Introduction
Glucocorticoids have been widely used in the
treatment of non-infectious inflammation and autoimmune disorders,
however, long-term glucocorticoid therapy has been demonstrated to
lead to irreversible bone injury (1). Weinstein (2) reported that the incidence of bone
fracture was 30–50% among patients receiving long-term
glucocorticoid therapy; and glucocorticoid-induced osteoporosis
(GIOP) is the most common type of secondary osteoporosis (3). Previous studies have suggested that
oral glucocorticoids may reduce the proliferation and increase the
apoptosis of osteoblasts and osteocytes (4), in addition to prolonging the survival
of osteoclasts (5). The balance
between bone formation and resorption is interrupted and the risk
of bone fracture is increased following glucocorticoid therapy
(6).
Bone marrow mesenchymal stem cells (BMSCs) are
essential in the maintenance of the dynamic homeostasis of bone
tissue, with studies demonstrating that when BMSC proliferation and
osteoblastic differentiation are defective, bone mass is reduced
(7,8). In addition, Zhou et al
(9) indicated that stem cell
injury and dysfunction are involved in the pathogenesis of
osteoporosis. However, whether human (h)BMSCs from patients with
GIOP are damaged and whether glucocorticoid is responsible for
these defective hBMSCs remains unclear.
Autophagy is a conserved cellular process that
involves the degradation and recycling of dysfunctional and
unnecessary cellular components, protein aggregates and
intracellular pathogens (10).
During the process of autophagy, cytoplasmic targets are enwrapped
within autophagosomes and fused with lysosomes, forming
autolysosomes. These cytoplasmic constituents are then degraded or
recycled (11). This physiological
process is essential for the regulation of cells and their
responses to diverse stimuli. Several studies have now demonstrated
that autophagy is able to regulate the function of osteoclasts
(12), osteoblasts (13) and osteocytes (14), suggesting this process to be
essential in bone homeostasis.
One study demonstrated that glucocorticoids had a
positive effect on osteocyte survival via the induction of
autophagy (15). Another study
observed that autophagy was able to regulate cell reprogramming.
This reduces the level of intercellular reactive oxygen species,
thus maintaining the ability of the cell to self-renew during stem
cell differentiation (16).
However, whether autophagy is involved in glucocorticoid-induced
damage to BMSCs remains to be elucidated.
The current study hypothesized that glucocorticoid
therapy impairs proliferation and induces autophagy in the BMSCs.
In addition, glucocorticoid-induced autophagy was hypothesized to
be involved in maintaining proliferation and preventing apoptosis
in BMSCs.
Materials and methods
Animals and experimental procedures
A total of 20 female Sprague-Dawley rats (4 months
old; average weight, 235±19.4 g) obtained from the Laboratory
Animal Centre of Fourth Military Medical University (Xi’an, China)
were randomly divided into two groups with five rats in each group.
Randomization was performed using a random number table. The groups
were divided as follows: i) Placebo, 10 ml/kg distilled water; and
ii) GIOP (prednis), 5 mg/kg prednisolone (Sigma-Aldrich, St. Louis,
MO, USA). All treatments were administered daily via oral gavage,
for 8 weeks prior to sacrifice by cervical vertebra luxation. All
experimental procedures were approved by the Institutional Ethics
Review Board of Xijing Hospital (permission code 20110405-5).
Cell culture
Bone marrow was obtained from the tibia and femur of
Sprague-Dawley rats and seeded into 75 cm2 culture
flasks with α-modified minimum essential media (α-MEM; Thermo
Fisher Scientific, Waltham, MA, USA), containing 10% fetal bovine
serum and 1% penicillin-streptomycin (all from Gibco Life
Technologies, Carlsbad, CA, USA) under conditions of 5%
CO2 and 37°C. When 80% confluence was reached, the cells
were detached using 0.25% trypsin-EDTA (Gibco Life Technologies)
and passaged at the ratio of 1:2. The number of nonadherent
hematopoietic cells reduced during consecutive passages. All cells
used in the experiment were used at passage 3.
Cell treatment
The BMSCs were treated with various concentrations
(0 M, 10−8 M, 10−7 M, 10−6 M) of
the glucocorticoid dexamethasone (Dex; Sigma-Aldrich) for 48 h. The
autophagy inhibitor, 3-methyladenine (3-MA; Sigma-Aldrich) was
added to the culture media with a final concentraion of 5 mM in
order to investigate the role and mechanism of autophagy in the
proliferation and apoptosis of BMSCs.
