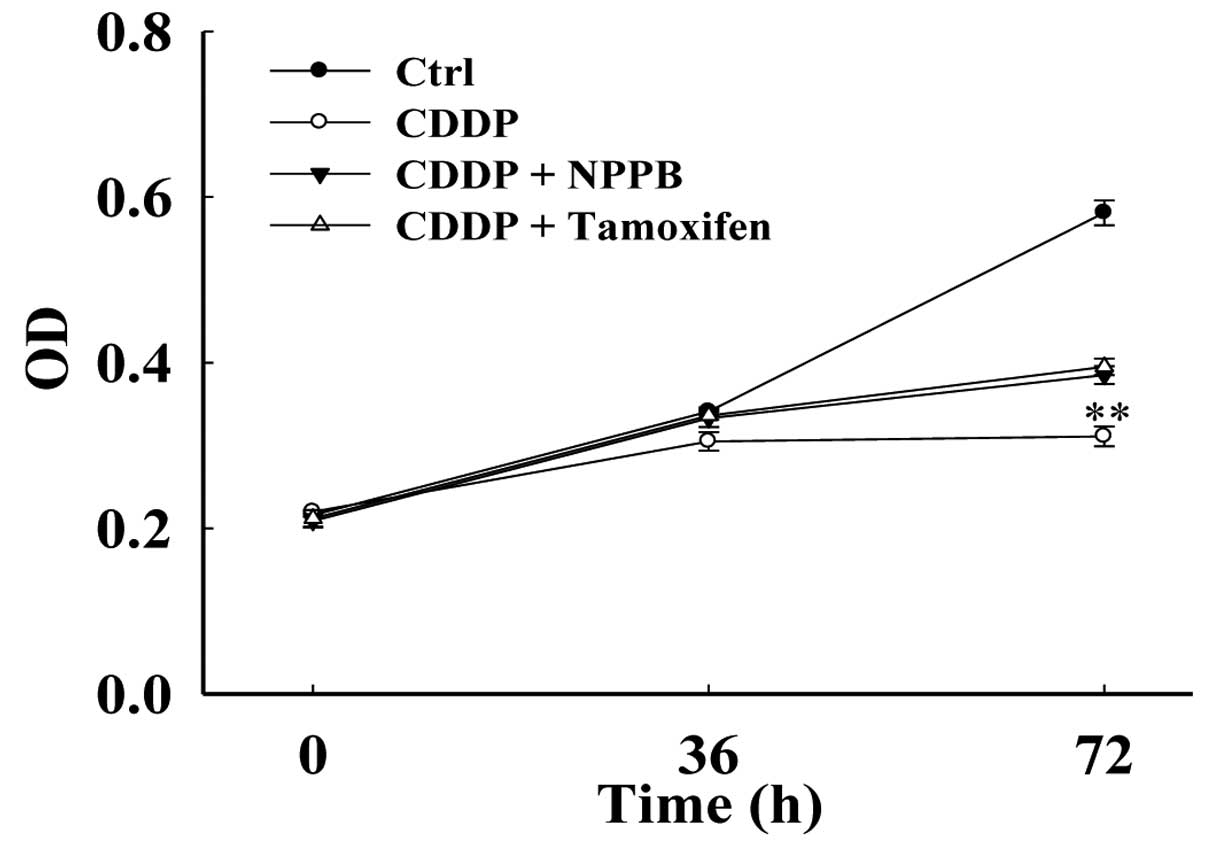
Chloride channel blockers prevent
cisplatin-induced suppression of cell proliferation
The results of the present study indicate that
chloride channel blockers may protect MG-63 cells against
cisplatin-induced apoptosis. Therefore, the present study aimed to
determine the effects of chloride channel blockers on the
proliferation of cisplatin-treated MG-63 cells. The proliferation
of MG-63 cells was evaluated by an MTT assay. The cells were
cultured under the following four conditions: Control (DMEM),
cisplatin (CDDP), CDDP+NPPB and CDDP+tamoxifen. The cells were
cultured for 36 and 72 h, and the optical density of the cells was
then measured using a microplate reader. The inhibition of
proliferation was 77.0±23.5% following treatment with cisplatin for
72 h (Fig. 2). Treatment with NPPB
and tamoxifen suppressed the cisplatin-induced inhibition of
proliferation. The inhibition rate was reduced to 30.51±8.30% and
45.21±7.28%, in response to treatment with NPPB and tamoxifen,
respectively (n=18, P<0.01).
