Introduction
Alzheimer’s disease (AD) is an age-associated
progressive neurodegenerative disorder that is characterized by the
presence of senile plaques, neurofibrillary tangles and loss of
synapses (1,2). This disease not only endangers health
and is detrimental to the life of patients with AD, but
additionally imposes a considerable burden on families, carers and
society (3). Effective treatment
for AD has been an ongoing challenge to clinical workers and
researchers (4,5). Current therapeutic interventions act
to relieve symptoms and delay the progression of AD; however, the
development of novel strategies is required to improve treatment
outcomes (6,7).
Neuroimmunological theory investigates the
association between the nervous and immune system and involves
numerous neuronal diseases, including Alzheimer’s, Parkinson’s and
Huntington’s disease (8,9). Analyzing the association between the
nervous and immune system may therefore generate valuable insight
leading to novel therapeutic approaches (10). Cytokines are small molecular
polypeptides secreted by immunocytes and other associated
immunological cells, including astrocytes, that mediate and
regulate the immune and inflammatory response (11,12).
Regulation of the immune response is associated with the
pathogenesis of AD (13).
Interleukin-1 beta (IL-1β) is a cytokine that has an important
modulatory effect on the pathology of AD. IL-1β functions as a
pro-inflammatory cytokine, and an increase in IL-1 expression has
been associated with AD (14).
Increased serum levels of IL-1β are used as a stage marker of the
ongoing brain neurodegeneration in the continuum between normal
ageing and AD (15). Although a
direct association between IL-1β and AD has been identified
(16,17), the most accepted view supports that
the pro-inflammatory effects of IL-1β promote a trend towards
disease deterioration, but this process involves numerous factors
and pathways. Few studies have further investigated the association
between IL-1β and other proteins and pathways involved in the
pathogenesis of AD. Synthetic analysis of the relationship between
these additional factors and IL-1β in AD may facilitate the
identification of the molecular mechanisms of IL-1β function in the
pathogenesis of AD (18,19).
Cytoscape (http://www.cytoscape.org/) (20) is an open source software project
for integrating biomolecular interaction networks with
high-throughput expression data and other molecular states into a
unified conceptual framework. This software has been extensively
used by researchers to investigate biological domains, the genome,
proteome and metabonomics. A large database of biological
interactions, including protein-protein and protein-DNA, are
displayed by the software in graphical format, displaying key nodes
and end-points (20–22). The Kyoto Encyclopedia of Genes and
Genomes (KEGG; www.kegg.jp) is a database resource for
systematic analysis of gene function, in terms of the networks of
genes and molecules. The central feature of the KEGG resource is
the PATHWAY database that consists of graphical diagrams of
biochemical pathways, including most of the known metabolic
pathways and some of the known regulatory pathways (23,24).
Two of the referenced pathways in KEGG are
associated with AD. One is a direct AD pathway and the other is a
WNT signaling pathway. Several factors which are associated with
IL-1β in AD have been identified in some of the pathways in the
KEGG database. By using Cytoscape, this study has identified the
associations between IL-1β and its associated proteins in AD. By
comparing these networks produced by Cytoscape with the KEGG
pathways, the mechanism and function of IL-1β in AD was
investigated.
Materials and methods
Data mining
The factors of interest were selected based on data
mining in the published literature. Factors were selected based
upon associations with IL-1β in AD (25–27).
The factor pool was constructed and is shown in Table I. Additional factors were
constituted from additional factor pools which included proteins
which have not previously been described to affect IL-1β but which
have a moderate association (Table
II).
 | Table IClassification of factors associated
with IL-1β in Alzheimer’s disease. |
Table I
Classification of factors associated
with IL-1β in Alzheimer’s disease.
| Upregulating
IL-1β | Upregulated by
IL-1β | Downregulating
IL-1β | Downregulated by
IL-1β |
|---|
| Specific proteins
with AD | Aβ, Aβ1–40, Aβ1–42,
Aβ25–35, APP, fAbeta (25–34) | Aβ, APP, βAPP,
sAPP, βAPP, αAPP, tau | | |
| Cytokines | IL-1β, IL-8, TNF-α,
IFN-γ, TGF-β, PGF (2α), CD40 | IL-1β, IL-6, IL-1α,
TNF-α | IL-4, IL-10,
IL-1Ra, NGF | GIF |
| Other immune
factors | C5a, LPS,
NALP3 | C3, C1s, C1r | | |
|
Neurotransmitters | Glutamate | ACh | ACh, AChE | |
| Signal factor | NF-κB, Wnt5a, TLR4,
JNK, ERK, MAPK-p38, PLC, Sp1, miRNA-146a, NO, DSP4 S100B,
α1-ACT | ICE, iNOS, TACE,
COX-2, cyclooxygenase, C/EBP, NO, S100Bβ, | MK-801 | |
| Hormone | insulin, Human
amylin, ET | CRF, PGE2,
ET-1 | Estrogen | |
| Lipid | apoE, apoE4, LXA
(4) | apoE,
clusterin | | |
| Globulin | |
α2-macroglobulin | | |
| Table IIClassification of other factors
associated with IL-1β in Alzheimer’s disease. |
Table II
Classification of other factors
associated with IL-1β in Alzheimer’s disease.
| Type | Factors |
|---|
| Cytokines | MCP-1, MIP-1β,
MIP-1α, CD36, G-CSF, TGF-β1, EGF, IL-13, oncostatin M, IL-2, IL-18,
sTNF-R1 |
| Other immune
factors | C4, C1-lnh,
NALP1 |
|
Neurotransmitters | BDNF, NT-3 |
| Signal factor | BACE1, BACE2,
IRAK-1, IRAK-2, Cox-IV, TLR, IKK, IκB, P65, FPR, MAChR, AP-1 |
| Hormone | PGE1,
norepinephrine
F2-isoprostane |
| Lipid | RAGE, DHA, EPA,
PUFAs |
| Globulin | Perlecan |
Data reduction
Cytoscape provides a convenient procedure to
construct functional interaction nets. Data were uploaded through
one of the supported formats, including Microsoft EXCEL. Data was
therefore constructed in tabular form in EXCEL, two columns
represented nodes and one column represented edges. The attributes
of these nodes and edges were compiled using Notebook (Smart
Software, Calgary, AB, Canada). The attributes of the nodes were
associated with these classifications of factors and the ones of
edges were associated with an interaction between these nodes
annotated as upregulating or downregulating. The map nodes and
edges, attributes and default visual properties for all elements
were therefore specified.