Cell proliferation assay
Following Dex treatment, the Cell Counting Kit-8
(CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was
used to measure cell growth, in accordance with the manufacturer’s
instructions, and cells were measured at wavelengths of 450 and 630
nm using a Thermo Labsystems Multiscan MK-3 enzyme-linked
microplate reader (Thermo Fisher Scientific).
Detection of autophagosomes by
transmission electron microscopy (TEM)
Cells were detached from the plates using 0.25%
trypsin trypsin-EDTA (Gibco Life Technologies) and fixed with 2%
paraformaldehyde/2% glutaraldehyde (Sigma-Aldrich) in 0.2 M sodium
cacodylate buffer (pH 7.4; Sigma-Aldrich). Cell pellets were
post-fixed with 1% (v/v) osmic acid (Sigma-Aldrich) in sodium
cacodylate buffer and were stained with 1% uranyl acetate (Amresco,
Solon, OH, USA). Following dehydration, the pellets were embedded
in Durcupan (Sigma-Aldrich). Ultrathin sections (50 nm) were
prepared using an Ultrotome Ultracut S (Leica Microsystems,
Wetzlar, Germany) and images were captured with a JEM-1230
transmission electron microscope (JEOL, Ltd., Tokyo, Japan).
Immunofluorescence
Cells were cultured in 4-well chamber slides (Thermo
Fisher Scientific) and treated in accordance with the
manufacturer’s instructions for immunostaining. Cells were fixed in
4% paraformaldehyde for 15 min, permeabilized with 100% methanol
(Sigma-Aldrich) for 10 min and were then incubated with the primary
antibody (1:200; monoclonal rabbit anti-rat LC3B antibody; Cell
Signaling Technology, Inc., Danvers, MA, USA) overnight, followed
by the secondary antibody (1:200; monoclonal goat anti-rabbit
DyLight 594; Abcam, Cambridge, MA, USA) for 1 h. Subsequent to
incubation with DAPI (0.5 μg/ml; KeyGEN, Nanjing, China) for 5 min,
the cells were analyzed using a FluoView FV1000 confocal laser
scanning microscope (Olympus Corporation, Tokyo, Japan). The
percentage of positively stained cells was calculated in three
random fields.
Western blot analysis
Subsequent to collection of the cells, the proteins
were extracted using lysis buffer (Beyotime, Shanghai, China), the
cell lysates were resolved using SDS-PAGE (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and were electrophoretically transferred
to nitrocellulose membranes (Bio-Rad Laboratories, Inc.).
Subsequent to blocking with 5% non-fat milk for 1 h at room
temperature, the membranes were incubated with primary antibodies,
including Monoclonal mouse anti-rat immunoglobulin G (IgG) β-actin
(1:10,000; Sigma-Aldrich) and anti-LC3B (1:1,000) followed by the
horseradish peroxidase-conjugated secondary antibodies (mouse
anti-mouse or mouse anti-rabbit IgG; 1:2000; Beyotime, Shanghai,
China). Proteins were visualized using a SuperSignal West Dura
Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford,
IL, USA).
Apoptosis assay
The culture medium was changed to serum-free α-MEM
