Introduction
Heterozygous missense mutations in β-tubulin isotype
III (TUBB3), which is exclusively expressed in neurons
(1), result in the TUBB3
syndrome, which includes congenital fibrosis of the extraocular
muscle type 3 (CFEOM3), intellectual impairments, facial paralysis
and/or an axonal sensorimotor neuropathy (2,3).
These findings suggested that TUBB3 has a role in mediating
axon guidance and maturation (4,5).
Brain magnetic resonance imaging (MRI) of patients with
TUBB3 mutations has demonstrated evidence of dysgenesis of
the corpus callosum, anterior commissure and internal capsule as
well as generalized loss of white matter (4). However, to the best of our knowledge,
there has been no report evaluating the features of lower limb MRIs
of patients with TUBB3 mutations.
Patients in the same family who harbored the D417N
mutation in TUBB3 exhibited a broad clinical spectrum,
including CFEOM3 only; mixed features of CFEOM3 with peripheral
neuropathy, learning disabilities and developmental delay;
peripheral neuropathy only or no clinical symptoms (3,4).
Therefore, the TUBB3 mutation may cause an isolated axonal
sensorimotor polyneuropathy and detailed descriptions of the
histopathology of sural nerve and lower extremity MRI results
require further evaluation.
The present study evaluated individuals exhibiting
an axonal sensorimotor polyneuropathy with the D417N mutation in
TUBB3, which was identified by whole exome sequencing (WES),
and reports the pathophysiological results observed.
Materials and methods
Patients
The present study enrolled five members of a Korean
family which was severely affected by axonal Charcot-Marie-Tooth
(CMT) (FC423), including two affected individuals. A healthy
45-year-old male was enrolled for nerve histology study. Written
informed consent was obtained from all participants according to
the protocol approved by the Institutional Review Board for Ewha
Womans University, Mokdong Hospital (approval no. ECT 11-58-37;
Seoul, Korea).
DNA preparation and whole exome
sequencing
DNA from peripheral blood was isolated using a
QIAamp blood DNA purification kit (Qiagen, Hilden, Germany). The
isolated DNA was prescreened for duplication of 17p12 (PMP22) and
mutations in the coding exons of GJB1, MPZ, NEFL and MFN2 as
previously described (6). WES and
subsequent filtering were performed in the affected members as
previously described (7).
Capillary sequencing
Mutations in TUBB3 were analyzed by capillary
sequencing. Exon four, harboring the mutation site, was amplified
using the following primers (Macrogen, Seoul, Korea): Forward,
5′-CATCCAGAGCAAGAACAGCA-3′ and reverse, 5′-TTCGTACATCTCGCCCTCTT-3′.
Polymerase chain reaction (PCR) amplifications was performed using
Platinum PCR SuperMix High Fidelity kit (Life Technologies, Grand
Island, NY, USA), with the above primers and the genomic DNA of the
patients. Amplification was performed as follows: 32 cycles of 95°C
for 30 sec, 56°C for 30 sec and 72°C for 60 sec. Following
amplification, the sequences were analyzed using a BigDye
terminator cycle sequencing kit and automatic genetic analyzer
(ABI3130XL; Applied Biosystems, Life Technologies, Foster City, CA,
USA).
Clinical assessments
Clinical assessments were performed by two
independent neurologists as previously described (8). Patients were evaluated by taking a
comprehensive medical history, including details of motor and
sensory impairments, deep tendon reflexes, muscle atrophy and gait
abnormalities, for example walking on heels or toes. The muscle
strength of the flexor and extensor muscles was measured manually
using the Medical Research Council scale (http://www.mrc.ac.uk). In order to determine physical
disability, three scales were used: The functional disability scale
(FDS) (9), CMT neuropathy score
(CMTNS) (10) and sensory
impairment, which was assessed with regard to the level and
severity of pain, temperature, vibration and position.
