Introduction
Pulmonary arterial hypertension (PAH) is a
life-threatening disease, which contributes to the morbidity and
mortality of patients with various lung and heart diseases
(1). PAH has a multifactorial
pathology and the pathogenesis remains to be fully elucidated. It
has been reported that a numerous cell types, including endothelial
cells, smooth muscle cells as well as inflammatory cells and
platelets, may be implicated in the progression of PAH (2).
Vascular smooth muscle cells (VSMCs) are located in
the medial wall of blood vessels; under normal conditions, these
cells are quiescent and express a differentiated phenotype in order
to maintain vascular tone (3).
However, under pathological conditions, VSMCs switch to a
‘synthetic’ phenotype, in which they secrete inflammatory cytokines
and contribute to vascular pathogenesis (4). VSMCs proliferation in the pulmonary
artery has been considered to be one of the primary causes of
pulmonary arterial remodeling (5).
Progressive pulmonary arterial remodeling is characteristic of PAH
and has an important role in the persistent deterioration involved
in PAH, as well as contributes to the difficult reversal of the
disease phenotype (6).
The role of the immune system in the progression of
PAH has been the focus of numerous studies in recent years
(7). However, the immunomodulatory
mechanisms which contribute to the pathogenesis of the disease
remain to be fully elucidated. CD4+CD25+
regulatory T cells (Tregs) are a specific subpopulation of T cells,
which have been reported to participate in the regulation of the
immune response as well as the progression of autoimmune diseases
(8). Therefore Treg deficiency or
dysfunction may disrupt immune homeostasis and lead to numerous
pathological conditions.
At present, few therapies have been developed for
the effective treatment of pulmonary arterial structure remodeling
and PAH. Tregs have been reported to exert beneficial effects on
the progression of numerous diseases (9,10),
including atherlerosclerosis (11), abdominal aortic aneurysms (12) and inflammatory bowel disease
(13). However, the role of Tregs
in the development of PAH remains to be elucidated. The present
study aimed to determine whether Tregs affected the development of
PAH, and to investigate the underlying mechanisms.
Materials and methods
Animals
A total of 60 male C57BL/6 J mice (10 weeks old)
were purchased from Beijing University Animal Research Center
(Beijing, China). The mice were randomly divided into three groups,
with 20 mice in each, as follows: Normoxia, hypoxia control and
Tregs. Mice in the normoxia group were maintained in air and
exposed to a normoxic environment. Mice in the hypoxia control and
Tregs groups were exposed to hypoxic conditions (10%
O2), as maintained using a litre ventilated chamber
(volume, 500 l; Flufrance apparatus, Cachan, France) for four
weeks. All animals were kept in the same room and had access to
standard mouse feed and water, the hypoxic group were kept in a
hypoxic chamber. All animal procedures were reviewed and approved
by the Animal Care and Use Committee of Shandong University (Jinan,
China).
Isolation and adoptive transfer
study
Ten C57BL/6 J (six weeks old) wild-type male mice,
also obtained from Beijing University Animal Research Centre,
served as Treg cells donors. These mice were housed in a
pathogen-free animal care facility at a constant temperature (24°C)
and a 12-h light/dark cycle, with free access to water. Mice were
euthanized by ketamine-xylazine (75 and 3 mg/kg, respectively;
Sigma-Aldrich, St. Louis, MO, USA) injection and then immersed in
75% ethanol (Annjet, Shandong, China) for 10 minutes and spleens
were then isolated from the mice under aseptic conditions. Spleens
were then gently mechanically disrupted and passed through a cell
strainer. phosphate-buffered saline (PBS; Sigma-Aldrich) was then
added to make a suspension up to 10 ml and the suspension was
centrifuged at 800 × g for 5 min at 4°C. The supernatant was then
discarded and the pellet was resuspended in 1 ml PBS. Purified
Tregs were then isolated using a CD4+CD25+
Regulatory T cell Isolation kit (Miltenyi Biotec, Bergisch
Gladbach, Germany). The average purity of Tregs was >97%, as
determined by fluorescence-activated cell sorting analysis (BD
FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA). Cells were
suspended in a total volume of 200 μl PBS for injection, as
previously described (9,10,14).
One day before exposure to hypoxic conditions, the mice in the
control and Tregs groups were injected with PBS or Tregs
(1×106 cells), respectively, into the tail vein.
Hemodynamic measurements
Mice were anesthetized via intraperitoneal injection
of ketamine (6 mg/100 g) and xylazine (1 mg/100 g). Mice were then
placed in a supine position and the trachea was cannulated. A
26-gauge needle (Sigma-Aldrich) was passed percutaneously into the
thorax via a subxyphoid approach and the right ventricular systolic
pressure (RVSP) was measured and recorded using a miniature
pressure transducer (MPCU-200; Millar, Houston, TX, USA) digitized
by a data acquisition system (Hitachi, Tokyo, Japan). During
surgery, the mice inhaled room air spontaneously and their heart
rate was maintained between 300 and 600 beats/min. Following
measurement of RVSP, the right ventricle (RV) was isolated from the
left ventricle (LV) and the septum (S) and each were weighed in
order to calculate the Fulton’s index [RV/(LV+S)].