for 12 h in order to induce apoptosis. Apoptotic cells were
identified by terminal deoxynucleotidyl transferase dUTP nick end
labeling (TUNEL) staining using an In Situ Cell Death
Detection Kit, Fluorescein (Roche Diagnostics, Indianapolis, IN,
USA) according to the manufacturer’s instructions. The cells were
incubated with DAPI (0.5 μg/ml) for 5 min and analyzed under a
confocal microscope (FluoView FV1000; Olympus Corp.). Green
indicated TUNEL-positive cells, and the percentage of positive
cells was calculated in three random fields
Statistical analysis
Analysis was performed using SPSS software Version
15.0 (SPSS Inc., Chicago, IL, USA). Quantitative data are presented
as the mean ± standard deviation and compared using Student’s
t-test to determine the significance between two groups or one-way
analysis of variance followed by a post hoc test among three or
more groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Autophagy was induced in BMSCs from GIOP
rats
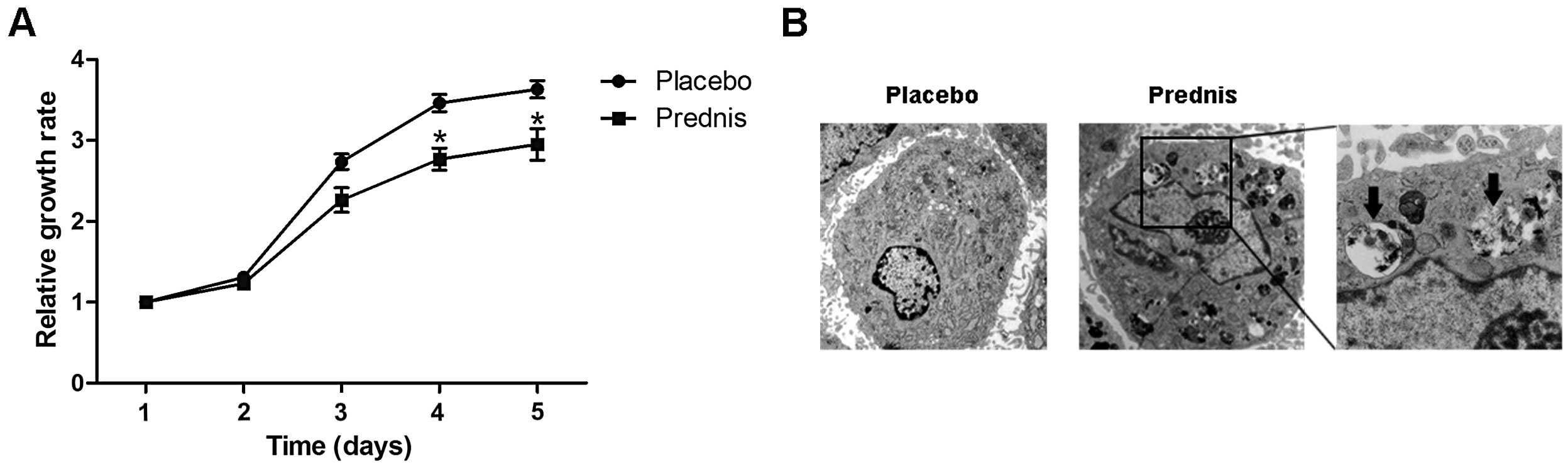
Prior to testing for autophagy, the proliferative
ability of BMSCs in GIOP rats was compared with that of control
rats. CCK-8 was used to determine cell number. The number of BMSCs
in the GIOP group was lower than in the placebo group, suggesting a
reduced proliferative ability (Fig.
1A). The relative growth rates on days four and five were
significantly lower than those in the placebo group (P<0.05).
Using a transmission electron microscope, autophagy was observed in
the BMSCs of the GIOP group, but not in those of the placebo group
(Fig. 1B).
Dex induced autophagy in BMSCs in
vitro
To further elucidate whether glucocorticoids induce
autophagy in BMSCs, the BMSCs were treated with three
concentrations of Dex (10−8 M, 10−7 M and
10−6 M). Following 48-h culture with Dex, autophagy was
detected by TEM. Autophagy was morphologically characterized by the
formation of autophagic vacuoles, also termed autophagosomes.
Thin-section electron microscopic analysis identified the
accumulation of autophagosomes in all Dex-treated groups, whereas
they were scarcely observed in the control group (Fig. 2A). When autophagy occurs,
microtubule-associated protein light chain 3 (LC3) is recruited and
aggregates in the cytoplasm, thus the expression of LC3 and the
increased level of LC3-II are important markers of autophagy.
Immunofluorescence analysis demonstrated that treatment with Dex
increased the number of LC3-positive cells (Fig. 2B and C). Compared with the control
group (3.7±1.2%), the percentage of LC3-positive cells was
significantly increased in all Dex-treated cell groups (9.3±2.3%,
8.7±2.6% and 10.8±3.6% in the 10−8, 10−7 and
10−6 M groups, respectively; P<0.05). The results
from the western blot analysis were consistent with the
immunofluorescence analysis, demonstrating a greater LC3-II:LC3-I
ratio in Dex-treated groups compared with the control (Fig. 2D).