Electrophysiological examination
Electrophysiological studies were performed using
the Sierra Wave EMG system (Cadwell Laboratories, Inc., Kennewick,
WA, USA) and the Toennies two-channel NeuroScreen system
(Jaeger-Toennies, Hochberg, Germany). Motor nerve conduction
velocities (MNCVs) of the median and ulnar nerves were determined
by electrical stimulation at the elbow or wrist, while recording
compound muscle action potentials (CMAPs) over the abductor
pollicis brevis and adductor digiti quinti, respectively.
Similarly, the MNCVs of the peroneal and tibial nerves were
determined by stimulation at the knee and ankle, while recording
CMAPs over the extensor digitorum brevis and adductor hallucis,
respectively. CMAP amplitudes were measured from baseline to
negative peak values. Sensory nerve conduction velocities were
measured over the finger-wrist segment from the median and ulnar
nerves by orthodromic scoring, and were also recorded for sural
nerves. Sensory nerve action potential (SNAP) amplitudes were
measured from positive to negative peaks. An electromyography (EMG)
was performed in the bilateral proximal and distal limb
muscles.
Lower extremity MRI
MRI study of the proband was performed using the
Siemens Vision 1.5-T system (Siemens, Erlangen, Germany). MRI was
performed in the axial (field of view, 24–32 cm; slice thickness,
10 mm and slice gap, 0.5–1.0 mm) and coronal planes (field of view,
38–40 cm; slice thickness, 4–5 mm and slice gap, 0.5–1.0 mm) using
the following protocol: T1-weighted spin-echo (SE) [repetition time
(TR)/echo time (TE) 570–650/14–20; 512 matrixes], T2-weighted SE
(TR/TE 2800-4000/96-99; 512 matrixes) and fat-suppressed
T2-weighted SE (TR/TE 3090-4900/85-99; 512 matrixes).
Histopathological studies
Histopathological analysis of the distal sural nerve
was performed on the proband. Semi-thin sections were stained with
0.1% Toluidine blue solution (Sigma-Aldrich, St. Louis, MO, USA)
for 3 min and then dehydrated with ethanol (Sigma-Aldrich). The
density of myelinated fibers (MFs), axonal diameter and myelin
thickness were determined using a computer-assisted image analyzer
(AnalySIS 3.0; Soft Imaging System, Münster, Germany). Ultrathin