Lipid profile
Mice were starved overnight and blood samples were
collected prior to sacrifice. Serum levels of total cholesterol
(TC), triglyceride (TG), low density lipoprotein cholesterol
(LDL-C) and high density lipoprotein cholesterol (HDL-C) were
determined using an enzymatic assay.
Cell culture
Human peripheral blood mononuclear cells (PBMCs)
were isolated from eight healthy volunteers (male:female, 5:3; aged
20–45). This study was approved by the ethics committee of Qilu
Hospital, Shandong University and written informed consent was
obtained from each patient for the use of their blood samples.
Tregs were isolated from PBMCs using a
CD4+CD25+ Regulatory T cell Isolation kit
(Miltenyi Biotec) according to the manufacturer’s instructions.
Human pulmonary artery smooth muscle cells (HPASMCs) were purchased
from the American Type Culture Collection (Manassas, VA, USA) and
cultured in Dulbecco’s modified Eagle’s medium (ScienCell Research
Laboratories, Carlsbad, CA, USA) containing 10% fetal bovine serum
(FBS; Gibco-BRL, Carlsbad, CA, USA) at 37°C in a 5% CO2
and 95% air atmosphere. Cells at up to passage 4 were used for the
subsequent experiments. HPASMCs were kept under hypoxic conditions
(1% O2, 5% CO2) in a cell culture without
Tregs (control group) or with Tregs (5×105/well; Tregs
group) in the presence of mouse monoclonal anti-CD3 antibody (50
ng/ml; #ab8671) at 37°C for 48 h, as previously described (15). At last, floating T cells were
discarded, HASMCs were collected.
Measurement of HPASMC proliferation
The proliferation of HPASMCs was determined using an
MTT assay. Briefly, HPASMCs were seeded into 96-well plates at a
density of 5,000 cells/well. Following exposure to hypoxic
conditions without or with Tregs treatment, HPASMCs were incubated
with 10 μl MTT (5 mg/ml)/well reagent for 4 h at 37°C. The
supernatant was carefully removed and 75 μl/well dimethylsulfoxide
(DMSO) was added to dissolve the formazan crystals. Samples were
then analyzed at 570 nm using a Varioskan Flash multifunction plate
reader (Thermo Scientific, Waltham, MO, USA).
For cell counting, HPASMCs were seeded into a
six-well plate (5,000 cells/well) and then treated as described
above. Cells were then washed with PBS, harvested with trypsin, and
counted using a hematocytometer (Beckman Coulter, Inc., Fullerton,
CA, USA).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
The lungs of the mice were isolated following
sacrifice and HPASMCs were harvested, as described above. RNA was
then prepared using TRIzol® reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA). RNA concentrations were
determined using standard spectrophotometric techniques (Varioskan
Flash; Thermo Fisher Scientific, St. Louis, MO, USA) and the mRNA
expression was analyzed. For the in vivo experiments, qPCR
was performed using the following primers (GenePharma, Shanghai,
China): forkhead/winged helix transcription factor (Foxp)3 sense,
5′-CCCATCCCCAGGAGTCTTG-3′ and antisense,
5′-ACCATGACTAGGGGCACTGTA-3′; monocyte chemotactic protein 1 (MCP-1)
sense, 5′-CAGCCAGATGCAGTTAACGC-3′ and antisense,
5′-GCCTACTCATTGGGATCATCTTG-3′; interleukin (IL)-1β sense,
5′-GCAACTGTTCCTGAACTCAACT-3′ and antisense,
5′-ATCTTTTGGGGTCCGTCAACT-3′; IL-6 sense,
5′-AGTCACAGAAGGAGTGGCTAAG-3′ and antisense,
5′-GAGGAATGTCCACAAACTGATA-3′; IL-10 sense,
5′-GCTCTTACTGACTGGCATGAG-3′ and antisense,
5′-CGCAGCTCTAGGAGCATGTG-3′; and β-actin sense,
5′-CACTGTGCCCATCTACGA-3′ and antisense,
5′-GTAGTCTGTCAGGTCCCG-3′.
For the in vitro experiment, the sequences of
primers were as follows: Cyclin D1 sense, 5′-CTC
CTCTCCGGAGCATTTTGATA3′ and antisense,
5′-TTAAAGACAGTTTTTGGGTAATCT3′; cyclin-dependent kinase (CDK)4
sense, 5′-ATGGCTACCTCTCGATATGAGCCA-3′ and antisense,
5′-TCACTCCGGATTACCTTCATCCTT-3′; p27 sense,
5′-CTTGGAGAAGCACTGCCGAGA-3′ and antisense,
5′CCCTGGACACTGCTCCGCTA-3′; and β-actin sense,
5′-CATGTACGTTGCTATCCAGGC-3′ and antisense,
5′-CTCCTTAATGTCACGCACGAT-3′. Amplification, detection and data
analysis were performed using the iCycler Real-Time PCR system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Relative
expression of genes was calculated using the 2−ΔΔct
method. Each sample was analyzed in triplicate, and expression was
normalized to that of β-actin.