Construction of networks using
Cytoscape
The data were imported into Cytoscape through the
‘IMPORT NETWORK FROM TABLE’ function, and the network of associated
factors with IL-1β in AD were generated automatically, only
defining the interaction of the list in EXCEL.
KEGG searches and reconstruction of
associated images by Cytoscape
In order to further determine the affected pathways
in AD through IL-1β, search terms including ‘Alzheimer’s disease’
or ‘IL-1β’ were used as keywords in the KEGG pathway analysis.
Following this, certain pathways, which were identified to be
associated with this research, were redrawn in Cytoscape in order
to better compare these factors.
Results
Interaction nets created in
Cytoscape
Three interaction nets were generated. The factors
were identified by querying the literature prior to collecting and
constituting the interactional net between IL-1β and other factors
involved in AD (Fig. 1A). Fig. 1B and C are subtypes of Fig. 1A. Fig.
1B includes the elements which could be affected by IL-1β in AD
and Fig. 1C includes the proteins
which could induce transformation of IL-1β in AD (see Fig. 1C).
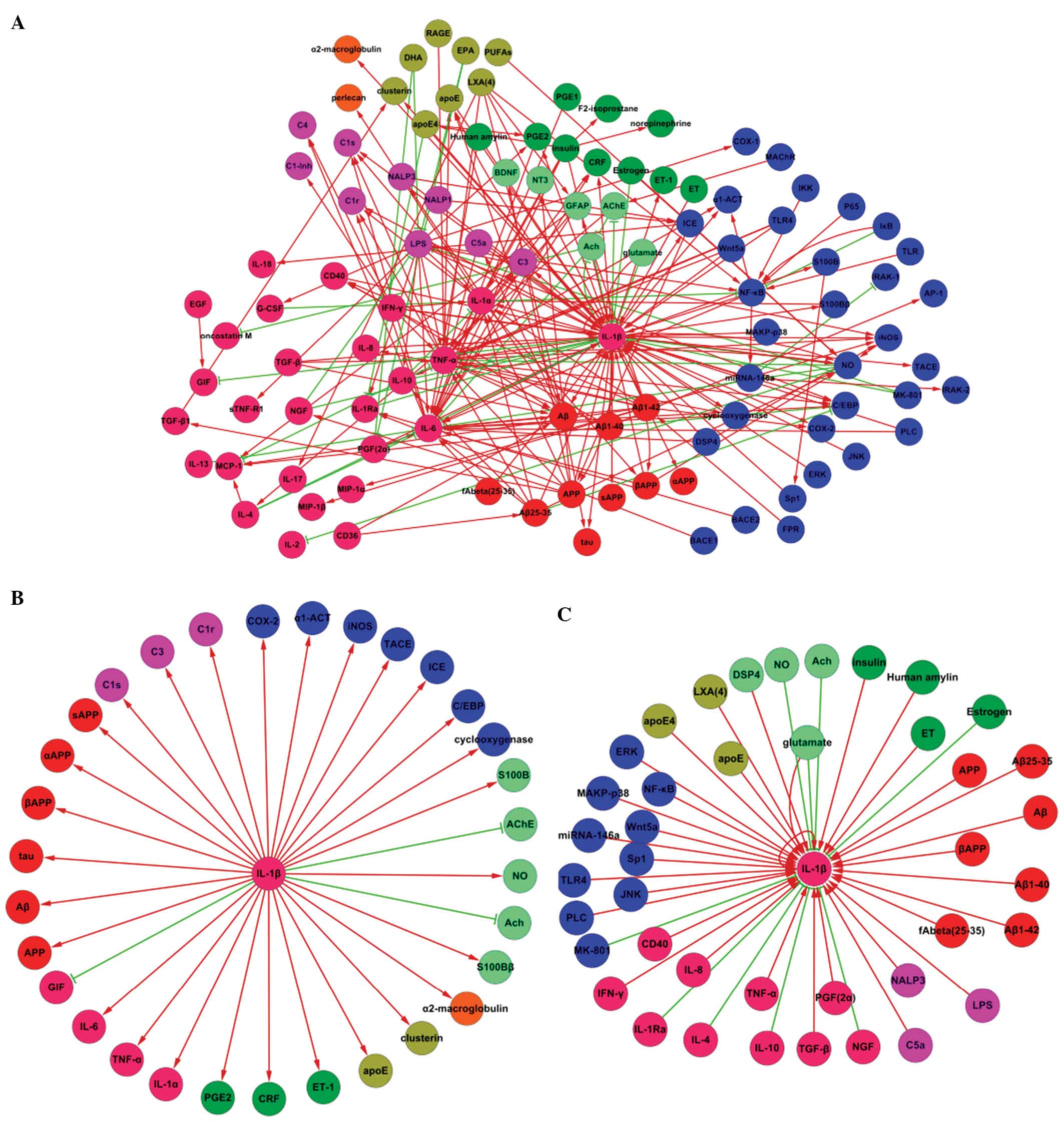 | Figure 1Association between IL-1β and other
factors in AD. (A) Protein interaction map describing the responses
implicated in the association between IL-1β and other factors.
Increases are reflected as straight arrows, shown as red lines and
decreases are reflected as inverted ‘T’ shapes, shown with green
lines. (B) A protein interaction map describing the responses
triggered in the factors affected by IL-1β in AD. (C) A protein
interaction map describing the responses triggered in the factors
affected by IL-1β in AD. The following colors were used in creating
the cytoscape nets: Pink, cytokines; green, hormones, blue, signal
factors; orange, globulin; purple, immune factors; blue-green,
neurotransmitters; red, specific proteins with AD; brown, lipid.
IL, interleukin; AD, Alzheimer’s disease. |
Analysis of the figures
Interaction net Fig. 1A
The association between IL-1β and other factors
involved in Alzheimer’s disease is shown in Fig. 1A. This network included 105 nodes
and 230 edges. Among these, 65 nodes and 75 edges were directly
connected with the node of IL-1β. The edges consisted of 63
positive (straight arrow) and 12 negative correlations (inverted
“T” shape). For the positive correlations, 35 of the edges
represented that IL-1β would be affected by other factors, whereas
28 edges indicated that IL-1β would affect other factors. This
applied to the negative correlations, where nine edges were
inhibited and three edges were inhibiting.
The nets indicated that IL-1β is associated with
numerous other factors involved in the pathology of AD. Several of
these associated factors were significantly correlated with AD,
including forms of beta amyloid (Aβ) and its precursor, amyloid
precursor protein (APP). These data indicated that IL-1β is a
central immunological factor which influences the formation of Aβ
and fibrous plaques.