sections were contrasted with uranyl acetate and lead citrate for
ultrastructural study using a transmission electron microscope
(H-7650; Hitachi, Tokyo, Japan).
Results
Genetic analysis
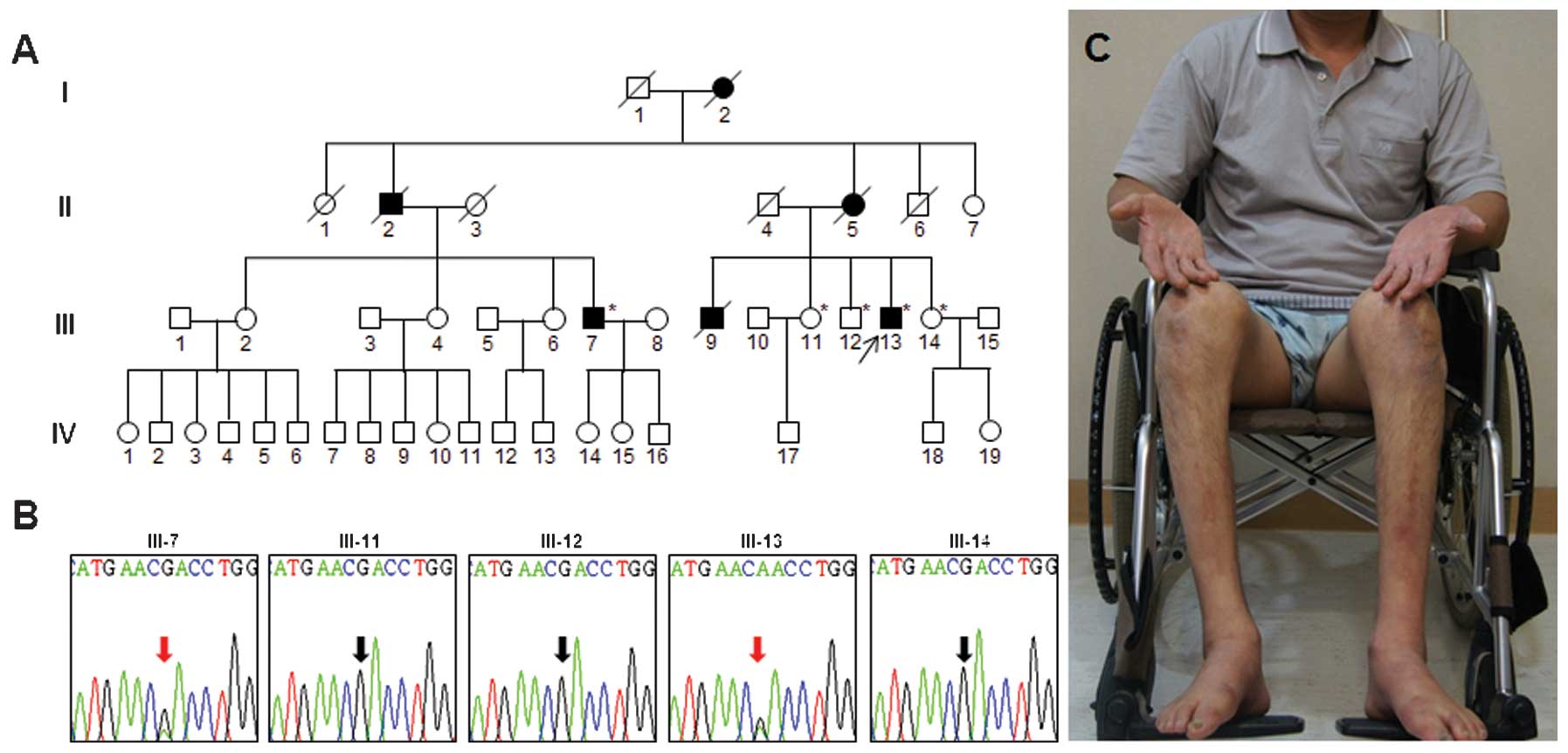
WES was performed in the two affected family members
(Fig. 1A; III-7 and III-13). The
total sequencing yields were 6.63 and 8.67 Gbp, respectively.
Subsequent filtering identified 11 variants, which were shared by
the affected family members (data not shown). Among the variants
identified, the heterozygous D417N (c.1249G>A) mutation in
TUBB3 was shown to be a cause of CFEOM3 (4). Since patients harboring the same
mutation exhibited peripheral neuropathy, it was concluded that the
D417N mutation was the putative cause of the clinical features
observed in the patients examined in the present study. Capillary
sequencing confirmed that the mutation was co-segregated within the
family members (Fig. 1B).
Clinical manifestations
The proband (Fig.
1A, III-13; 48 years old) initially exhibited gait disturbance
at 12 years of age. Progressive gait impairment was identified and
the proband became wheelchair bound at 40 years of age (Fig. 1C). At 48 years, significant muscle
weakness and atrophy of the distal lower limbs was observed. All
sensory modalities were severely impaired and the vibration sense
was more markedly affected than the pinprick sense. Deep tendon
reflexes were absent and pathological reflexes were not present in
all limbs. However, the proband did not demonstrate
ophthalmoplegia, blepharoptosis or strabismus (Fig. 2). Mini-mental state examination and
all cranial nerve examinations were normal.
By contrast, a 60-year-old cousin of the proband
(Fig. 1A; III-7) began to
experience gait disabilities when he was 45 years old and, unlike
the proband, was still able to walk using ankle-foot devices.
Neurological examination at 60 years of age revealed bilateral pes
cavus and distal muscle atrophy of the upper and lower limbs. Based
on the medical history notes taken, the deceased family members
(I-2, II-2, II-5 and III-9) had exhibited distal lower limb
weakness and gait disturbance. Furthermore, none had experienced
any eye symptoms.