Western blot analysis
Total proteins were extracted from the lung of the
mice or HPASMCs. Protein samples were separated using a 10–12%
SDS-polyacrylamide gel (Beyotime, Jiangsu, China) and
electrophoretically transferred onto a nitrocellulose membranes
(Millipore, Billerica, MA, USA). Following blocking with 5% non-fat
milk for 2 h at room temperature, the membranes were washed three
times with Tris-buffered saline with Tween 20 (TBS-T; Boster,
Hubei, China) for 10 min and then incubated with primary antibodies
for rabbit polyclonal anti-cyclin D1 (1:1,000; #2922; Cell
Signaling Technology, Danvers, MA, USA), rabbit polyclonal
anti-CDK4 (1:500; #ab7955; Abcam, Cambridge, MA, USA), rabbit
polyclonal anti-p27 (1:500; #ab7961; Abcam), rabbit monoclonal
anti-Akt (1:1,000; #4685; Cell Signaling Technology, rabbit
monoclonal p-Akt (1:1,000; #13038; Cell Signaling Technology),
rabbit polyclonal anti-extracellular signal-regulated kinase (ERK;
#9102; Cell Signaling Technology), rabbit monoclonal anti-p-ERK
(1:1,000; #4376; Cell Signaling Technology) and rabbit polyclonal
anti-β-actin (1:1,000; #4967; Cell Signaling Technology) at 4°C
overnight. Following washing three times in TBS-T, the membranes
were incubated with a horseradish peroxidase-conjugated secondary
antibody (Jackson Immunoresearch, West Grove, PA, USA). The bands
were detected using an enhanced chemiluminescence method
(Millipore) and analyzed using Image-Pro Plus 6.0
(MediaCybernetics, Rockville, MD, USA).
Statistical analysis
SPSS 13.0 (SPSS Inc., Chicago, IL, USA) was used to
analyze the data. All statistical comparisons were tested using an
unpaired Student’s t-test or one-way analysis of variance. Values
are presented as the mean ± standard error and P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
Body weight and lipid profile
studies
Following four weeks of treatment, only one mouse
succumbed in the control group and no adverse effects were observed
in each group during the experiment. No significant differences
were observed in the body weight and serum levels of TC, TG, LDL-C
and HDL-C among the three groups (data not shown). These results
suggested that Tregs had no effect on the above parameters in
mice.
Adoptive transfer of Tregs enhances Foxp3
expression
Foxp3 is a specific marker for Tregs lineage, which
is exclusively expressed in Tregs and is a requisite factor for the
maturation and function of Tregs; in addition, deficiency of Foxp3
may result in Treg disfunction (16). In the present study, Foxp3 mRNA
expression in the lungs of the mice was measured in order to assess
whether the intravenously injected Tregs were succeful in
infiltrating the lung tissues. The results showed that Foxp3 mRNA
was significantly increased in mice treated with Tregs compared
with that of the control group (data not shown). These results
confirmed that the exogenous Tregs were present in the lung
tissues.
Adoptive transfer of Tregs improves
hypoxia-induced pulmonary hypertension and vascular remodeling
The hemodynamic parameters of mice were measured
prior to euthanasia. Compared with that of the normoxia group,
hypoxia-exposed mice demonstrated a significant increase in RVSP;
however, following Tregs treatment, RVSP was significantly reduced
compared with that of the hypoxia control group (P<0.05)
(Fig. 1A). Chronic severe PAH is a
cardiopulmonary disease which may affect RV hypertrophy, which is
promoted through exposure to chronic hypoxia (4). In the present study, the Fulton’s
index was measured for the assessment of RV hypertrophy. The
results showed that exposure to hypoxia was associated with an
increase in the Fulton’s index; however, the increased index
observed in the hypoxic control group was partially reversed in the
Tregs group (P<0.05) (Fig. 1B).
This therefore indicated that Tregs may improve hypoxia-induced
pulmonary hypertension and vascular remodeling.