Numerous cytokines have been observed to alter in
function or expression in AD, including tumor necrosis factor
(TNF), interferon (IFN) and complement component 3 (C3). This has
demonstrated that the process of immunity is involved in the
pathology of AD. Furthermore, some of these factors are correlated
with IL-1β, which may prove to be a central factor in AD-associated
immunological reactions.
The pathology of AD is often comorbid with hormone
disorders, thus the neuroendocrine system is additionally
considered to participate in the pathology of AD. Hormonal
secretions have been observed to be interrelated with IL-1β and the
nervous, immune and the endocrine systems, which contribute to the
pathology of AD.
The expression and accumulation of starch in the
nervous system due to AD lesions has an effect on neuronal
activity. Numerous neurotransmitters have been shown to be involved
in this pathology of AD, in which certain aspects have been
correlated with IL-1β.
The activation or inhibition of signaling pathways
maintains the determination of gene expression and protein
synthesis. IL-1β is dependent upon multiple pathways in order to
exert effects on the pathology of AD. The interaction net created
in Fig. 1A indicated that various
signaling molecules may be associated with IL-1β, thus forming a
signaling pathway between IL-1β and AD.
Interaction net Fig. 1B
The net shown in Fig.
1B was composed of 31 edges and 32 nodes. These represent
factors that were increased or decreased by IL-1β. Of the 32 edges,
28 were upregulated and three were downregulated.
The interaction net indicated that IL-1β could
promote the secretion of Aβ, which originates from APP cleavage
products, including αAPP, βAPP, sAPP. IL-1β may therefore induce Aβ
assembly through the upregulation of APP. Aggregates of Aβ in the
central nervous system (CNS), including the hippocampus, would lead
to neuronal toxicity, synapse damage and neuronal apoptosis.
The interaction net indicated that numerous
inflammatory mediators may contribute to the pathology of AD. IL-1β
acts as an inflammatory mediator that drives the secretion of other
inflammatory factors, including IL-6, tumor necrosis factor (TNF)-α
and complement C1s subcomponent (C1s), complement C1r subcomponent
(C1r) and C3. The activity of astrocytes and microglia in the CNS
has been previously observed in AD (28), which supports that
neuroimmunomodulation participates in the process of AD. IL-1β,
IL-6, TNF-α as well as complements C1 and C3 are pivotal factors in
inflammation. The interaction net indicated that IL-1β can
upregulate these inflammatory factors, thereby aggregating
inflammation, which results in the generation of adverse conditions
for immunological disorders of AD.
Neurological diseases are often accompanied by
deterioration of the endocrine system (29). It has been reported that low levels
of estrogen may be one cause of AD, which is prevalent in females
following the menopause (30). The
interaction net identified that IL-1β may upregulate the internal
secretions of factors including corticotropin-releasing factor
(CRF), prostaglandin E2 (PGE2) and endothelin (ET)-1. An increase
in estrogen levels may change the cerebral blood supply and
metabolism and result in abnormal neuronal viability.
According to the interaction map in Fig. 1B, IL-1β may affect the secretion of
various neurotransmitters. IL-1β could enhance nitric oxide (NO)
and S100B, which cause neuronal damage, whilst suppressing
acetylcholin (ACh), which usually acts to protect neurons. It may
be concluded that IL-1β expression has a disadvantageous function
in AD, inducing neuronal loss and suppression of protective
factors.
Pathway analysis was conducted to determine the
influence of IL-1β and the affected signaling molecules in the
interaction map. Fig. 1B shows
that IL-1β may impact the production of prostaglandin-endoperoxide
synthase 2 (COX-2), thus functioning in the respiratory chain and
disturbing mitochondrial activity. Disturbance of mitochondrial
function could be attributed to disturbance of neural structure and
function, which ultimately results in apoptosis.
Interaction net Fig. 1C
The interaction map in Fig. 1C is composed of 43 edges and 42
nodes, which represent the factors that increase or decrease IL-1β
expression. With the exception of eight nodes, 34 nodes including
IL-1β itself, could upregulate IL-1β. Of the factors identified,
glutamate was shown to both activate and inhibit IL-1β, thus the
total number of nodes that could upregulate IL-1β was 35. These
data showed that the majority of factors in AD may be attributed to
the activation of immune cells, including monocytes, lymphocytes,
astroglia and microglia in the CNS, to secret IL-1β. High levels of
IL-1β would aggravate the inflammatory damage caused to the nervous
system.
The interaction map indicated that Aβ and its
degradation products are able to initiate the production of IL-1β.
Accumulation of Aβ and the formation of fibrous plaques are typical
characteristics of AD. The induced secretion of IL-1β by these
products can aggravate the pathological changes that occur in AD.
The secretion of various inflammatory substances, including
cytokines IFN-γ, TNF-α and factors such as lipopolysaccharide
(LPS), NACHT leucine-rich repeat protein (NALP3) and complement
C5a, is a major component of the pathology of AD. An increase in
these substances is associated with the production of IL-1β by
immune cells, which stimulates an inflammatory reaction, thus
leading to inflammation of the neuron. Lesions in the CNS develop
cellular hypoxia and changes to the cytoskeleton, which further
induce apoptosis. Despite the existence of numerous negative
factors, protective elements, including IL-4 and IL-10, could
inhibit IL-1β and prevent further pathological damage.
It is known that the absence of insulin can induce
diabetes. Conversely, high levels of insulin have emerged to be
able to upregulate IL-1β, and thus may be associated with AD.
Estrogen is considered to be a protective factor which may prevent
the occurrence of AD through inhibition of IL-1β. It has been shown
that the immune system affects the CNS following endocrinium
influences on the immune system. This evidence follows a review of
what is known about the association between the nervous, immune and
endocrine systems, which is a major focus in the medical treatment
of AD (31). Accordingly, this
interaction net illustrates that few neurotransmitters are
associated with IL-1β, including ACh, which may be a protective
factor in AD. There is conflicting evidence regarding the effects
of glutamate, suggesting it could therefore increase or decrease
IL-1β expression.
Signaling molecules, including the toll-like
receptor (TLR)4 receptor for the TOLL signaling pathway and
mitogen-activated protein kinase (MAPK)-P38 in the MAPK pathway,
have been observed to affect IL-1β. These molecules contribute to
induce the gene expression of IL-1β, therefore resulting in
aggravation of inflammation of the CNS.