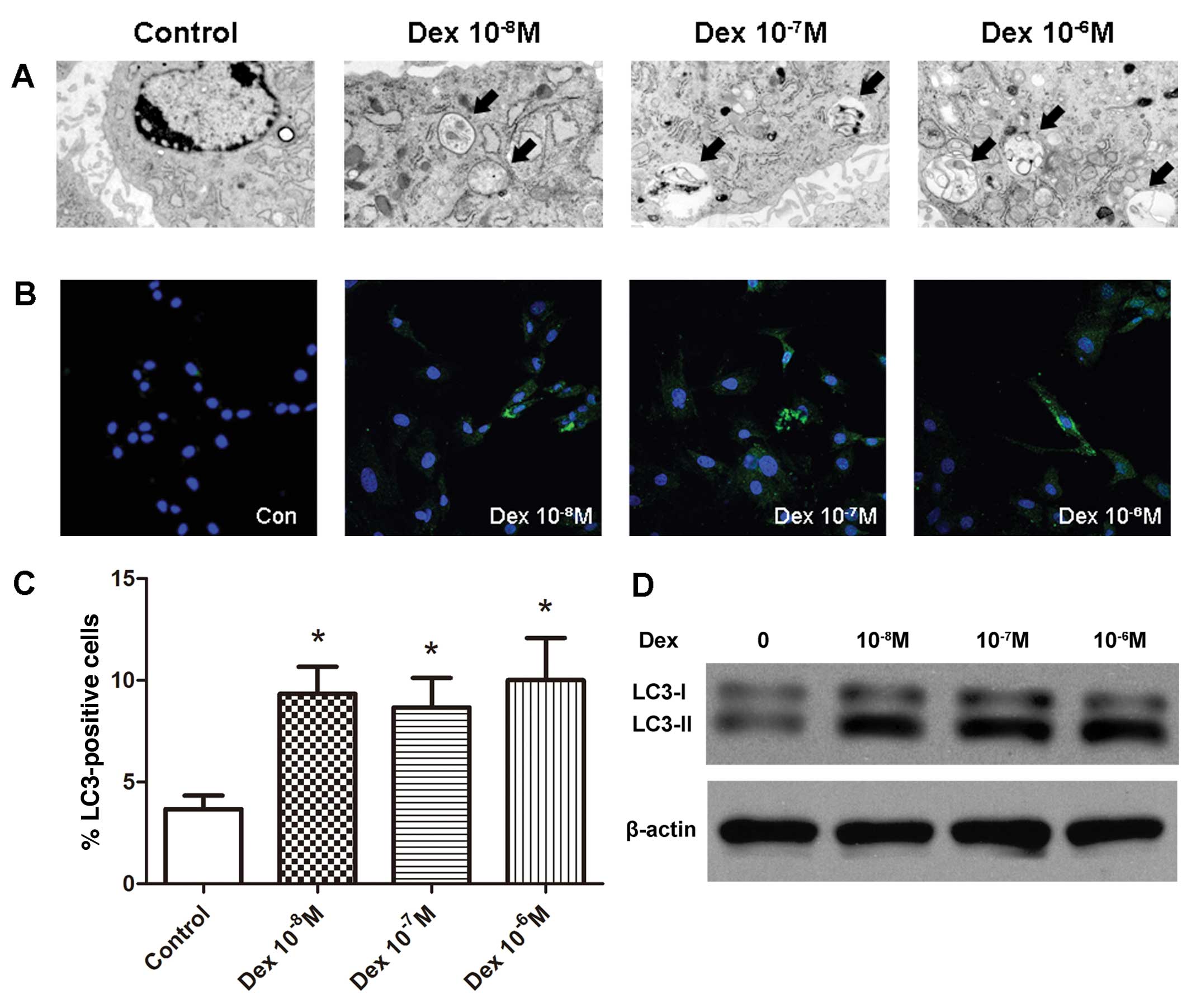 | Figure 2Induction of autophagy in BMSCs in
vitro by Dex. (A) Representative micrographs demonstrating an
increased number of autophagosomes in all Dex-treated groups
compared with the control group. The arrows indicate the
autophagosomes (magnification, ×6,000). (B) Confocal microscopic
analysis of BMSCs following immunofluorescent staining using an
anti-LC3 antibody and labeled with the nuclear marker DAPI
(magnification, ×400). LC3, green; nuclei, blue. (C) Number of
LC3-positive cells as a percentage of the positively stained BMSCs.