Axonal neuropathies identified by
electrophysiological analysis
Nerve conduction studies were performed on the two
patients aged 48 and 60 years, respectively (Table I). The values of the median MNCV
were from 39.1–40.6 m/s, and those of the ulnar MNCV were from
40.0–53.7 m/s. Right CMAPs of the median nerves were below normal
range, whereas those of the ulnar nerve were normal. Tibial and
peroneal MNCVs were not elicited and SNAPs of the median, ulnar and
sural nerves were absent. A needle EMG identified fibrillation
potentials, positive sharp waves and neurogenic motor unit action
potentials.
 | Table IElectrophysiological features of
patients with the D417N mutation in the TUBB3 gene. |
Table I
Electrophysiological features of
patients with the D417N mutation in the TUBB3 gene.
| Features | III-7 | III-13 | Normal value |
|---|
| Age at exam
(years) | 60 | 48 | |
| Side | Right | Left | Right | Left | |
| Median nerve |
| TL (ms) | 6.8 | 5.5 | 6.0 | 5.4 | <3.9 |
| CMAP (mV) | 5.6 | 6.9 | 0.8 | 2.2 | >6.0 |
| MNCV (m/s) | 39.1 | 40.6 | 39.7 | 39.7 | >50.5 |
| Ulnar nerve |
| TL (ms) | 4.4 | 3.8 | 3.6 | 3.7 | <3.0 |
| CMAP (mV) | 10.9 | 8.8 | 12.6 | 12.0 | >8.0 |
| MNCV (m/s) | 40.0 | 45.7 | 52.3 | 53.7 | >51.1 |
| Peroneal nerve |
| TL (ms) | A | A | A | A | <5.3 |
| CMAP (mV) | A | A | A | A | >1.6 |
| MNCV (m/s) | A | A | A | A | >41.2 |
| Tibial nerve |
| TL (ms) | A | A | A | A | <5.4 |
| CMAP (mV) | A | A | A | A | >6.0 |
| MNCV (m/s) | A | A | A | A | >41.1 |
| Median sensory
nerve |
| SNAP (μV) | A | A | A | A | >8.8 |
| SNCV (m/s) | A | A | A | A | >39.3 |
| Ulnar sensory
nerve |
| SNAP (μV) | A | A | A | A | >7.9 |
| SNCV (m/s) | A | A | A | A | >37.5 |
| Sural nerve |
| SNAP (μV) | A | A | A | A | >6.0 |
| SNCV (m/s) | A | A | A | A | >32.1 |
Lower extremity MRI results are
length-dependent
MRIs of the lower extremities of the proband
revealed hyperintense signal abnormalities in the lower leg muscles
(Fig. 3A), whereas brain MRIs
exhibited normal features (data not shown). T1-weighted images
identified marked muscle atrophy with signal alterations observed
in the lower leg muscles compared to those of the hip and thigh
muscles. The predominant atrophy of the distal muscles was
consistent with the length-dependent neuropathy hypothesis. At the
hip (Fig. 3B) and thigh (Fig. 3C) levels, the fatty involvement of
compartment muscles was not observed. By contrast, almost all
muscles in the lower extremities demonstrated diffuse atrophies and
fatty hyperintense signal changes (Fig. 3D).