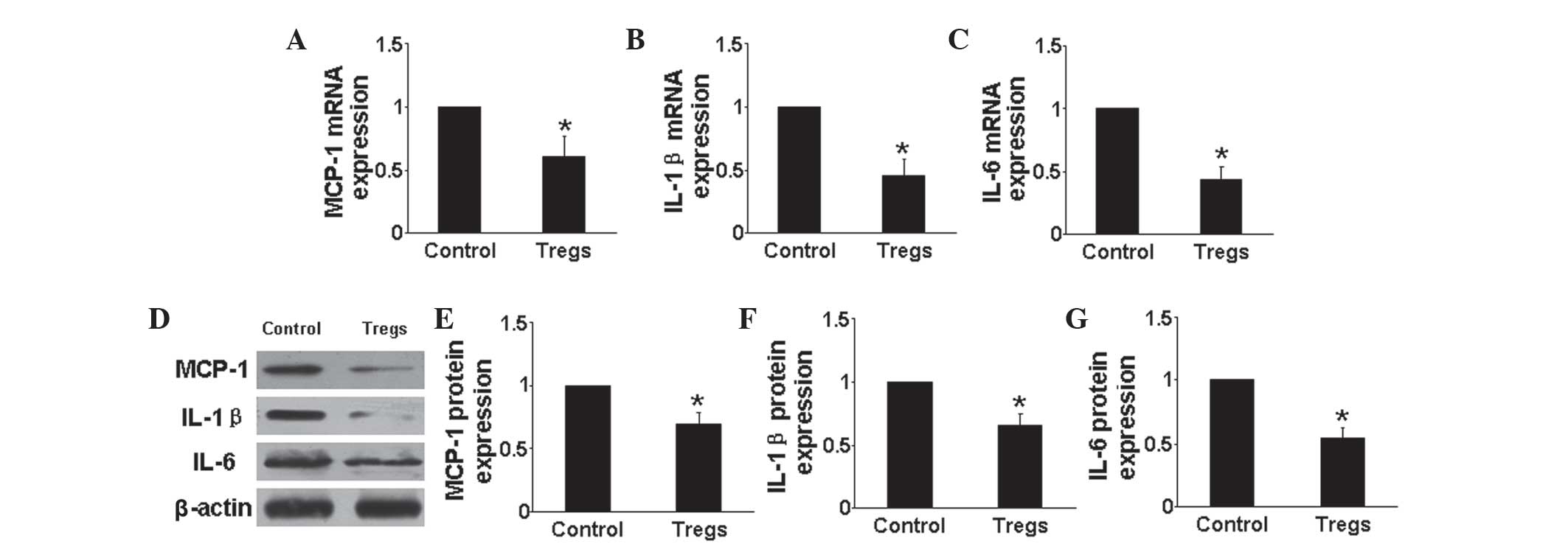
Tregs downregulate proinflammatory
cytokine expression and upregulate anti-inflammatory cytokine
expression in vivo
RT-qPCR and western blot analysis were used to
determine the mRNA and protein expression, respectively, of
pro-inflammatory cytokines, including MCP-1, IL-1β and IL-6
(Fig. 2), as well as the
anti-inflammatory cytokine IL-10 in the lungs of mice in each
group. The results showed that mRNA and protein expression levels
of each of the pro-inflammatory cytokines were significantly
downregulated in the Tregs group compared with those of the hypoxia
control group (P<0.05) (Fig.
2A–G). In addition, the mRNA and protein expression levels of
IL-10 were found to be significantly upregulated in the Tregs
groups compared with that of the hypoxia control group (P<0.05)
(Fig. 3). These results indicated
that Tregs regulated the inflammatory response through reducing the
expression of proinflammatory cytokines and increasing that of
anti-inflammatory cytokines.
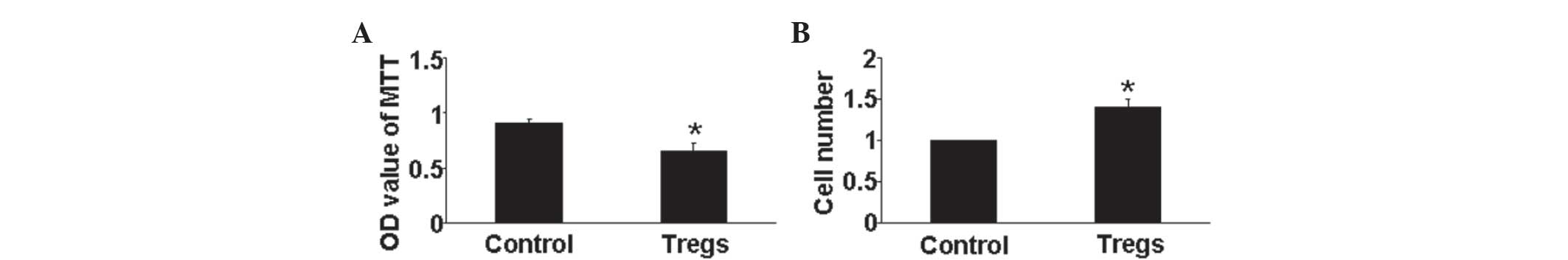
Tregs reduce hypoxia-induced HPASMCs
proliferation in vitro
The effect of Tregs on HPASMCs proliferation under
hypoxic conditions was determined using an MTT assay. The results
showed that Tregs significantly inhibited HPASMCs proliferation
compared with that of the hypoxia control group (P<0.05)
(Fig. 4A). In addition, a cell
counting assay demonstrated a significant increase in cell number
in the Treg groups (P<0.05) (Fig.
4B). Therefore, the regulation of HPASMC proliferation by Tregs
may be an important anti-PAH mechanism.