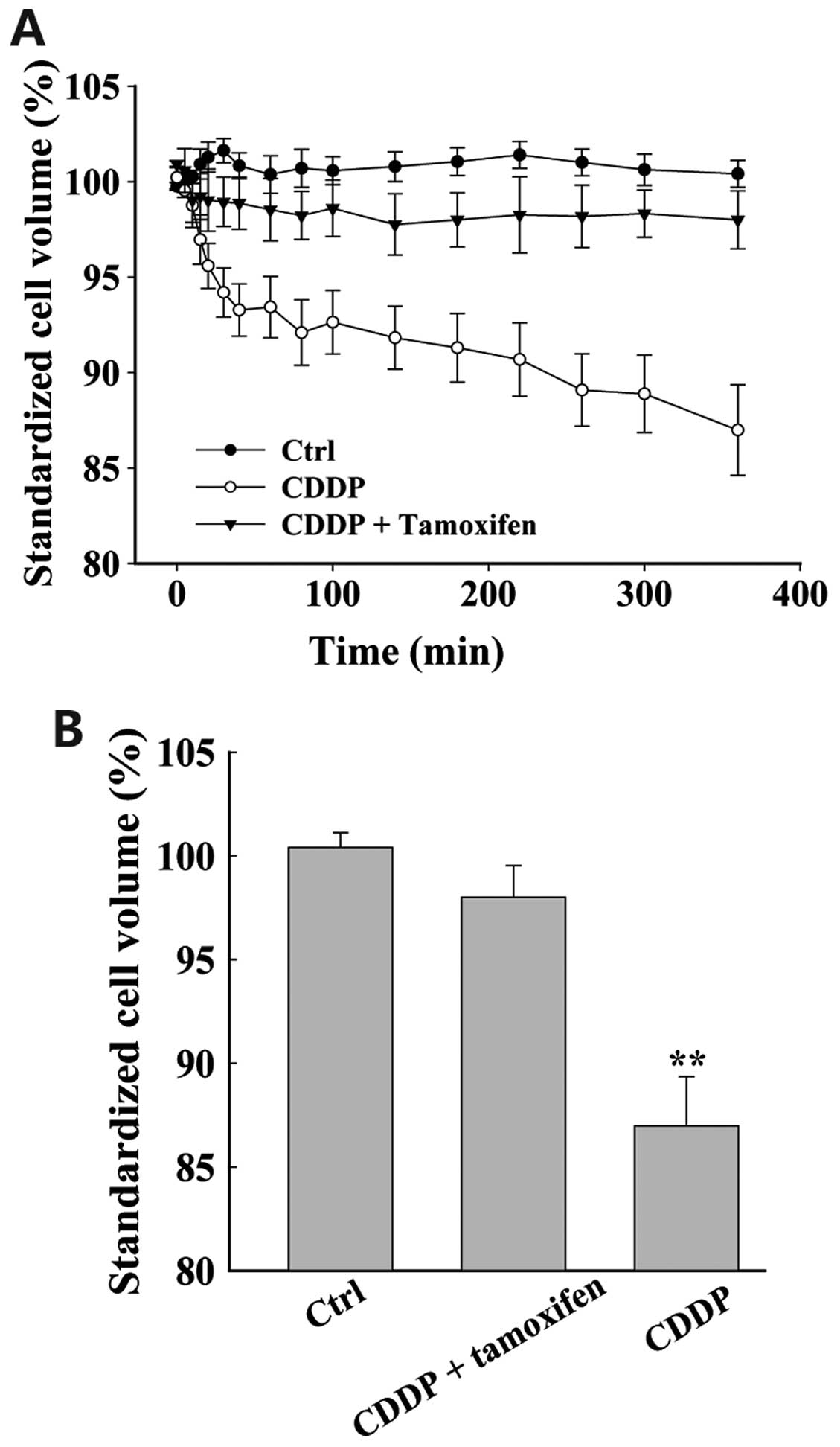
Chloride channel blockers inhibit
cisplatin-induced AVD
Live cell imaging was used to detect cell volume.