Pathways collected from KEGG
analysis
Five results were obtained when using ‘Alzheimer’s
disease’ as a key word. Among these results, map05010 and map04310
were objective pathways which could be used to compare with the
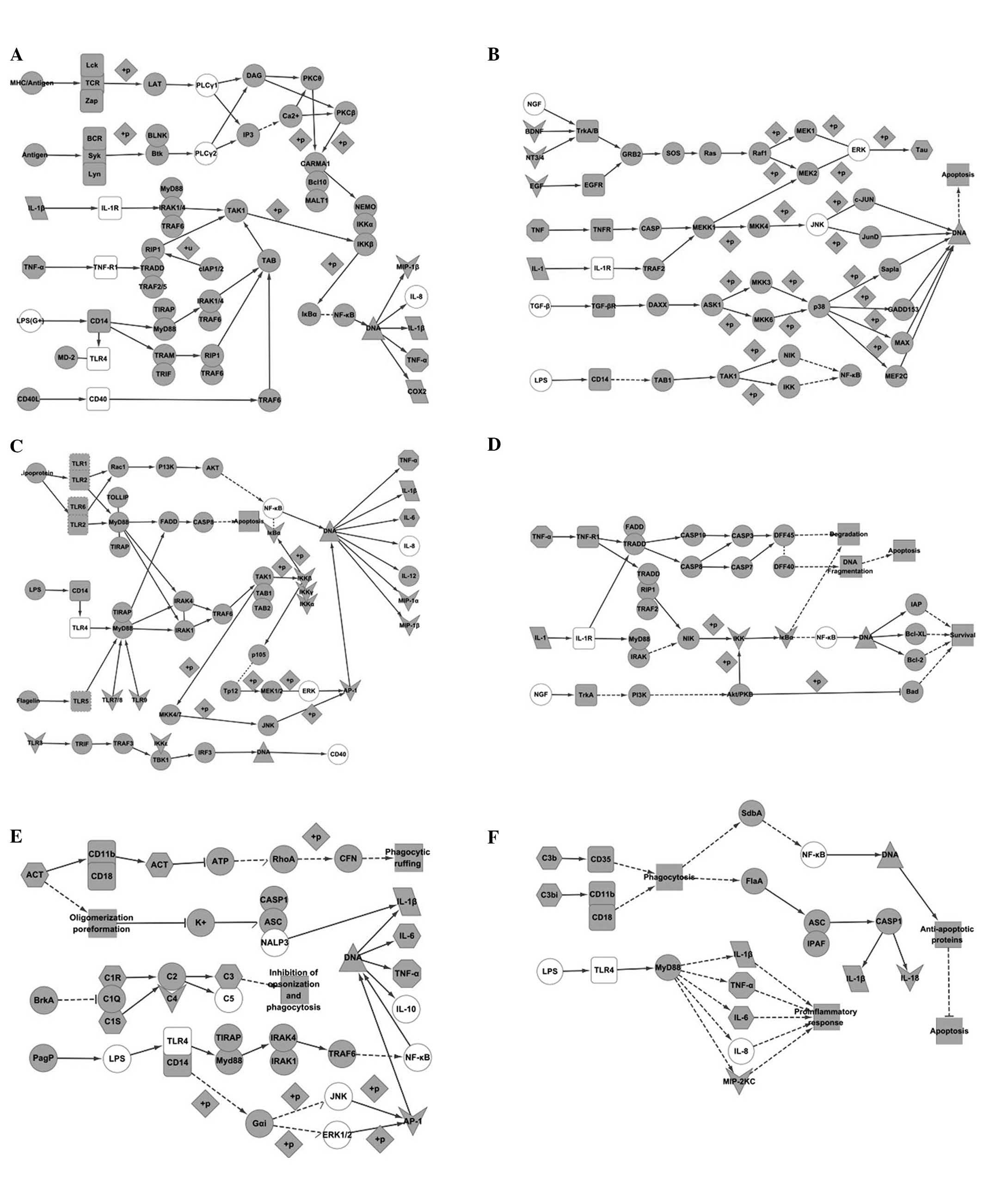
interaction nets produced in Cytoscape (Fig. 2). Using ‘IL-1β’ as the key word, 27
results were obtained. These pathways included: i) The nuclear
factor (NF)-κB signaling pathway, originating from map04064 in KEGG
(Fig. 3A); ii) the MAPK signaling
pathway, originating from map04010 in KEGG (Fig. 3B); iii) the TLR signaling pathway,
originating from map04620 in KEGG (Fig. 3C); iv) apoptosis signaling pathway,
originating from map04210 in KEGG (Fig. 3D); v) pertussis signaling pathway,
originating from map05133 in KEGG (Fig. 3E); and vi) legionellosis signaling
pathway, originating from map05134 in KEGG (Fig. 3F).
Comparison of the AD pathway in KEGG with
IL-1β and analysis of associated factors in AD
Comparison of Fig. 1B and Fig. 2A
Fig. 1B illustrates
that IL-1β is able to upregulate NO. In addition, increases of NOS
are driven by the accumulation of Ca2+ in the cytoplasm
(Fig. 2A). Binding of NOS
effectively causes secretion of NO-ONOO−, inducing
protein oxidation, mitochondrial dysfunction, apoptosis, DNA
damage, inflammation and lipid peroxidation. High levels of
Ca2+ in the cytoplasm may be attributed to activating
receptors, including FAS/TNFR, G protein-coupled receptor (GPCR),
N-methyl-d-aspartate receptor and voltage-dependent calcium
channels, whose ligands involve Aβ that can be increased by IL-1β.
Accordingly, Fig. 1B shows that
IL-1β can upregulate COX-2, which is effective in enhancing
mitochondrial function. IL-1β can therefore cause an increase in
Aβ, which would be associated with upregulation of COX-4 and
Aβ-binding alcohol dehydrogenase. Mitochondrial dysfunction could
therefore occur, which would increase reactive oxygen species
production to induce cell death. It is widely recognized that APP
is one of key factors in AD. Fig.
2A shows that the existence of APP has the potential to alter
the expression levels of signaling molecules in the cytoplasm,
including upregulation of Fe65, APP-binding protein 1 and
downregulation of GAPD. These changes may lead to apoptosis and
decreased energy production. This process is additionally
associated with IL-1β, which may increase APP (Fig. 1B). In addition to Aβ and APP, IL-1β
has been observed to upregulate apolipoprotein (Apo)E and Tau
(Fig. 1B.) The former is connected
with lipoprotein lipase, which can trigger LPR to cause aggregation
of Aβ. The latter may cause formation of neurofibrillary tangles,
which is a consequence of the activation of p25 and
cyclin-dependent kinase (Cdk)5.