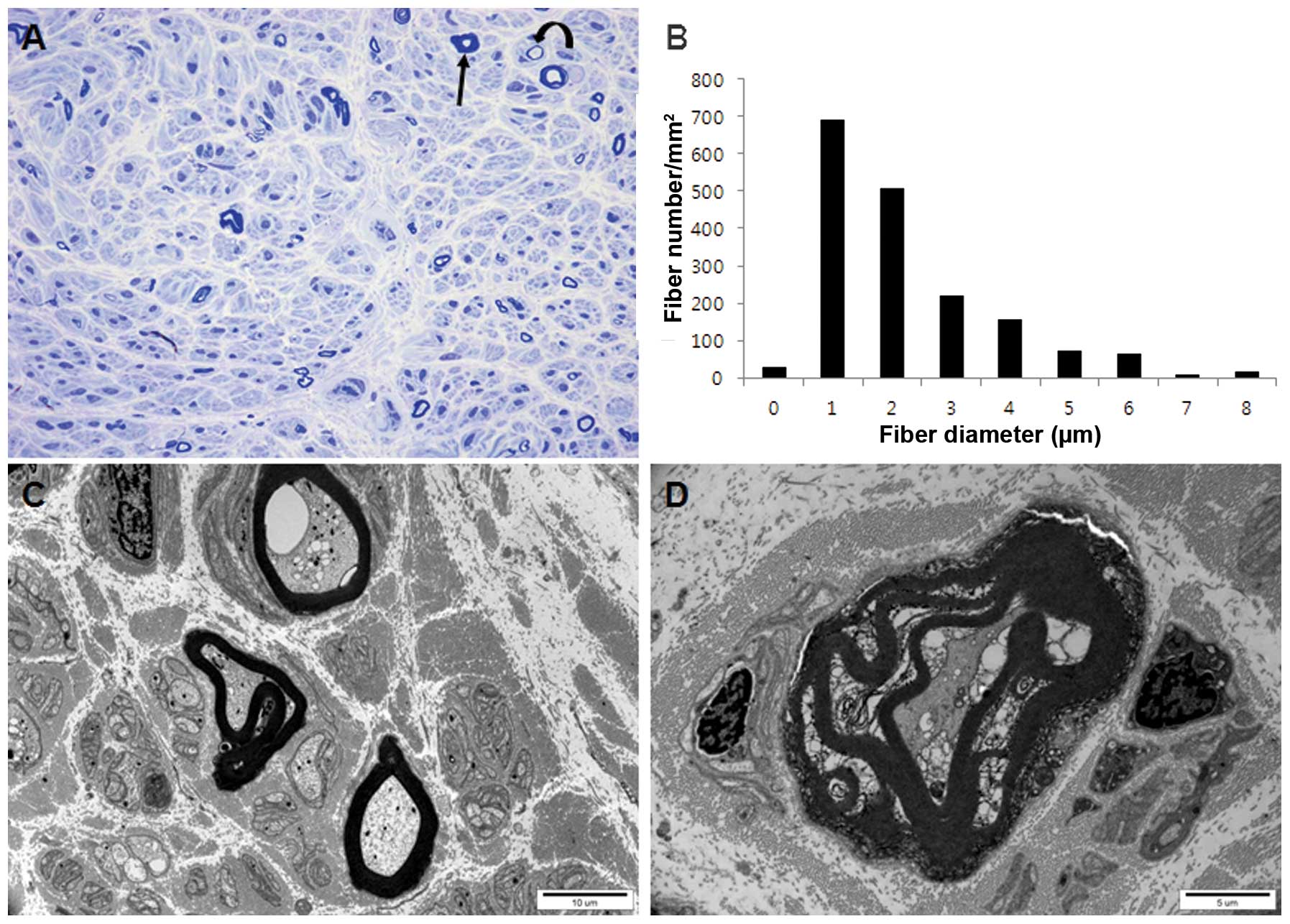
Histopathological observations
Histopathological examination of the distal sural
nerve of the proband (III-13) aged 48 years revealed an absence of
large MFs, with the presence of small- or medium-sized and thinly
scattered MFs (Fig. 4A).
Regenerating axonal clusters and thick MFs were rarely identified.
The number of remaining MFs (1,770/mm2) was markedly
lower than that of a healthy 45-year-old male volunteer
(7,300/mm2). The range and average MF diameter were
smaller than age-matched controls and the histogram identified a
unimodal distribution pattern (Fig.
4B). Electron microscopic examination revealed scattered
myelinated and unmyelinated axons with swelling or vacuolization of
the axoplasm as well as degenerated abnormal mitochondria and
membranous structures (Fig. 4C).
Degenerating MFs with the breakdown of myelin and axons were rarely
noted (Fig. 4D). Although there
were thinly scattered MFs, no further evidence of demyelination,
including demyelinated axons or onion bulb formation, were
present.