Under isotonic conditions (control), the cell volume was stable
(Fig. 3; n=18). However, when the
cells were treated with cisplatin, cell volume gradually decreased.
Cell shrinkage was detected as early as 10 min after application of
cisplatin. After 6 h, cell volume had decreased by 15.1±2.2% (n=23,
P<0.01). Cell shrinkage was alleviated in response to treatment
with chloride channel blockers. Incubation with NPPB (data not
shown) and tamoxifen suppressed the cisplatin-induced decrease in
cell volume. In addition, the cell volume was not significantly
different in the cells treated with chloride channel blockers, as
compared with the control cells (n=32, P>0.05).
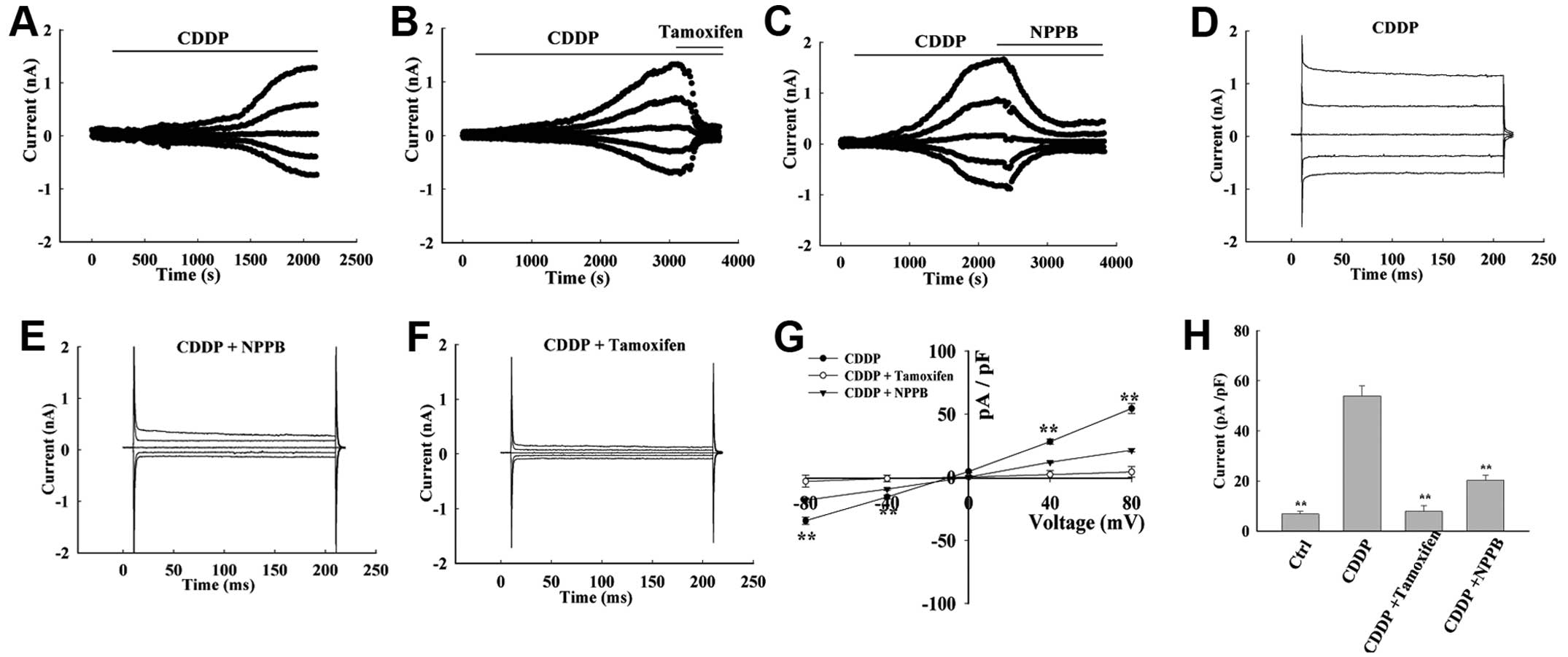
Activation of chloride currents by
extracellular application of cisplatin
The results of the present study indicate that
cisplatin may induce AVD, and it was hypothesized that chloride
channels may be involved in this process. Whole cell patch-clamp
recordings were taken, in order to determine the effects of
cisplatin on chloride channel currents in MG-63 cells. The
background chloride current in isotonic solution was weak and
stable, with a density of 6.81±1.02 pA/pF at +80 mV; and −5.81±1.45
pA/pF at −80 mV (n=15, Fig. 4). In
the majority of cells (7/10 cells), the currents were significantly
increased in response to treatment with cisplatin for 10–15 min.
The currents reached a plateau with mild outward-rectification at
30–50 min (Fig. 4D). The current