Comparison of Fig. 1C and Fig. 2A
Initiating factors, including Aβ, ApoE and APP
(Fig. 2A) may increase IL-1β
expression (Fig. 3C). The
activation of neuronal signaling molecules has been associated with
immunological functions, thus resulting in neuronal damage. The
activation of Gq occurs through GPCR, which is the next molecule of
oligomeric Aβ to which alterations would induce phospholipase C
(PLC) to cause endoplasmic reticulum stress and cell injury by
caspase (Casp)12. PLC was identified to be able to upregulate IL-1β
(Fig. 1C).
Comparison of Fig. 1C and Fig. 2B
c-Jun N-terminal kinase (JNK) and PLC have been
identified in the Cytoscape net and the KEGG pathway. G protein,
which could activate PLC, would be expressed through signaling
between Wnt5 and Frizzled, and Ca2+ influx that could
trigger Ca2+/calmodulin-dependent protein kinase II, CaN
and protein kinase C (PKC). These signaling molecules would
dephosphorylate nuclear factor of activated T cells transcription
factor, which would have effects on DNA expression (Fig. 2B). In addition, Wnt11 is
responsible for the function of JNK through Rac or Ras homolog gene
family, member A. Both PLC and JNK could increase the expression
levels of IL-1β (Fig. 1C). The Wnt
pathway not only participates in the pathology of AD, but can
stimulate secretion of IL-1β, which in turn enhances neural immune
damage.
Comparison of the NF-κB signaling pathway
with IL-1β and analysis of associated factors in AD
Comparison of Fig. 1B and Fig. 3A
The NF-κB signaling pathway is crucial for
understanding the mechanism of inflammation. It was observed by
analyzing Fig. 1B that IL-1β is
both an initiation factor and objective factor. The connection of
IL-1β and IL-1R initiates interleukin-1 receptor-associated kinase
(IRAK)1/4-transforming growth factor β-activated kinase 1
(TAK1)-binding proteins (TAB)-inhibitor of nuclear factor kappa-B
kinase subunit alpha (IKK) signaling, resulting in continuous
phosphorylation of inhibitor of kappa B (IκB)α by IKK, which would
expose the DNA binding site for NF-κB (p50/p65), ultimately
inducing IL-1β, TNF-α and COX-2 expression. It was observed that
IL-1β itself, TNF-α and COX-2 are upregulated by IL-1β in AD
(Fig. 3A). It is therefore
predicted that an inflammatory reaction in AD would be equivalent
to the activation of the NF-κB signaling pathway.
Comparison of Fig. 1C and Fig. 3A
Factors, including IL-1β, IL-8, TNF-α, CD40, TLR4,
PLC and LPS were observed in both the interaction net and KEGG
pathway. Communication between TNF-α and TNF-R1 may activate TNF
receptor type 1-associated DEATH domain protein
(TRADD)/receptor-interacting serine/threonine protein (RIP1)/TNF
receptor associated factor (TRAF)2/5, and the downstream factor
RIP1 would stimulate TAK1. Finally, secretion of IL-1β would be
induced by IKK-IκB-NF-κB. The function of TLR4 is possibly through
CD14 activated by LPS, which would cause NF-κB stimulation of DNA
through Toll-interleukin 1 receptor (TIR) domain-containing adapter
protein (TIRAP)/MyD88 or translocating chain-associating membrane
protein (TRAM)/TIR domain-containing adaptor protein inducing
interferon β (TRIF). A connection between major histocompatibility
complex/antigen and T-cell receptor also addresses the activation
of NF-κB through PLC-PKC. CD40 and may affect NF-κB through TRAF6.
Furthermore IL-8 is consistent with IL-1β in its secretion through
the activation of NF-κB (see Fig.
3A). IL-1β may be upregulated by TNF-α, CD40, LPS and TLR4 in
AD (Fig. 1C). This is evidence to
support the involvement of the NF-κB signaling pathway in AD.
Comparison of Table II with Fig. 3A
Macrophage inflammatory protein (MIP)-1β is the
conjunct factor shown in Table II
and Fig. 3A. MIP-1β is secreted by
the activation of NF-κB, which would be stimulated by IL-1β
(Fig. 3A). Thus it is predicted
that IL-1β may upregulate MIP-1β through the NF-κB signaling
pathway in AD.
Comparison of the MAPK signaling pathway
with IL-1β and analysis of the associated factors in AD
Comparison of Fig. 1B and Fig. 3B
Two factors, TNF-α and Tau, were identified in both
figures. The function can be initiated by a connection between
IL-1, IL-1R and TAB1-TAK1, leading to mitogen-activated protein
kinase kinase (MEK)1/MEK2-mediated phosphorylation of tau.
Hyperphosphorylated tau is a competitor of tau protein, which
occupies the sites of microtubules, which would damage the normal
structure of the cytoskeleton and lead to apoptosis. TNF-α would
stimulate Casp or TRAF2 in this pathway.
Comparison of Fig. 1C and Fig. 3B
Six factors were identified when comparing the two
figures produced by Cytoscape and KEGG. These factors included
TNF-α, TGF-β, JNK, extracellular-signal regulated kinase (ERK), LPS
and nerve growth factor (NGF), which would upregulate IL-1β.
However, all of these are causative factors in the MAPK pathway. It
has been shown that TGF-β and TNF-α effect TAK1, which can
phosphorylate NF-κB-inducing kinase NIK/IKK and NF-κB (Fig. 3B), leading to secretion of IL-1β
(Fig. 3A). LPS would connect to
CD14, which regulates NF-κB. It was identified that c-fos would be
secreted when TrkA/TrkB is signaling with NGF or brain-derived
neurotrophic factor (BDNF) (see Fig.
3B); however, an association has not been identified with
NF-κB. Furthermore, ERK, a downstream factor of transforming
tyrosine kinase protein (Trk)A/TrkB, would up-regulate IL-1β
(Fig. 1C), thus inferring an
association between this signaling pathway and AD. JNK would
increase IL-1β (Fig. 1C), but the
nexus with IL-1β (Fig. 3B) was not
identified.
Comparison of Table II with Fig. 3B
It was identified that EGF, BDNF and neurotrophin
(NT)-3 were included in Table II
and the MAPK pathway, which take effects as ligands but do not
affect IL-1β.