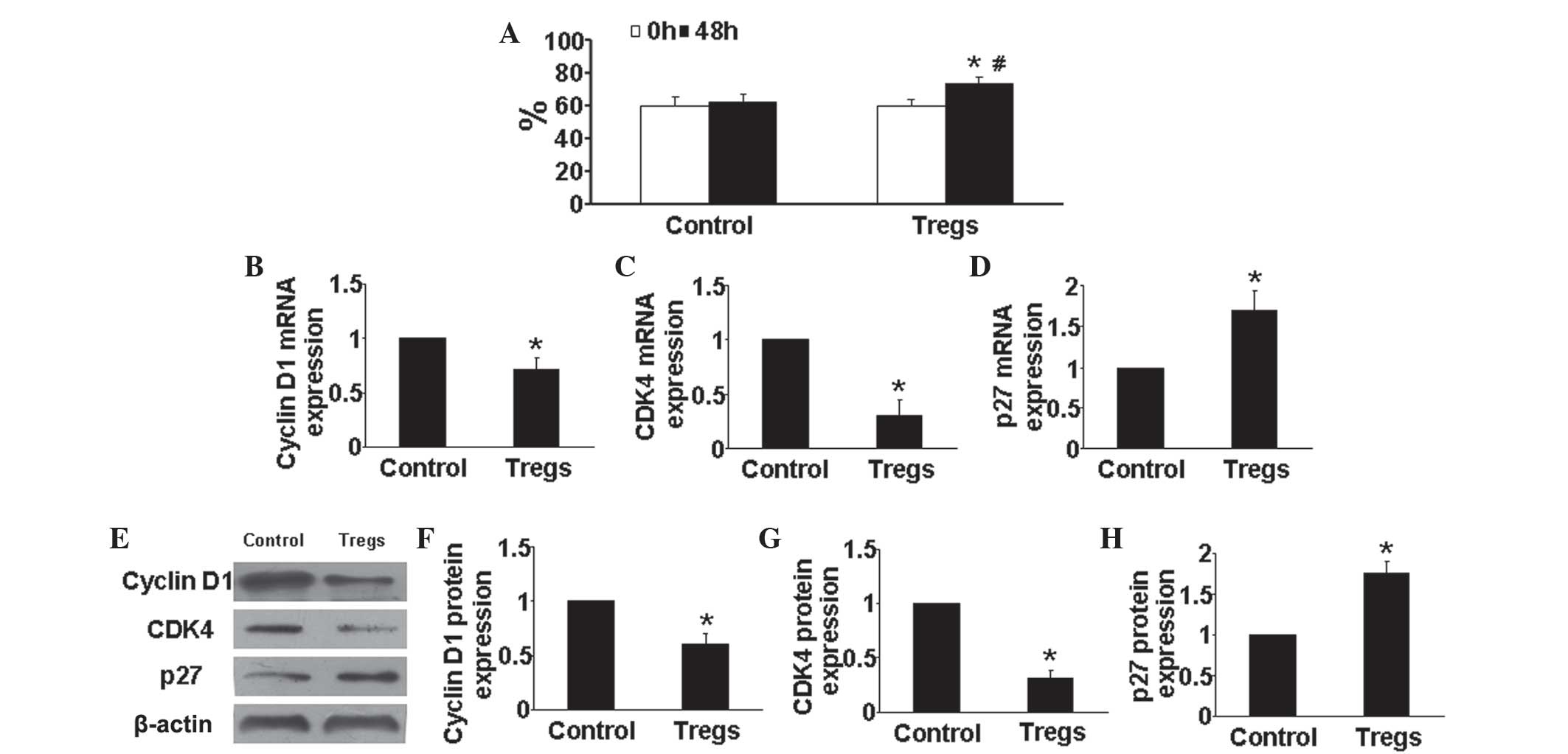
Tregs arrest HPASMCs in G1/G0-phase under
hypoxic conditions
Cell proliferation is dependent on cell cycle
transition from G1/G0 to G2/S-phase (17); therefore, in the present study, the
effects of Tregs on cell cycle of HPASMCs was evaluated. As shown
in Fig. 5A, compared with that of
the hypoxia control group, the Tregs group demonstrated as
significantly increase percentage of HPASMCs in the
G1/G0 phase (P<0.05).
Tregs reduce cyclin D1 and CDK4
expression as well as increase p27 expression
Previous studies shown that cyclin D1, CDK4 and p27
have key roles in proliferation and the cell cycle. In the present
study, the expression levels of cyclin D1, CDK4 and p27 in HPASMCs
were determined in vitro. Compared with those of the control
group, the mRNA and protein expression levels of cyclin D1 and CDK4
were markedly reduced in Tregs-treated HPASMCs (P<0.05)
(Fig. 5B, C, E and G), whereas
Tregs treatment significantly enhanced p27 mRNA and protein
expression compared with that of the hypoxia control group
(P<0.05) (Fig. 5D and H). These
results suggested that the effect of Tregs on cell cycle regulation
may be another mechanism by which it exerts anti-PAH effects.