displayed an almost linear current-voltage relationship, with an
outward current of 53.96±4.01 pA/pF at +80 mV, and an inward
current of −41.9±3.451 pA/pF at −80 mV (n=12, P<0.01, Fig. 4A). There was no time-dependent
inactivation observed at ±40 and ±80 mV. The cisplatin-activated
current reversed at −3.74±1.43 mV (n=15), which is close to the
calculated Cl− equilibrium potential (−0.9 mV). There
was no potassium shown to be present in either the pipette or bath
solutions. In addition, equilibrium potentials for Na+
and Ca2+ were predicted to be >+200 mV. Therefore,
these results support the conclusion that the cisplatin-activated
current is generated primarily by Cl−.
Chloride channel blockers inhibit
cisplatin-activated chloride currents
Once the cisplatin-activated currents had reached
the maximum, the bath solution was changed to cisplatin solution
containing NPPB and tamoxifen. Extracellular application of NPPB
and tamoxifen inhibited the current (Fig. 4B and C). The outward and inward
currents were almost equally inhibited, and the inhibition rate was
70.21±3.08% and 97.88±1.50% at +80 mV, and 68.321±4.98% and
98.36±2.04% at −80 mV, respectively (n=5, Fig. 4G and H). In these experiments, DMSO
was used to prepare the NPPB solutions, and the final concentration
of DMSO in the bath solutions did not exceed 0.1% (v/v). At this
concentration, it did not affect cell volume or current, and was
not cytotoxic within the 24 h period of analysis.