Comparison of the TLR signaling pathway
with IL-1β and analyzing associated factors in AD
Comparison of Fig. 1B and Fig. 3C
Two factors, IL-6 and TNF-α, were found in both the
Cytoscape net and the KEGG pathway. Both factors are increased when
the initiation of the TLR lead to the weakening of the
transcription factors, AP-1 or interferon regulatory factor (IRF)5.
This process is additionally required for expression of IL-1β.
However when TLR1/TLR2 connect to their appropriate activators,
NF-κB can function in this pathway through AKT. It is known that
IL-1β can activate NF-κB, therefore this pathway may be one
mechanism for upregulating IL-6 and TNF-α through IL-1β.
Comparison of Fig. 1C and Fig. 3C
Eight factors were identified when comparing the
Cytoscape interaction net in Fig.
1C with the KEGG pathway in Fig.
3C. These factors included IL-8, TNF-α, CD40, NF-κB, TLR4, JNK,
ERK and LPS. The secretion of IL-1β may be upregulated through AP-1
upon interaction between TLR4 with LPS. LPS activates TIRAP/MyD88,
of which the downstream targets are TAB1/2/TAK1-mitogen-activated
protein kinase kinase (MKK)33/4/6/7-p38/JNK or
TAB1/2/TAK1-MKK3/4/6/7-IKK-ERK (Fig.
3C). IL-1β can be increased by LPS, TLR4, JNK and ERK (Fig. 1C). This has supported that the
Toll-like signaling pathway, specifically AP-1 trigged by LPS-TLR4,
could result in an imbalance of IL-1β in AD. This pathway predicts
that IL-8, TNF-α and CD40 are only effectors through activating the
TLR.
Comparison of Table II and Fig. 3C
Six factors were identified when comparing Table II and the Toll-like pathway
(Fig. 3C), including MIP-1α,
MIP-1β, TLR, IKK, IκB and AP-1. IL-1β could be produced when TLR4
is activated (Fig. 3C). It was
therefore inferred that AP-1 would increase IL-1β expression during
AD, through activation of the Toll-like signaling pathway. IκB can
be activated by IKK, exposing the DNA binding site for NF-κB, which
is able to promote expression of IL-1β. IκB and IKK are positive
regulators of IL-1β in AD. However, MIP-1α and MIP-1β may be
expressed when triggering the TLR in this pathway (Fig. 3C).
Comparison of the apoptosis signaling
pathway with IL-1β and analysis of associated factors in AD
Comparison of Fig. 1B and Fig. 3D
TNF-α was identified in both the Cytoscape
interaction net (Fig. 1B) and the
KEGG pathway (Fig. 3D). Both TNF-α
and IL-1β are apoptotic factors in this pathway, with objective
enzyme Casp3 activated through TRADD/Fas-associated protein with
death domain (FADD)-Casp10/Casp8 signaling. This results in
cleavage of the Casp substance. The activity of Casp3 is a typical
change in AD (Fig. 2A), thus it
could be inferred that TNF-α and IL-1β may lead to apoptosis
through the Casp3 pathway in AD.
Comparison of Fig. 1C and Fig. 3D
Three factors, including TNF-α, NF-κB and NGF, were
identified when comparing Fig. 1C
with Fig. 3D. In these pathways,
NGF connects with TrkA which would in turn activate NF-κB through
Akt/PKB. Other factors, including inhibitor of apoptosis (IAP),
B-cell lymphoma-2 (Bcl-2) extra large protein (Bcl-XL) and Bcl-2
could inhibit this pathway. It is indicated that NGF may
up-regulate IL-1β through this signaling pathway in AD.
Comparison of Table II and Fig. 3D
Only IKK was present in both Table II and the apoptosis pathway
(Fig. 3D), which is associated
with NF-κB.
Comparison of the pertussis signaling
pathway with IL-1β and analysis of associated factors in AD
Comparison of Fig. 1B and Fig. 3E
Pertussis is an infectious disease in which the
pathological changes were analyzed through the signaling pathways
shown in Fig. 1B and Fig. 3E. The pertussis signaling pathway
was compared in order to further understand the molecular
mechanisms contributing to AD. Six factors were identified in
common between both figures, including IL-6, TNF-α, C3, C1r, C1s
and actin assembly-inducing protein (ACT). The complement proteins
C1r, C1s, and C3 would be activated upon triggering BrkA, which
would inhibit opsonization and phagocytosis of macrophages. It was
not identified whether these complements would affect IL-1β, IL-6
and TNF-α (Fig. 3E). Conversely,
NF-κB would be weakened through cyclic adenosine monophosphate-p38
when activating ACT by cyanobacterial adenylate cyclase (CyaC)
(Fig. 3E), which would ultimately
result in the secretion of IL-1β, IL-6 and TNF-α.
Comparison of Fig. 1C and Fig. 3E
Nine factors were identified in common in Fig. 1C and Fig. 3E. These included IL-1β, IL-8,
TNF-α, IL-10, TLR4, ERK, JNK, LPS, NALP3. The association between
impact factors, including LPS, TLR4, ERK, JNK, and objective
factors, including IL-10, IL-1β, IL-6 and TNF-α, are described in
the preceding discussion. IL-1β would be expressed upon connection
between Casp1 with ASC and NALP3, while this may be suppressed by
high concentrations of K+ (Fig. 3E). This pathway also showed that
IL-8 may inhibit the recruitment of immune cells and it is not
conductive to inflammation elimination.
Comparison of Table II and Fig. 3E
Two factors, C4 and AP-1, were present in both
Table II and the pertussis
pathway. The function of AP-1 has been described in the Toll-like
signaling pathway and C4 is a signaling molecule between C1 and
C3/C5.
Comparison of the legionellosis signaling
pathway with IL-1β and analysis of associated factors in AD
Comparison of Fig. 1B and Fig. 3F
Legionellosis is another infectious disease which
involves various factors that are associated with IL-1β in AD.
These correlated elements include IL-6, TNF-α and C3. IL-1β would
be secreted through NF-κB, which is downstream of Legk1 or SdbA, as
well as through FlaA-Casp1. However, both signaling pathways are
activated through a connection between C3 with CD35 or CD11b/CD18
(Fig. 3F). It could not be shown
that C3 is upregulated by IL-1β in this pathway.
Comparison of Fig. 1C and Fig. 3F
Six factors, including IL-8, TNF-α, NF-κB, TLR4, LPS
and IL-1β, were identified in both Fig. 1C and Fig. 3F. IL-1β, TNF-α and IL-8 would be
upregulated by LPS and TLR4, which belong to the Toll-like receptor
signaling pathway.