Discussion
The present study revealed that a Korean family with
a TUBB3 mutation exhibited an axonal sensorimotor
neuropathy. Histopathological analysis of the sural nerve biopsy of
the proband revealed features characteristic of an axonal
neuropathy, including a decreased number of MFs, the absence of
large MFs and abnormally thick MFs. However, CFEOM3,
ophthalmoplegia and intellectual impairment were not observed in
the two patients. According to previous studies, the clinical
features of the D417N mutation in TUBB3 are heterogeneous
(4). Tischfield et al
(4) compared the phenotypes of 15
individuals from four families harboring the mutation: Three
patients exhibited CFEOM3 only, four patients exhibited CFEOM3 with
peripheral neuropathy, two patients exhibited CFEOM3 with
peripheral neuropathy and developmental delay, three patients
exhibited peripheral neuropathy only, one patient exhibited CFEOM3
with peripheral neuropathy and learning disability, one patient
exhibited CFEOM3 with learning disability and one patient was a
non-penetrant carrier. Therefore, one-fifth of the patients
exhibited a peripheral neuropathy phenotype only. An alternate
amino acid change at the same site (D417H) also resulted in a
heterogeneous phenotype within a family: All three affected family
members exhibited CFEOM, wrist and finger contractures and facial
weakness. However, one family member exhibited peripheral
neuropathy, while another exhibited peripheral neuropathy with
additional developmental delay and the third did not exhibit
peripheral neuropathy (3). These
results supported the hypothesis that mutations at the D417
position result in a broad phenotypic spectrum, suggesting that the
clinical features may depend on the genetic background of each
individual. In the present study, in comparison to the proband, his
cousin experienced late-onset symptoms and a mild phenotype.
Therefore, these patients exhibited clinical diversity within the
family.
Application of WES was found to be a cost- and
time-effective method used to reveal the underlying genetic causes
of rare human diseases (11–13).
Previous studies by our group have also applied WES and
successfully isolated the causative mutations of patients with
peripheral neuropathy (7,14–16).
In the present study, the sole genetic cause of the symptoms
observed was not isolated. However, certain evidence has suggested
that the TUBB3 mutation may be the underlying causative gene
of CMT in the patients examined in the present study. Only the
TUBB3 D417N mutation has been reported to cause peripheral
neuropathy and only four of those genes (APPL2, LMO7, TNRC6C
and TUBB3) were expressed in the sural nerve according to
transcriptome sequencing (data not shown). The TUBB3 D417N
mutation was not found in the Korean controls (n=300) or reported
in the Exome Variant Server (http://evs.gs.washington.edu/EVS/). Therefore, it was
concluded that the TUBB3 mutation was the genetic cause of
the axonal neuropathy observed.
To the best of our knowledge, to date, pathological
features in the sural nerve have not been reported in patients with
TUBB3 mutations. In the present study, a distal sural nerve
biopsy was performed on a patient harboring a TUBB3
mutation. The results identified marked axonal loss, but no other
evidence of demyelination, including demyelinated axons or onion
bulb formation, were present. In addition, the nerve conduction
studies indicated that the median and ulnar nerve conduction
velocities were >38 m/s, and electromyography revealed
neurogenic motor unit action potentials and fibrillation
potentials. These results were compatible with those expected in
axonal neuropathy.
MRI analysis revealed a distinct pattern of muscular
involvement, where markedly greater muscle atrophy with
hyperintense signal changes was identified in the lower leg muscles
than that in the thigh or hip muscles. This was compatible with the
hypothesis of length-dependent axonal degeneration. Previously, a
type-specific pattern of fatty infiltration in muscles was
reported: In demyelinating hereditary motor and sensory neuropathy
(HMSN type 1A) the tibilalis anterior and peronei muscles were
predominantly involved. However, late-onset axonal HMSN type 2A
revealed predominant involvement of the soleus muscle, whereas
early-onset HMSN type 2A exhibited whole calf and muscle
involvements, including moderate to severe fatty changes in the
thigh muscle (17). In the present
study, the proband exhibited whole-compartment calf muscle
involvement with no involvement of the thigh muscles. The proband
therefore exhibited a pattern which differed from those identified
in axonal HMSN type 2A. These findings are likely due to the
variable pathophysiologies of axonal neuropathies.