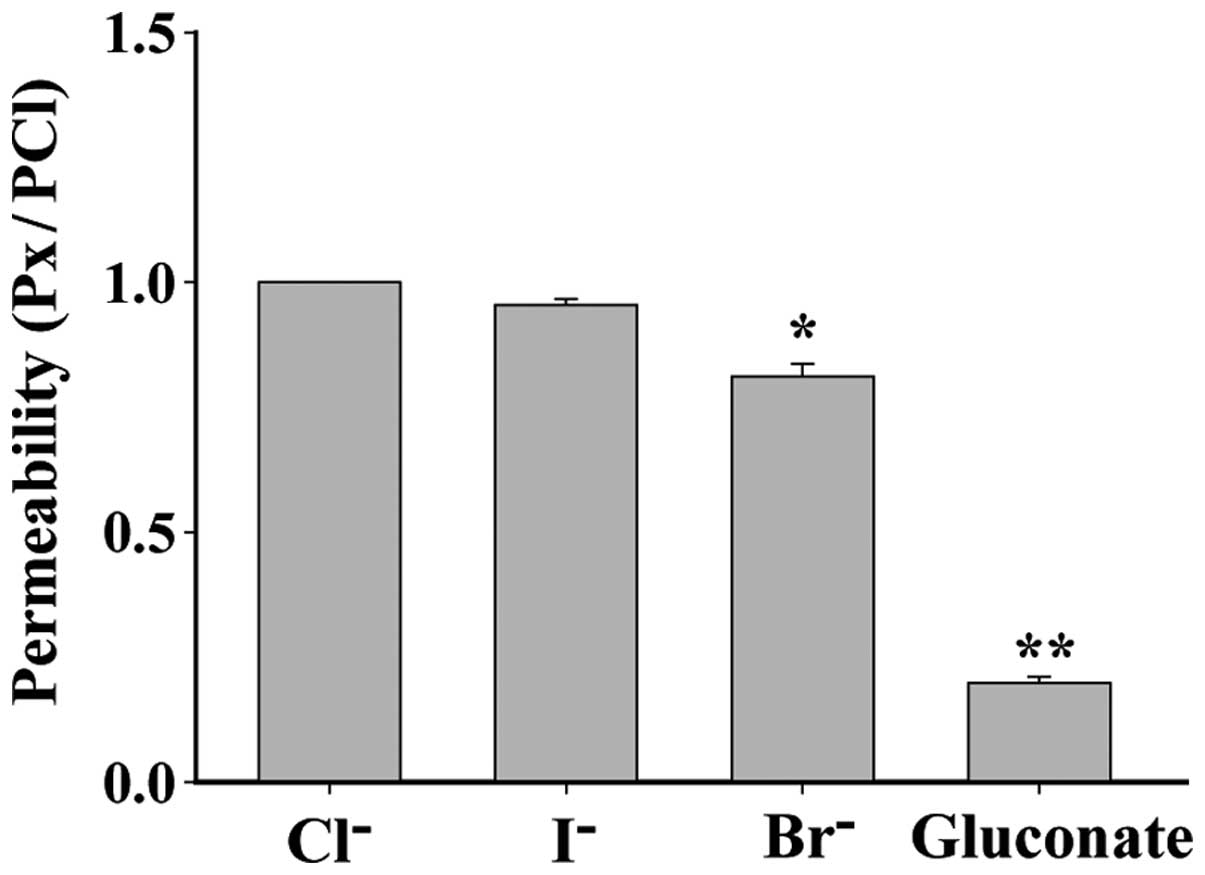
Anion selectivity of the
cisplatin-activated chloride channel
Anion selectivity of the cisplatin-activated current
was determined by replacing 70 mM NaCl with equimolar Na (X), where
X represents the substituted anion I−, Br−,
or gluconate. The permeability of I−, Br− and
gluconic acid ion, as compared with Cl−, which was
defined as 1, was: I−, 0.95±0.01 (P>0.05, n=5);
Br−, 0.81±0.03 (P<0.05, n=6), gluconic acid ion,
0.20±0.01 (P<0.01, n=6). Therefore, the order of the anion
permeability of the cisplatin-activated channel was Cl−=
I−>Br−>gluconate acid ion (Fig. 5).
Discussion
Osteosarcoma is the most common primary malignant
bone tumor in children and adolescents. There are numerous
chemotherapeutic drugs that exert different pharmacological
effects; however, the current obstacle to clinical chemotherapy is
the development of treatment strategies that kill tumor cells,
without harming normal healthy cells (1). Cisplatin is often used as a single
agent, or in combination with other drugs, and is effective in the
treatment of various types of tumor (13).