Comparison of Table II and Fig. 3F
Two factors, MIP and IL-18, were identified in both
Table II and the legionellosis
pathway. The function of MIP has been described in the Toll-like
signaling pathway. IL-18 and IL-1β are produced by Casp1, which is
directly stimulated by C3 (see Fig.
3F).
Integration of networks created by
Cytoscape and KEGG
Through comparison of the networks created by
Cytoscape and KEGG, the present study identified that certain
factors may affect IL-1β in AD, which is contradictory to previous
reports, which did not show that these factors up- or downregulated
IL-1β. These factors included: i) MIP-1β, which is secreted by
IL-1β; ii) IKK and IκB, which can both affect IL-1β through the
NF-κB pathway; and iii) AP-1, which can increase the expression
levels of IL-1β.
Discussion
A growing body of evidence indicated that IL-1β
expression is one of the earliest and most important
neuropathological factors in numerous diseases of the nervous
system, including AD (32,33). The present study therefore
collected numerous factors which are known to be associated with
IL-1β in AD and constructed an associated net using Cytoscape.
Furthermore, pathways that are associated with AD or IL-1β were
analyzed in order to determine the predominant functional
mechanisms.
IL-1β was shown to connect with IL-1R, which would
activate numerous pathways and subsequently lead to various
pathological changes in AD. This would cause an energy metabolism
disorder of the neurons and changes to the cytoskeleton. This would
precipitate neuronal apoptosis and the formation of fibrous
plaques. Certain important pathways would be employed in this
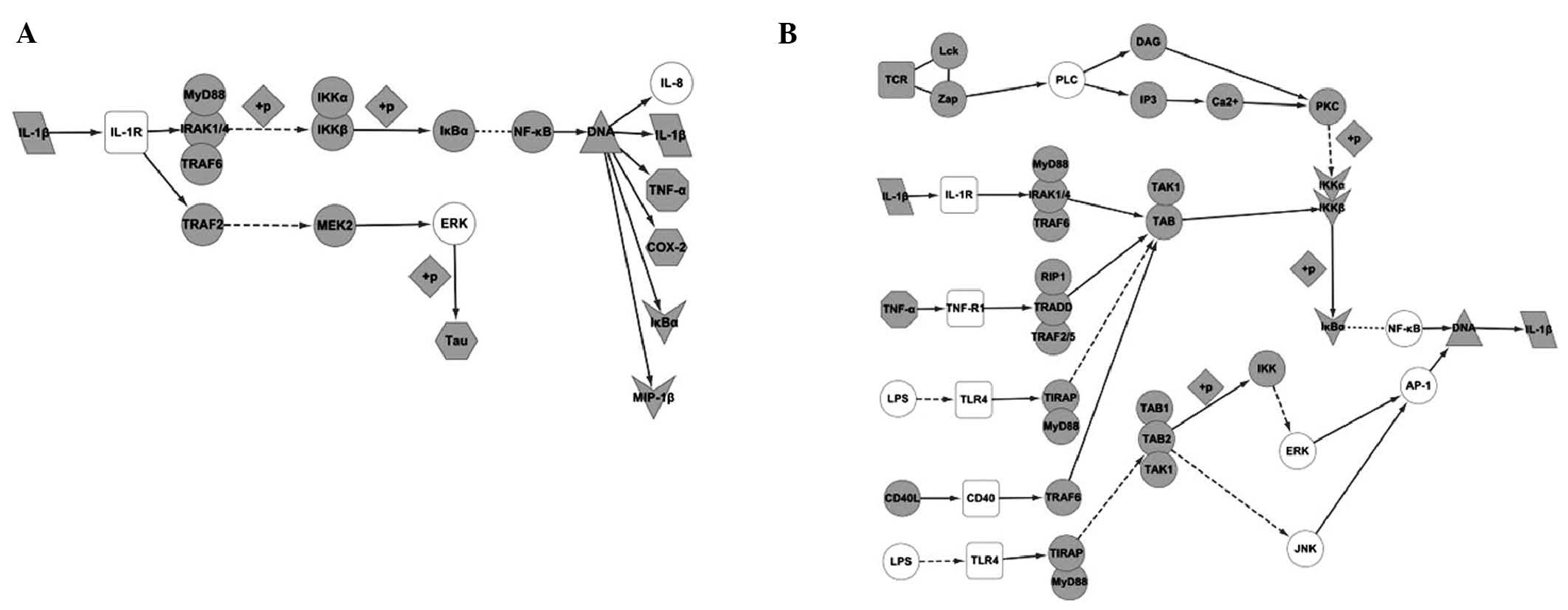
change, including: i) the NF-κB signaling pathway, which would be
activated when IL-1β is connecting with IL-1R. This would induce
expression of IL-1β itself as well as TNF-α, Cox-2 and MIP-1β, and
ii) the MAPK signaling pathway, with the main effect in this
pathway being the secretion of Tau (Fig. 4A).
IL-1β is one of numerous effectors in AD, which
would be upregulated by various signaling molecules to promote
neuronal damage. These pathways include: i) the NF-κB signaling
pathway - besides activation by IL-1β itself, the NF-κB pathway
could be activated by TNF-α, LPS, PLC, CD40, TLR4 and TGF-β, to
induce secretion of IL-1β. ii) The MAPK signaling pathway - factors
in this pathway include NGF, BDNF, RAS and C-fos. iii) The
Toll-like receptor signaling pathway - this pathway would be
activated by LPS and TLR4, with JNK, ERK and AP-1 being affected by
this activation. iv) The caspase signaling pathway, which would be
activated by NGF (Fig. 4B).
In conclusion, the present study aimed to analyze
the association between known factors and IL-1β in AD, and predict
the mechanisms of action by comparing these factors with pathways
generated in KEGG. It was identified that molecules may modulate
IL-1β, or be modulated by IL-1β, in AD, in which IL-1β is the core
factor in the inflammatory pathology. Numerous factors may increase
IL-1β expression, thus leading to immunological damage. However,
certain factors were identified to decrease IL-1β and therefore
have protective effects on neuronal damage.
Abbreviations:
|
AD
|
Alzheimer’s disease
|
|
IL
|
interleukin
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
TNF-α
|
tumor necrosis factor-alpha
|
References
|
1
|
Scheff SW and Price DA: Synaptic pathology
in Alzheimer’s disease: a review of ultrastructural studies.
Neurobiol Aging. 24:1029–1046. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Serý O, Povová J, Míšek I, Pešák L and
Janout V: Molecular mechanisms of neuropathological changes in
Alzheimer’s disease: a review. Folia Neuropathol. 51:1–9. 2013.