In the future, specific molecular diagnoses will be
important for the development of personalized therapies for axonal
neuropathy. In this context, the results of the present study may
aid in the development of genetic testing for axonal
neuropathypatients.
Acknowledgements
The present study was supported in part by the
Korean Health Technology R&D Project, Ministry of Health &
Welfare (Sejong, Korea; no. A120182).
References
|
1
|
Katsetos CD, Legido A, Perentes E and Mörk
SJ: Class III beta-tubulin isotype: a key cytoskeletal protein at
the crossroads of developmental neurobiology and tumor
neuropathology. J Child Neurol. 18:851–866. 2003. View Article : Google Scholar
|
|
2
|
Poirier K, Saillour Y, Bahi-Buisson N, et
al: Mutations in the neuronal β-tubulin subunit TUBB3 result in
malformation of cortical development and neuronal migration
defects. Hum Mol Genet. 19:4462–4473. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Demer JL, Clark RA, Tischfield MA and
Engle EC: Evidence of an asymmetrical endophenotype in congenital
fibrosis of extraocular muscles type 3 resulting from TUBB3
mutations. Invest Ophthalmol Vis Sci. 51:4600–4611. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tischfield MA, Baris HN, Wu C, et al:
Human TUBB3 mutations perturb microtubule dynamics, kinesin
interactions, and axon guidance. Cell. 140:74–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang YQ and Oblinger MM: Differential
regulation of beta III and other tubulin genes during peripheral
and central neuron development. J Cell Sci. 103:643–651.
1992.PubMed/NCBI
|
|
6
|
Choi BO, Kim J, Lee KL, Yu JS, Hwang JH
and Chung KW: Rapid diagnosis of CMT1A duplications and HNPP
deletions by multiplex microsatellite PCR. Mol Cells. 23:39–48.
2007.PubMed/NCBI
|
|
7
|
Choi BO, Koo SK, Park MH, et al: Exome
sequencing is an efficient tool for genetic screening of
Charcot-Marie-Tooth Disease. Hum Mutat. 33:1610–1615. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung KW, Hyun YS, Lee HJ, et al: Two
recessive intermediate Charcot-Marie-Tooth patients with GDAP1
mutations. J Peripher Nerv Syst. 16:143–146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Birouk N, LeGuern E, Maisonobe T, et al:
X-linked Charcot-Marie-Tooth disease with connexin 32 mutations:
clinical and electrophysiologic study. Neurology. 50:1074–1082.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shy ME, Blake J, Krajewski K, et al:
Reliability and validity of the CMT neuropathy score as a measure
of disability. Neurology. 64:1209–1214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi M, Scholl UI, Ji W, et al: Genetic
diagnosis by whole exome capture and massively parallel DNA
sequencing. Proc Natl Acad Sci USA. 106:19096–19101. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bamshad MJ, Ng SB, Bigham AW, Tabor HK,
Emond MJ, Nickerson DA and Shendure J: Exome sequencing as a tool
for mendelian disease gene discovery. Nat Rev Genet. 12:745–755.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ng SB, Buckingham KJ, Lee C, et al: Exome
sequencing identifies the cause of a Mendelian disorder. Nat Genet.
42:30–35. 2010. View
Article : Google Scholar :
|
|
14
|
Lee SS, Lee HJ, Park JM, et al: Proximal
dominant hereditary motor and sensory neuropathy with proximal
dominance association with mutation in the TRK-fused gene. JAMA
Neurol. 70:607–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakhro K, Park JM, Hong YB, et al: SET
binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease
type 4B3. Neurology. 81:165–173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HJ, Hong YB, Park JM, et al: Mutations
in the PLEKHG5 gene is relevant with autosomal recessive
intermediate Charcot-Marie-Tooth disease. Orphanet J Rare Dis.
8:1042013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chung KW, Suh BC, Shy ME, et al: Different
clinical and magnetic resonance imaging features between
Charcot-Marie-Tooth disease type 1A and 2A. Neuromuscul Disord.
18:610–618. 2008. View Article : Google Scholar : PubMed/NCBI
|