Cisplatin is a platinum complex that exhibits
profound cytotoxic effects. It has a strong broad-spectrum
anticancer effect, and is the most common drug used to treat
nasopharyngeal carcinoma, osteosarcoma and other solid tumors
(14,15). Cisplatin can damage DNA and cause
apoptosis, and is capable of acting synergistically with various
chemotherapeutic drugs to increase effectiveness; however, the
mechanisms by which this occurs are not fully understood.
Furthermore, cytotoxicity and drug resistance to cisplatin reduce
its effectiveness. Understanding how cisplatin exerts its effects
is crucial in the development of improved treatment strategies. The
present study hypothesized that cisplatin may exert its functions
through chloride channels.
Chloride channels are widely distributed throughout
mammalian tissues. In addition to regulating cell volume,
volume-sensitive chloride channels have been shown to correlate
with cell cycle regulation, and proliferation, migration and
apoptosis of cells (4,16–22).
AVD marks the early stages of apoptosis, and is closely associated
with the activation of chloride channels (23). The present study explored the role
of chloride channels in cisplatin treatment of osteosarcoma, with
the aim of identifying novel targets for osteosarcoma therapy.
The present study examined the role of chloride
channels in cisplatin-induced apoptosis of MG-63 cells. Flow
cytometry and an MTT assay were used to detect the rate of
apoptosis and proliferation, respectively. In the cells treated
with cisplatin, cell proliferation was suppressed and the rate of
apoptosis was increased. Furthermore, co-treatment of cisplatin
with chloride channel blockers NPPB and tamoxifen resulted in a
reduction in the rate of cell apoptosis and inhibition of cell
proliferation, which had initially been induced by cisplatin. These
results suggest that chloride channels are activated and involved
in cisplatin-induced apoptosis.
To demonstrate that activation of chloride channels
is a key signal controlling the early stages of apoptosis, live
cell imaging and whole cell patch-clamp recordings were used to
detect cisplatin-induced changes in cell volume and chloride
currents, in osteosarcoma cells. Live cell imaging analysis showed
that cell volume gradually decreased in response to treatment with
cisplatin; this type of continuous cell volume decrease has
previously been shown to activate apoptosis (24,25).
Whole cell patch-clamp analysis indicated that chloride channel
currents were significantly increased upon treatment with
cisplatin, and that the cisplatin-activated current was reversed at
a potential close to the calculated chloride equilibrium potential
(−0.9 mV). The typical current traces indicate that the currents
were not time-dependent. In addition, chloride channel blockers
were shown to inhibit cisplatin-activated chloride currents.
Further experiments were then conducted to determine anion
selectivity of the channel, and the order of anion permeability was
shown to be Cl−=I−
>Br−>gluconate acid anion. These results suggest
that cisplatin activates chloride channels, opening them and
thereby driving the flow of chloride ions out of the cell, causing
the cells to shrink, and thus resulting in a decrease in cell
volume and ultimately induction of cell apoptosis (26,27).
It is possible that other ion channels may also be
involved in osteosarcoma therapy. A previous study showed that
capacitative Ca2+ entry influx may activate transient
receptor potential channels, which may affect cell proliferation of
osteosarcoma cells (28). Kv1.3 is
another channel that may be involved in the treatment of
osteosarcoma. Previous research demonstrated that a knockdown of
Kv1.3 significantly inhibited the growth of MG-63 xenografts
(29). In addition, other groups
have reported that treatment with trichostatin A can restore
functional expression of VSOR chloride channels, and that this may
lead to a decrease in the cisplatin resistance of KCP-4 human
epidermoid cancer cells. These results suggests that impaired
activity of VSOR chloride channels may be involved in the
acquisition of cisplatin resistance in this type of cancer
(30). Ransom et al
(31) demonstrated that glioma
cell migration through brain tissue may require volume-activated
chloride currents, which participate in cell shape and volume
changes. Furthermore, tumor necrosis factor-mediated liver cell
death has been shown to be triggered by the activation of
K+ and Cl− channels, which is an early signal
in apoptotic pathways (32). These
results further demonstrate that numerous types of ion channels,
particularly chloride channels, are involved in tumor growth,
apoptosis and migration.