View Article : Google Scholar
|
|
3
|
Cummings JL, Vinters HV, Cole GM and
Khachaturian ZS: Alzheimer’s disease: etiologies, pathophysiology,
cognitive reserve, and treatment opportunities. Neurology.
51:S2–S17. 1998. View Article : Google Scholar
|
|
4
|
Bredesen DE and John V: Next generation
therapeutics for Alzheimer’s disease. EMBO Mol Med. 2013.
View Article : Google Scholar
|
|
5
|
Ryan TM, Caine J, Mertens HD, et al:
Ammonium hydroxide treatment of Abeta produces an aggregate free
solution suitable for biophysical and cell culture
characterization. Peer J. 1:e732013. View
Article : Google Scholar
|
|
6
|
Aloe L, Rocco ML, Bianchi P and Manni L:
Nerve growth factor: from the early discoveries to the potential
clinical use. J Transl Med. 10:2392012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brady R and Weinman J: Adherence to
cholinesterase inhibitors in Alzheimer’s disease: a review. Dement
Geriatr Cogn Disord. 35:351–363. 2013. View Article : Google Scholar
|
|
8
|
Chorsky RL, Yaghmai F, Hill WD and Stopa
EG: Alzheimer’s disease: a review concerning immune response and
microischemia. Med Hypotheses. 56:124–127. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reale M, Greig NH and Kamal MA: Peripheral
chemo-cytokine profiles in Alzheimer’s and Parkinson’s diseases.
Mini Rev Med Chem. 9:1229–1241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maguire-Zeiss KA and Federoff HJ:
Immune-directed gene therapeutic development for Alzheimer’s,
prion, and Parkinson’s diseases. J Neuroimmune Pharmacol.
4:298–308. 2009. View Article : Google Scholar
|
|
11
|
Wiese S, Karus M and Faissner A:
Astrocytes as a source for extracellular matrix molecules and
cytokines. Front Pharmacol. 3:1202012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rubio-Perez JM and Morillas-Ruiz JM: A
review: inflammatory process in Alzheimer’s disease, role of
cytokines. ScientificWorldJournal. 2012:7563572012. View Article : Google Scholar
|
|
14
|
Di Bona D, Plaia A, Vasto S, et al:
Association between the interleukin-1beta polymorphisms and
Alzheimer’s disease: a systematic review and meta-analysis. Brain
Res Rev. 59:155–163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Forlenza OV, Diniz BS, Talib LL, et al:
Increased serum IL-1beta level in Alzheimer’s disease and mild
cognitive impairment. Dement Geriatr Cogn Disord. 28:507–512. 2009.
View Article : Google Scholar
|
|
16
|
Hamajima N and Yuasa H: Genetic
polymorphisms related to interleukin-1 beta production and disease
risk. Nihon Koshu Eisei Zasshi. 50:194–207. 2003.(In Japanese).
PubMed/NCBI
|
|
17
|
Mitroulis I, Skendros P and Ritis K:
Targeting IL-1beta in disease; the expanding role of NLRP3
inflammasome. Eur J Intern Med. 21:157–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Del Bo R, Angeretti N, Lucca E, De Simoni
MG and Forloni G: Reciprocal control of inflammatory cytokines,
IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci
Lett. 188:70–74. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blume AJ and Vitek MP: Focusing on
IL-1-promotion of beta-amyloid precursor protein synthesis as an
early event in Alzheimer’s disease. Neurobiol Aging. 10:406–408;
discussion 412–404. 1989. View Article : Google Scholar
|
|
20
|
Shannon P, Markiel A, Ozier O, et al:
Cytoscape: a software environment for integrated models of
biomolecular interaction networks. Genome Res. 13:2498–2504. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vertommen A, Møller AL, Cordewener JH, et
al: A workflow for peptide-based proteomics in a poorly sequenced
plant: a case study on the plasma membrane proteome of banana. J
Proteomics. 74:1218–1229. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pahl MC, Derr K, Gäbel G, et al: MicroRNA
expression signature in human abdominal aortic aneurysms. BMC Med
Genomics. 5:252012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ogata H, Goto S, Fujibuchi W and Kanehisa
M: Computation with the KEGG pathway database. Biosystems.
47:119–128. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aoki-Kinoshita KF and Kanehisa M: Gene
annotation and pathway mapping in KEGG. Methods Mol Biol.
396:71–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu L, Aboud O, Jones RA, Mrak RE, Griffin
WS and Barger SW: Apolipoprotein E expression is elevated by
interleukin 1 and other interleukin 1-induced factors. J
Neuroinflammation. 8:1752011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao J, O’Connor T and Vassar R: The
contribution of activated astrocytes to Abeta production:
implications for Alzheimer’s disease pathogenesis. J
Neuroinflammation. 8:1502011. View Article : Google Scholar
|
|
27
|
He FQ, Qiu BY, Zhang XH, et al:
Tetrandrine attenuates spatial memory impairment and hippocampal
neuroinflammation via inhibiting NF-kappaB activation in a rat
model of Alzheimer’s disease induced by amyloid-beta(1–42). Brain
Res. 1384:89–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Serrano-Pozo A, Muzikansky A, Gómez-Isla
T, et al: Differential relationships of reactive astrocytes and
microglia to fibrillar amyloid deposits in Alzheimer disease. J
Neuropathol Exp Neuro. 72:462–471. 2013. View Article : Google Scholar
|
|
29
|
Palm R, Ayala-Fontanez N, Garcia Y, et al:
Neuroendocrinology-based therapy for Alzheimer’s disease.
Biofactors. 38:123–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Henderson VW and Rocca WA: Estrogens and
Alzheimer disease risk: is there a window of opportunity?
Neurology. 79:1840–1841. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aloe L, Rocco ML, Bianchi P and Manni L:
Nerve growth factor: from the early discoveries to the potential
clinical use. Journal of Translational Medicine. 10:2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di Bona D, Plaia A, Vasto S, et al:
Association between the interleukin-1 beta polymorphisms and
Alzheimer’s disease: A systematic review and meta-analysis. Brain
Research Reviews. 59:155–163. 2008. View Article : Google Scholar
|
|
33
|
Azizi G and Mirshafiey A: The potential
role of proinflammatory and antiinflammatory cytokines in Alzheimer
disease pathogenesis. Immunopharmacology and Immunotoxicology.
34:881–895. 2012. View Article : Google Scholar : PubMed/NCBI
|