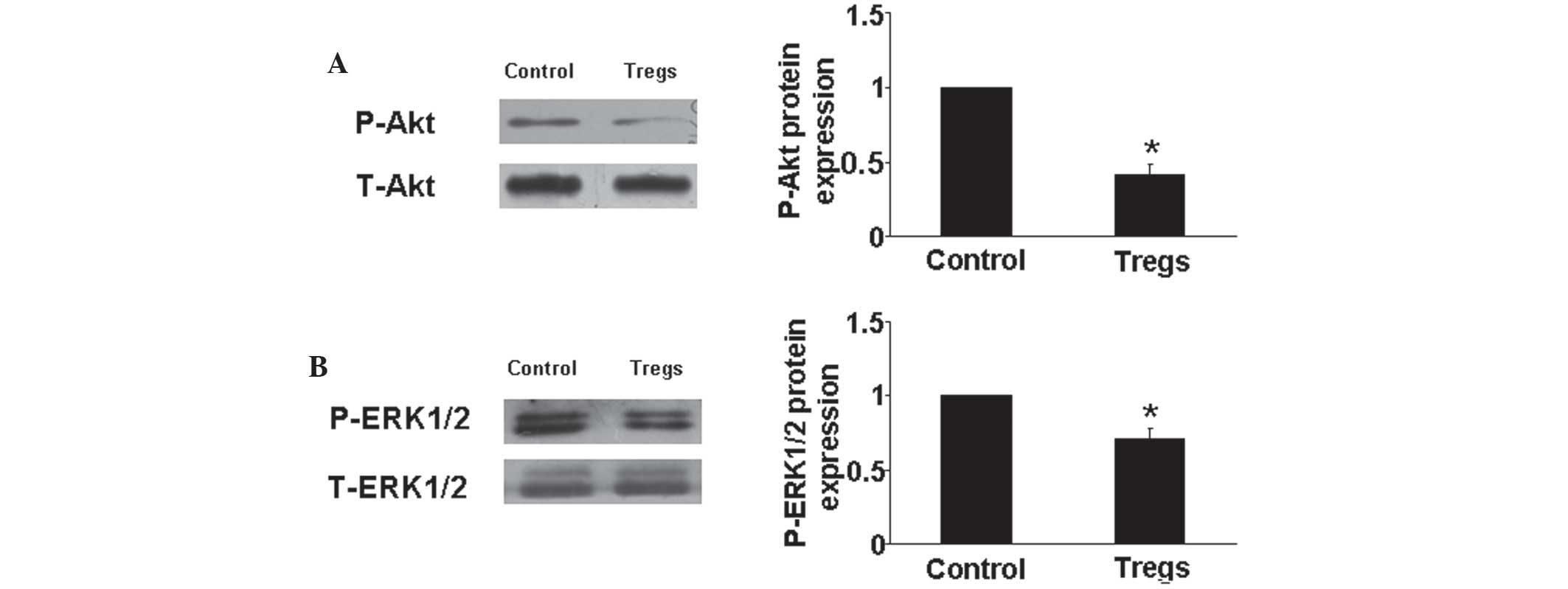
Tregs decrease Akt and ERK1/2
phosphorylation
It has been previously reported that the Akt and ERK
pathways were involved in HPASMCs proliferation and the progression
of PAH (4). Therefore, in the
present study, western blot analysis was used to determine Akt and
ERK protein expression in vitro. The results showed that
Tregs significantly downregulated the phosphorylation of Akt and
ERK compared with those of the hypoxia control group (P<0.05)
(Fig. 6). This may therefore be a
further protective mechanism of Tregs against PAH.
Discussion
PAH is a fatal disease with unknown etiology.
Numerous studies have focused on the development and treatment of
PAH; however, there remains to be few therapies which are effective
in treating the disease. Tregs suppress the activation and
proliferation of effector T cells, prevent autoimmunity and control
autoimmune diseases (18). In
recent years studies have increasingly focus on the role of immune
mechanisms in modulating the disease process. However, whether
Tregs have a beneficial and protective effect on PAH remained to be
elucidated. The present study provided direct evidence that Tregs
treatment prevented the progression of PAH in an animal model. To
the best of our knowledge, the present study was the first to show
that Tregs significantly suppressed the inflammatory response
through enhancing anti-inflammatory cytokine levels, inhibiting
HPASMCs proliferation and regulating their cell cycle.
It was hypothesized that exposure to chronic hypoxia
may increase vasomotor tone and structural remodeling of the
pulmonary vascular bed in PAH patients, leading to pulmonary
hypertension. Previous in vivo experiments have shown that
hypoxia was able to induce pulmonary hyperation (6); in addition, increased right
ventricular pressure confirmed the successfully established PAH
animal models. In the present study, mice in control group
developed a higher RVSP compared with that of those in the normoxia
group, which was in accordance with a prior study (6). However, in the present study, RVSP
was significantly reduced following Tregs treatment, which
suggested that Tregs exerted a beneficial on pulmonary
hypertension. Increased pulmonary artery remodeling is
characteristic of PAH. An increased afterload resulted in a degree
hypertrophy of RV (14). The
results of the present study showed that Tregs partially reversed
the increased Fulton’s index induced by hypoxia, therefore
improving the hemodynamic parameters and vascular remodeling in
PAH. Furthermore, Tregs therapy did not affect lipid parameters in
the animal model, suggesting that the effects of Tregs on PAH were
lipid-independent.
Inflammatory processes involved the pathogenesis of
PAH are increasingly considered as major pathogenic components of
pulmonary vascular remodeling (19). Infiltration of inflammatory cells
and increased levels of inflammatory cytokines have been observed
in the vascular lesions of PAH (20). Previous studies have shown that in
comparison with healthy populations, the circulating and pulmonary
expression of inflammatory cytokines was significantly increased in
patients with PAH (21,22). Another study demonstrated that IL-6
knockout mice did not develop pulmonary hypertension, while the
overexpression of IL-6 accelerated spontaneous pulmonary vascular
remodeling and progression in vivo (23). IL-10, known as a pleiotropic
anti-inflammatory cytokine, has been reported to inhibit
inflammatory cell infiltration and pro-inflammatory cytokine
secretion (24). A previous study
reported that overexpression of IL-10 prevented the development of
monocrotaline-induced PAH in animal models (25). Furthermore, IL-10, as a potent
immunomodulator, was shown to mediate the effects of Tregs
(26). The results of the present
study showed that Tregs suppressed the mRNA and protein expression
of pro-inflammatory cytokines, including MCP-1, IL-1β and IL-6, as
well as upregulated the expression of anti-inflammatory cytokine
IL-10. This therefore indicated that Tregs modulated the balance of
the inflammatory response, which may a mechanism by which Tregs
exert a protective effect against PAH.