*P<0.05 vs. control, all data are presented as the
mean ± standard deviation and represents the results of three
separate experiments.. (D) BMSCs were subject to immunoblotting
analysis using anti-LC3 or anti-β-actin antibodies, and the
autophagic marker LC3-II was induced by various concentrations of
Dex. BMSC, bone marrow mesenchymal stem cell; Dex, dexamethasone;
LC3, light chain 3. |
Autophagy maintained the proliferation
ability of BMSCs
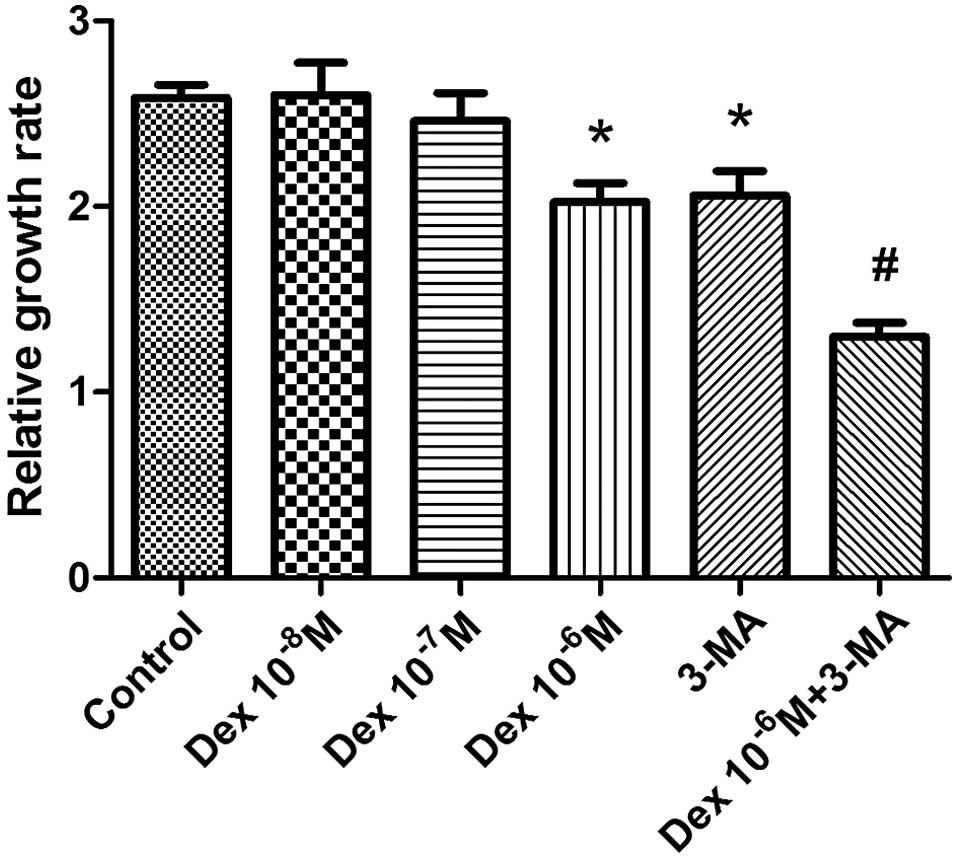
To determine the effect of glucocorticoid
administration on the proliferation of BMSCs, the BMSCs were
treated with three concentrations of Dex (10−8 M,
10−7 M or 10−6 M) for 48 h and then
proliferation was evaluated by CCK-8 assay. The 10−6 M
concentration of Dex was observed to significantly inhibit the
proliferation and number of BMSCs (P<0.05), while
10−8 and 10−7 M Dex did not produce
significant alterations in proliferation, compared with the control
group (Fig. 3). The autophagy
inhibitor 3-MA was used to investigate the involvement of autophagy
in the damage resulting from Dex treatment of BMSCs. The addition
of 3-MA was observed to further reduce proliferation compared with
the control group (P<0.05), indicating that autophagy partially
maintained the proliferation of BMSCs. In addition, the relative
growth rate was significantly reduced when BMSCs were co-treated
with 10−6 M Dex and 3-MA, suggesting that autophagy
protected BMSCs from the negative effects on proliferation
resulting from Dex treatment.
Autophagy protects against apoptosis in
BMSCs
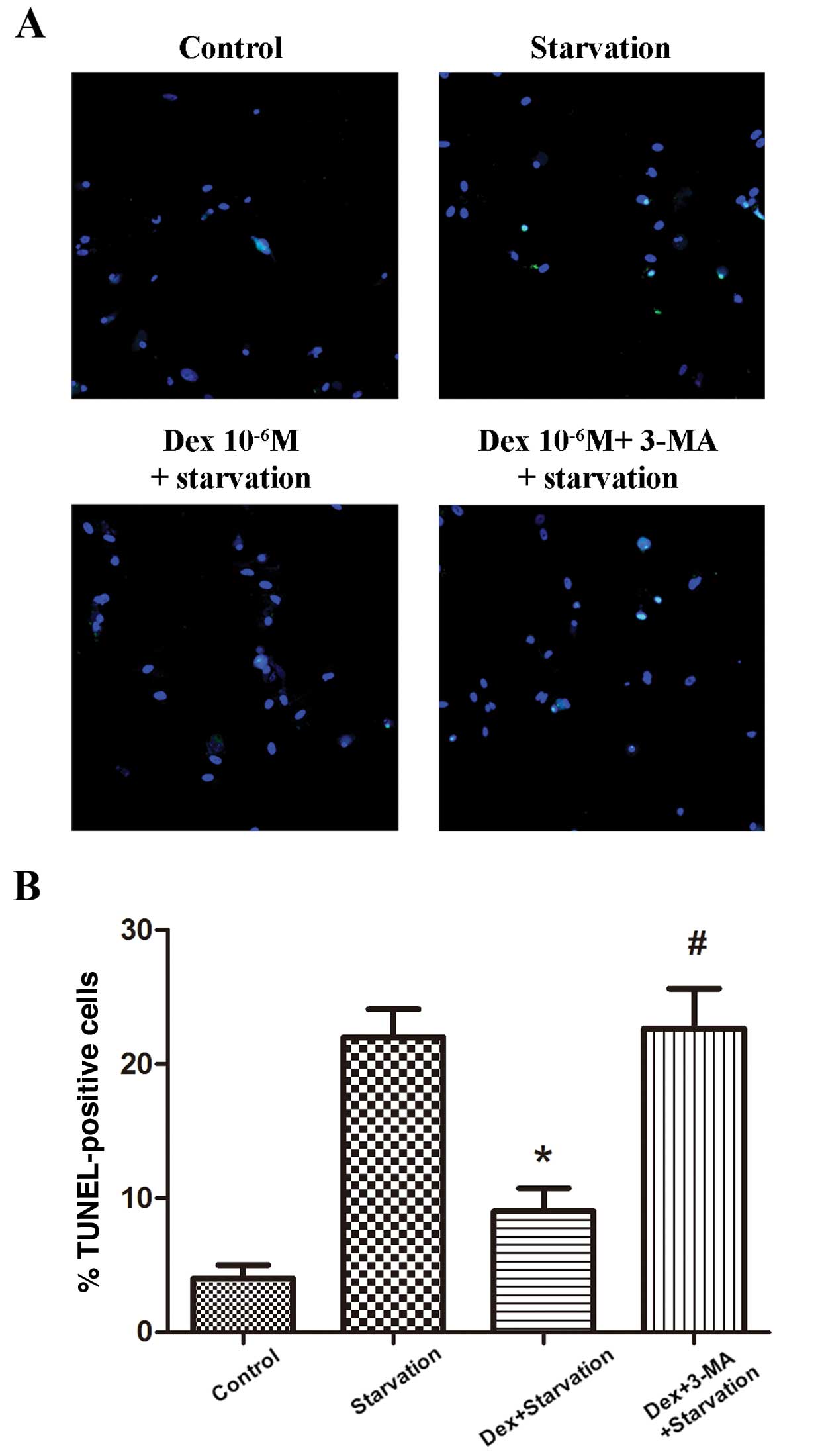
To investigate the effect of autophagy induced by
Dex on apoptosis in BMSCs, the culture medium was changed to
serum-free α-MEM for 12 h to induce apoptosis subsequent to culture
with 10−6 M Dex in the absence or presence of 3-MA
(Fig. 4). TUNEL assay was used to