The results of the present study demonstrate that
cisplatin activates chloride channels, causing a cell volume
decrease, which may lead to apoptosis of osteosarcoma cells
(26,27). It may therefore be concluded that
chloride channel activation is an early signal in pathways leading
to cisplatin-mediated suppression of proliferation and induction of
apoptosis in MG-63 cells. Identification of the specific chloride
channel activated by cisplatin may be a potential focus of future
research.
Acknowledgements
The present study was supported by the Health
Development Research Funds of Peking (grant no. 201302007).
References
|
1
|
Reedijk J: New clues for platinum
antitumor chemistry: kinetically controlled metal binding to DNA.
Proc Natl Acad Sci USA. 100:3611–3616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Woźniak K and Błasiak J: Recognition and
repair of DNA-cisplatin adducts. Acta Biochim Pol. 49:583–596.
2002.
|
|
4
|
Lang F, Busch GL, Ritter M, et al:
Functional significance of cell volume regulatory mechanisms.
Physiol Rev. 78:247–306. 1998.PubMed/NCBI
|
|
5
|
Shen MR, Droogmans G, Eggermont J, Voets
T, Ellory JC and Nilius B: Differential expression of
volume-regulated anion channels during cell cycle progression of
human cervical cancer cells. J Physiol. 529:385–394. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zheng YJ, Furukawa T, Tajimi K and Inagaki
N: Cl− channel blockers inhibit transition of quiescent
(G0) fibroblasts into the cell cycle. J Cell Physiol.
194:376–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang L, Chen L and Jacob TJ: The role of
ClC-3 in volume-activated chloride currents and volume regulation
in bovine epithelial cells demonstrated by antisense inhibition. J
Physiol. 524:63–75. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimizu T, Numata T and Okada Y: A role of
reactive oxygen species in apoptotic activation of volume-sensitive
Cl− channel. Proc Natl Acad Sci USA. 101:6770–6773.
2004. View Article : Google Scholar
|
|
9
|
Rubera I, Duranton C, Melis N, Cougnon M,
Mograbi B and Tauc M: Role of CFTR in oxidative stress and suicidal
death of renal cells during cisplatin-induced nephrotoxicity. Cell
Death Dis. 4:e8172013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Min XJ, Li H and Hou SC: Dysfunction of
volume-sensitive chloride channels contributes to cisplatin
resistance in human lung adenocarcinoma cells. Exp Biol Med
(Maywood). 236:483–491. 2011. View Article : Google Scholar
|
|
11
|
Su J, Xu Y, Zhou L, et al: Suppression of
chloride channel 3 expression facilitates sensitivity of human
glioma U251 cells to cisplatin through concomitant inhibition of
Akt and autophagy. Anat Rec (Hoboken). 296:595–603. 2013.
View Article : Google Scholar
|
|
12
|
Yang L, Ye D, Ye W, et al: ClC-3 is a main
component of background chloride channels activated under isotonic
conditions by autocrine ATP in nasopharyngeal carcinoma cells. J
Cell Physiol. 226:2516–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muggia F: Platinum compounds 30 years
after the introduction of cisplatin: implications for the treatment
of ovarian cancer. Gynecol Oncol. 112:275–281. 2009. View Article : Google Scholar
|
|
14
|
Iizuka N, Hirose K, Noma T, Hazama S,
Tangoku A, Hayashi H, et al: The nm23-H1 gene as a predictor of
sensitivity to chemotherapeutic agents in oesophageal squamous cell
carcinoma. Br J Cancer. 81:469–475. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quan YH, Kim B, Park JH, Choi Y, Choi YH
and Kim HK: Highly sensitive and selective anticancer effect by
conjugated HA-cisplatin in non-small cell lung cancer overexpressed
with CD44. Exp Lung Res. 40:475–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lang F, Föller M, Lang K, et al: Cell
volume regulatory ion channels in cell proliferation and cell
death. Methods Enzymol. 428:209–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mao J, Li X, Chen W, et al: Cell
cycle-dependent subcellular distribution of ClC-3 in HeLa cells.