The principal phenotype of VSMCs is contraction,
which preserves vasodilation and blood flow regulation under
physiological conditions. However, VSMCs have been shown to have a
‘synthetic’ phenotype under pathological conditions, in which they
have an increased capacity to proliferate and generate the matrix
components of the blood vessel wall, which contributes to vascular
remodeling (27). In addition,
aberrant HPASMC proliferation has been reported to lead to
pulmonary arterial remodeling and contribute to the progression of
PAH. Effective inhibition of the aberrant HPASMCs may delay and
even cease the deteriorative progress of PAH (28). In the present study, the role of
Tregs in hypoxia-induced HPASMCs proliferation was evaluated and
the results showed that Tregs effectively inhibited HPASMCs
proliferation. Therefore, the anti-PAH properties of Tregs in
treated-mice may be attributed to their role in HPASMCs
proliferation.
Under hypoxic conditions, an increase number of
HPASMCs enter the mitosis phase of the cycle; in addition, the
acceleration of the cell cycle is an initial factor in cell
proliferation. Hypoxia retains small cell numbers in G0/G1 phase
and promotes HPASMCs to enter G2/S phase. In the present study, the
effects of Tregs on the cell cycle of HPASMCs was assessed and the
results demonstrated that Tregs reversed the effects of hypoxia on
the cell cycle. A previous study reported that the balance between
cell quiescence and proliferation was regulated by CDKs and CDK
inhibitors (29). Cyclin D1 and
CDKs, primarily CDK4, are key cell cycle control genes, which were
associated with cell proliferation and facilitated the transition
of cells from the G1 phase into the S phase (30). Overexpression of CDK4 promotes cell
proliferation and inhibition of CDK4 expression may lead to the
arrest of the G1 phase and suppression of cell proliferation
(31). The present study showed
that Tregs significantly reduced Cyclin D1 and CDK4 expression
in vitro. P27, as one of the key CDK inhibitors, effectively
inhibits Cyclin D1-CDK4 protein kinase activity and negatively
regulates G1 progression in cells; in addition, the overexpression
of p27 induces G1 arrest and decreases HPASMCs proliferation
(32). The results of the present
study revealed that Tregs significantly increased p27 mRNA and
protein expression, therefore indicating that Tregs promoted G1
phase arrest, which may be the direct mechanism of Tregs against
HPASMCs proliferation and PAH.
Akt and ERK are activated via diverse extracellular
signals, which trigger cell cascade responses, including cell
growth, proliferation, survival and motility. Therefore, in the
present study, the expression of Akt and ERK were investigated
their in HPASMCs and the results showed that Tregs blocked the
activation of Akt and ERK pathway induced through hypoxia.
In conclusion, the results of the present study
demonstrated that Tregs protected against hypoxia-induced PAH in
mice. These findings provided evidence for a possible targeted
therapy for the treatment of pulmonary hypertensive disorders.
References
|
1
|
Gaine SP and Rubin LJ: Primary pulmonary
hypertension. Lancet. 352:719–725. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Humbert M, Morrell NW, Archer SL, Stenmark
KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O,
Voelkel NF and Rabinovitch M: Cellular and molecular pathobiology
of pulmonary arterial hypertension. J Am Coll Cardiol. 43(12 Suppl
S): 13S–24S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Orlandi A, Bochaton-Piallat ML, Gabbiani G
and Spagnoli LG: Aging, smooth muscle cells and vascular
pathobiology: Implications for atherosclerosis. Atherosclerosis.
188:221–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen B, Calvert AE, Meng X and Nelin LD:
Pharmacologic agents elevating cAMP prevent arginase II expression
and proliferation of pulmonary artery smooth muscle cells. Am J
Respir Cell Mol Biol. 47:218–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling: Cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nicolls MR, Taraseviciene-Stewart L, Rai
PR, Badesch DB and Voelkel NF: Autoimmunity and pulmonary
hypertension: a perspective. Eur Respir J. 26:1110–1118. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakaguchi S: Naturally arising
Foxp3-expressing CD25+CD4+ regulatory T cells in immunological
tolerance to self and non-self. Nat Immunol. 6:345–352. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng X, Li W, Yang J, Zhang K, Qin W, An
G, Gao F, Wang Y, Zhang C and Zhang Y: Regulatory T cells prevent
plaque disruption in apolipoprotein E-knockout mice. Int J Cardiol.
168:2684–2692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ait-Oufella H, Wang Y, Herbin O, et al:
Natural regulatory T cells limit angiotensin II-induced aneurysm
formation and rupture in mice. Arterioscler Thromb Vasc Biol.