detect the apoptotic cells. The results demonstrated that, compared
with the starvation group (22.8±3.6%), 10−6 M Dex
significantly reduced the size of the population of apoptotic BMSCs
(9.3±2.9%; P<0.01). The use of 3-MA abrogated the anti-apoptotic
effects of Dex, as identified by the increase in apoptotic cells
(22.6±5.2%; P<0.05). These results suggest that Dex protected
BMSCs from the serum deprivation-induced apoptosis, and that
autophagy induced by Dex may be a possible mechanism for this.
Discussion
Glucocorticoids have been widely used in the
treatment of non-infectious inflammation and autoimmune disorders.
However, the side effects of glucocorticoid therapy contribute to
low bone mass and the occurrence of bone fracture, via disruption
to the metabolism and homeostasis of bone tissue. The current study
demonstrated that the proliferative ability of BMSCs derived from
GIOP rats and Dex-treated BMSCs was impaired. In addition,
autophagy was observed in the two types of BMSCs, and the
inhibition of autophagy resulted in a further reduction in
proliferation. Furthermore, Dex treatment alleviated the levels of
apoptosis in starvation-induced BMSCs. These results suggest that
autophagy, induced by glucocorticoid therapy, was self-protective
in the maintenance of BMSCs.
A previous study using similar methods established
that the bone density, the trabecular bone mass and the capacity to
form new bone were reduced in GIOP model rats compared with
controls (17). In the current
study, the proliferative ability of GIOP-BMSCs was observed to be
impaired, suggesting that long-term oral glucocorticoid therapy
impairs the functional activity of BMSCs. This was hypothesized to
be the cause of the reduction in bone formation in the GIOP model.
Also, the appearance of autophagy in GIOP-BMSCs indicated that the
application of glucocorticoids put BMSCs under stressful
conditions.