Histochem Cell Biol. 137:763–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mao J, Yuan J, Wang L, et al: Tamoxifen
inhibits migration of estrogen receptor-negative hepatocellular
carcinoma cells by blocking the swelling-activated chloride
current. J Cell Physiol. 228:991–1001. 2013. View Article : Google Scholar
|
|
19
|
Okada Y, Sato K and Numata T:
Pathophysiology and puzzles of the volume-sensitive outwardly
rectifying anion channel. J Physiol. 587:2141–2149. 2009.PubMed/NCBI
|
|
20
|
Zhang H, Zhu L, Zuo W, et al: The ClC-3
chloride channel protein is a downstream target of cyclin D1 in
nasopharyngeal carcinoma cells. Int J Biochem Cell Biol.
45:672–683. 2013. View Article : Google Scholar
|
|
21
|
Zhu L, Yang H, Zuo W, et al: Differential
expression and roles of volume-activated chloride channels in
control of growth of normal and cancerous nasopharyngeal epithelial
cells. Biochem Pharmacol. 83:324–334. 2012. View Article : Google Scholar
|
|
22
|
Zhu L, Zuo W, Yang H, et al: Involvement
of volume-activated chloride channels in H2O2
preconditioning against oxidant-induced injury through modulating
cell volume regulation mechanisms and membrane permeability in PC12
cells. Mol Neurobiol. 48:205–216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Okada Y, Maeno E, Shimizu T, Dezaki K,
Wang J and Morishima S: Receptor-mediated control of regulatory
volume decrease (RVD) and apoptotic volume decrease (AVD). J
Physiol. 532:3–16. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen LX, Zhu LY, Jacob TJ and Wang LW:
Roles of volume-activated Cl− currents and regulatory
volume decrease in the cell cycle and proliferation in
nasopharyngeal carcinoma cells. Cell Prolif. 40:253–267. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Okada Y and Maeno E: Apoptosis, cell
volume regulation and volume-regulatory chloride channels. Comp
Biochem Physiol A Mol Integr Physiol. 130:377–383. 2001. View Article : Google Scholar
|
|
26
|
Wang L, Chen L, Zhu L, et al: Regulatory
volume decrease is actively modulated during the cell cycle. J Cell
Physiol. 193:110–119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okada Y: Cell and volume-sensitive
chloride channels: phenotypic properties and molecular identity.
Contrib Nephrol. 152:9–24. 2006. View Article : Google Scholar
|
|
28
|
Labelle D, Jumarie C and Moreau R:
Capacitative calcium entry and proliferation of human
osteoblast-like MG-63 cells. Cell Prolif. 40:866–884. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu J, Zhong D, Wu X, Sha M, Kang L and
Ding Z: Voltage-gated potassium channel Kv1.3 is highly expressed
in human osteosarcoma and promotes osteosarcoma growth. Int J Mol
Sci. 14:19245–19256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee EL, Shimizu T, Ise T, Numata T, Kohno
K and Okada Y: Impaired activity of volume-sensitive Cl−
channel is involved in cisplatin resistance of cancer cells. J Cell
Physiol. 211:513–521. 2007. View Article : Google Scholar
|
|
31
|
Ransom CB, O’Neal JT and Sontheimer H:
Volume-activated, chloride currents contribute to the resting
conductance and invasive migration of human glioma cells. J
Neurosci. 21:7674–7683. 2001.PubMed/NCBI
|
|
32
|
Nietsch HH, Roe MW, Fiekers JF, Moore AL
and Lidofsky SD: Activation of potassium and chloride channels by
tumor necrosis factor alpha. Role in liver cell death. J Biol Chem.
275:20556–20561. 2000. View Article : Google Scholar : PubMed/NCBI
|