33:2374–2379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mor A, Planer D, Luboshits G, et al: Role
of naturally occurring CD4+CD25+ regulatory T cells in experimental
atherosclerosis. Arterioscler Thromb Vasc Biol. 27:893–900. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin M, Zhang J, Wang Y, et al: Deficient
CD4+CD25+ T regulatory cell function in patients with abdominal
aortic aneurysms. Arterioscler Thromb Vasc Biol. 30:1825–1831.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guidi L, Felice C, Bonanno G, et al: FOXP3
T regulatory cell modifications in inflammatory bowel disease
patients treated with anti-TNFα agents. Biomed Res Int.
2013:2863682013. View Article : Google Scholar
|
|
14
|
Oka M, Homma N, Taraseviciene-Stewart L,
Morris KG, Kraskauskas D, Burns N, Voelkel NF and McMurtry IF: Rho
kinase-mediated vasoconstriction is important in severe occlusive
pulmonary arterial hypertension in rats. Circ Res. 100:923–929.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu X, Murphy TC, Nanes MS and Hart CM:
PPAR{gamma} regulates hypoxia-induced nox4 expression in human
pulmonary artery smooth muscle cells through NF-{kappa}B. Am J
Physiol Lung Cell Mol Physiol. 299:L559–L566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim JM, Rasmussen JP and Rudensky AY:
Regulatory T cells prevent catastrophic autoimmunity throughout the
lifespan of mice. Nat Immunol. 8:191–197. 2007. View Article : Google Scholar
|
|
17
|
Fang K, Fu W, Beardsley AR, Sun X, Lisanti
MP and Liu J: Overexpression of caveolin-1 inhibits endothelial
cell proliferation by arresting the cell cycle at G0/G1 phase. Cell
Cycle. 6:199–204. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kraaij MD, Savage ND, van der Kooij SW,
Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, Kuijpers TW,
Holmdahl R, van Kooten C and Gelderman KA: Induction of regulatory
T cells by macrophages is dependent on production of reactive
oxygen species. Proc Natl Acad Sci USA. 107:17686–17691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hassoun PM, Mouthon L, Barberà JA, et al:
Inflammation, growth factors, and pulmonary vascular remodeling. J
Am Coll Cardiol. 54(1 Suppl): S10–S19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
El Chami H and Hassoun PM: Inflammatory
mechanisms in the pathogenesis of pulmonary arterial hypertension.
Compr Physiol. 1:1929–1941. 2011.PubMed/NCBI
|
|
21
|
Schober A and Zernecke A: Chemokines in
vascular remodeling. Thromb Haemost. 97:730–737. 2007.PubMed/NCBI
|
|
22
|
Itoh T, Nagaya N, Ishibashi-Ueda H,
Kyotani S, Oya H, Sakamaki F, Kimura H and Nakanishi N: Increased
plasma monocyte chemoattractant protein-1 level in idiopathic
pulmonary arterial hypertension. Respirology. 11:158–163. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steiner MK, Syrkina OL, Kolliputi N, Mark
EJ, Hales CA and Waxman AB: Interleukin-6 overexpression induces
pulmonary hypertension. Circ Res. 104:236–244. 2009. View Article : Google Scholar
|
|
24
|
Asadullah K, Sterry W and Volk HD:
Interleukin-10 therapy - review of a new approach. Pharmacol Rev.
55:241–69. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ito T, Okada T, Miyashita H, Nomoto T,
Nonaka-Sarukawa M, Uchibori R, Maeda Y, Urabe M, Mizukami H, Kume
A, Takahashi M, Ikeda U, Shimada K and Ozawa K: Interleukin-10
expression mediated by an adeno-associated virus vector prevents
monocrotaline-induced pulmonary arterial hypertension in rats. Circ
Res. 101:734–741. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lavasani S, Dzhambazov B, Nouri M, et al:
A novel probiotic mixture exerts a therapeutic effect on
experimental autoimmune encephalomyelitis mediated by IL-10
producing regulatory T cells. PLoS One. 5:e90092010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Owens GK: Regulation of differentiation of
vascular smooth muscle cells. Physiol Rev. 75:487–517.
1995.PubMed/NCBI
|
|
28
|
Luo Y, Xu DQ, Dong HY, Zhang B, Liu Y, Niu
W, Dong MQ and Li ZC: Tanshinone iia inhibits hypoxia-induced
pulmonary artery smooth muscle cell proliferation via
akt/skp2/p27-associated pathway. PLoS One. 8:e567742013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu L, Quinn DA, Garg HG and Hales C: Gene
expression of cyclin-dependent kinase inhibitors and effect of
heparin on their expression in mice with hypoxia-induced pulmonary
hypertension. Biochem Biophys Res Commun. 345:1565–1572. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong Y, Sui L, Sugimoto K, Tai Y and
Tokuda M: Cyclin d1-cdk4 complex, a possible critical factor for
cell proliferation and prognosis in laryngeal squamous cell
carcinomas. Int J Cancer. 95:209–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakamoto K, Ohki K, Saito M, Nakahara T
and Ishii K: Small molecule cyclin-dependent kinase inhibitors
protect against neuronal cell death in the ischemic-reperfused rat
retina. J Ocul Pharmacol Ther. 27:419–425. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Toyoshima H and Hunter T: P27, a novel
inhibitor of g1 cyclin-cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|