Dex is a potent glucocorticoid that has been
demonstrated to induce autophagy in BMSCs in several experiments
(18), the present study examined
LC3 in BMSCs using immunostaining and western blot analysis. LC3
was observed to accumulate in the cytoplasm, and the ratio of
LC3-II:LC3-I increased with the addition of Dex. LC3-II and LC3-I
are important indicators of autophagy.
The results of the current study demonstrated that
all three concentrations of Dex (10−8 M, 10−7
M and 10−6 M) were able to significantly increase the
occurrence of autophagy, but not in a dose-dependent manner. This
observation was in line with the results of a study by Xia et
al (15), which identified
that Dex was able to induce autophagy in osteocytes.
Dex was used to stimulate the differentiation of
BMSCs. However, the regulatory effects of Dex on BMSC proliferation
have been reported to be diverse, depending on the different dosage
and species used (19,20). The results of the current study
demonstrated that 10−8 and 10−7 M doses of
Dex did not affect the proliferation of BMSCs, however,
10−6 M Dex resulted in a significant reduction. This
observation indicates that a high dosage of Dex inhibited the
proliferation of BMSCs, which is consistent with the observations
in the GIOP-BMSCs.
A study by Oliver et al (21) demonstrated that hBMSCs exhibited a
high level of constitutive autophagy and that the knockdown of
B-cell lymphoma-XL suppressed autophagy, influencing the survival
and differentiation of the hBMSCs. To confirm the role of autophagy
in the maintenance of BMSC proliferation, 3-MA (an inhibitor of
autophagy) was used. The application of 3-MA was observed to result
in a reduction in BMSC survival, suggesting a protective effect of
autophagy. In the current study, autophagy alleviated the negative
effect of Dex on BMSC proliferation.
Dex has been previously demonstrated to exhibit an
effect on the apoptosis of cells (22,23),
and it has also been identified to inhibit confluence-induced
apoptosis of hBMSCs and negatively regulate the expression of
apoptosis-associated genes (24,25).
The crosstalk between autophagy and apoptosis is complex and
crucial in the determination of cell fate (26). A study by Zhang et al
(27) reported that autophagy was
able to protect MSCs from apoptosis induced by low oxygen, in
addition to serum deprivation. The results of the current study
demonstrated that Dex treatment protected against
starvation-induced apoptosis to a certain degree. However, Wang
et al (28) observed an
exacerbation of starvation-induced apoptosis with the
administration of 10−6 M Dex. This discrepancy may be
due to the different time-courses of Dex treatment and cell types
used in the different studies.
Autophagy, as mentioned, has been reported to
maintain cell activity through removing damaged cellular
components. However, a study reported that Dex-induced autophagy
leads to cell death due to the destruction of certain components
necessary for cell survival (29).
The end result of autophagy may depend on the type of cells
involved and the severity or time-course of stimuli (30). The results of the current study
suggested that upon stimulation with 10−6 M Dex,
autophagy protected BMSCs from apoptosis. However, whether
autophagy remained protective or became detrimental when the
glucocorticoid dose was increased, or the time-course was
prolonged, remains unclear. Whether or not autophagy-induced cell
death is involved in the pathogenesis of osteoporosis requires
further investigation.
In summary, for the first time, to the best of our
knowledge, glucocorticoid therapy was demonstrated to induce
autophagy in BMSCs and maintain the proliferative ability of BMSCs.
Autophagy induced by Dex may protect BMSCs from starvation-induced
apoptosis, suggesting this as a survival mechanism against
cell-death. Thus, the regulation of autophagy should be considered
as a novel strategy to enhance the activity of BMSCs and increase
bone mass in GIOP.
Acknowledgements
The current study was supported by the Ministry of
Science and Technology of China (grant no. 2011CB964703); National
High Technology Research and Development Program 863 (grant no.
2012AA020502); and the China Postdoctoral Science Foundation (grant
nos. 20100480093 and 2012T50856).
References
|
1
|
Saag KG, Shane E, Boonen S, et al:
Teriparatide or alendronate in glucocorticoid-induced osteoporosis.
N Engl J Med. 357:2028–2039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weinstein RS: Glucocorticoid-induced
osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am.
41:595–611. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fraser LA and Adachi JD:
Glucocorticoid-induced osteoporosis: treatment update and review.
Ther Adv Musculoskelet Dis. 1:71–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O’Brien CA, Jia D, Plotkin LI, et al:
Glucocorticoids act directly on osteoblasts and osteocytes to
induce their apoptosis and reduce bone formation and strength.
Endocrinology. 145:1835–1841. 2004. View Article : Google Scholar
|
|
5
|
Jia D, O’Brien CA, Stewart SA, et al:
Glucocorticoids act directly on osteoclasts to increase their life
span and reduce bone density. Endocrinology. 147:5592–5599. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kanis JA, Johansson H, Oden A, et al: A
meta-analysis of prior corticosteroid use and fracture risk. J Bone
Miner Res. 19:893–899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Long MW: Osteogenesis and
bone-marrow-derived cells. Blood Cells Mol Dis. 27:677–690. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miura M, Chen XD, Allen MR, et al: A
crucial role of caspase-3 in osteogenic differentiation of bone
marrow stromal stem cells. J Clin Invest. 114:1704–1713. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou Z, Apte SS, Soininen R, et al:
Impaired endochondral ossification and angiogenesis in mice
deficient in membrane-type matrix metalloproteinase I. Proc Natl
Acad Sci USA. 97:4052–4057. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong AS, Cheung ZH and Ip NY: Molecular
machinery of macroautophagy and its deregulation in diseases.
Biochim Biophys Acta. 1812:1490–1497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burman C and Ktistakis NT: Autophagosome
formation in mammalian cells. Semin Immunopathol. 32:397–413. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hocking LJ, Mellis DJ, McCabe PS, Helfrich
MH and Rogers MJ: Functional interaction between sequestosome-1/p62
and autophagy-linked FYVE-containing protein WDFY3 in human
osteoclasts. Biochem Biophys Res Commun. 402:543–548. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Darcy A, Meltzer M, Miller J, et al: A
novel library screen identifies immunosuppressors that promote
osteoblast differentiation. Bone. 50:1294–1303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Onal M, Piemontese M, Xiong J, et al:
Suppression of autophagy in osteocytes mimics skeletal aging. J
Biol Chem. 288:17432–17440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xia X, Kar R, Gluhak-Heinrich J, et al:
Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res.
25:2479–2488. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vessoni AT, Muotri AR and Okamoto OK:
Autophagy in stem cell maintenance and differentiation. Stem Cells
Dev. 21:513–520. 2012. View Article : Google Scholar
|
|
17
|
Folwarczna J, Pytlik M, Sliwiński L, et
al: Effects of propranolol on the development of
glucocorticoid-induced osteoporosis in male rats. Pharmacol Rep.
63:1040–1049. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Both SK, van der Muijsenberg AJ, van
Blitterswijk CA, de Boer J and de Bruijn JD: A rapid and efficient
method for expansion of human mesenchymal stem cells. Tissue Eng.
13:3–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong L, Sultana H, Paulius K and Zhang G:
Steroid regulation of proliferation and osteogenic differentiation
of bone marrow stromal cells: a gender difference. J Steroid
Biochem Mol Biol. 114:180–185. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oliver L, Hue E, Priault M and Vallette
FM: Basal autophagy decreased during the differentiation of human
adult mesenchymal stem cells. Stem Cells Dev. 21:2779–2788. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oh HY, Namkoong S, Lee SJ, et al:
Dexamethasone protects primary cultured hepatocytes from death
receptor-mediated apoptosis by upregulation of cFLIP. Cell Death
Differ. 13:512–523. 2006. View Article : Google Scholar
|
|
23
|
Dorscheid DR, Low E, Conforti A, Shifrin
S, Sperling AI and White SR: Corticosteroid-induced apoptosis in
mouse airway epithelium: effect in normal airways and after
allergen-induced airway inflammation. J Allergy Clin Immunol.
111:360–366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song IH, Caplan AI and Dennis JE:
Dexamethasone inhibition of confluence-induced apoptosis in human
mesenchymal stem cells. J Orthop Res. 27:216–221. 2009. View Article : Google Scholar
|
|
25
|
Xiao Y, Peperzak V, van Rijn L, Borst J
and de Bruijn JD: Dexamethasone treatment during the expansion
phase maintains stemness of bone marrow mesenchymal stem cells. J
Tissue Eng Regen Med. 4:374–386. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Q, Yang YJ, Wang H, et al: Autophagy
activation: a novel mechanism of atorvastatin to protect
mesenchymal stem cells from hypoxia and serum deprivation via
AMP-activated protein kinase/mammalian target of rapamycin pathway.
Stem Cells Dev. 21:1321–1332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Pang B, Li Y, Zhu D, Pang T and
Liu Y: Dexamethasone has variable effects on mesenchymal stromal
cells. Cytotherapy. 14:423–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laane E, Tamm KP, Buentke E, et al: Cell
death induced by dexamethasone in lymphoid leukemia is mediated
through initiation of autophagy. Cell Death Differ. 16:1018–1029.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Codogno P and Meijer AJ: Autophagy and
signaling: their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|