Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide (1) and ~85% of cases of lung cancer are
classified as non-small cell lung cancer (NSCLC) (2). Despite improvements in diagnostic and
therapeutic strategies, the prognosis for patients with NSCLC
remains poor, with a 5-year survival rate of 8–14% (3). The primary cause of lung
cancer-associated mortality is metastasis, and the majority of
patients with NSCLC have begun to develop metastatic disease by the
time they are diagnosed (1,2).
Thus, effective NSCLC therapies must include strategies to control
metastatic disease. Such strategies may be improved by a more
thorough understanding of the underlying mechanisms of NSCLC
metastasis.
Epithelial-mesenchymal transition (EMT) is an early
event in the metastatic progression of a number of types of
epithelial cancer, such as lung cancer (4–10).
EMT is the process by which epithelial cells transition from a
typical epithelial phenotype (polarized and adherent) to a
mesenchymal phenotype (spindle-shaped and motile). EMT results in
clear alterations in the morphology, adhesive properties and gene
expression of cells, including the upregulation of vimentin,
N-cadherin and fibronectin, in addition to the downregulation of
E-cadherin and cytokeratin (4,5).
Additionally, the mesenchymal state during EMT is associated with a
higher capacity for migration and invasion (11).
The process of EMT is regulated by a complex system
of signal transduction pathways. One key regulator of EMT in lung
cancer is the transforming growth factor-β (TGF-β) signaling
pathway (11,12). In addition to TGF-β, the Hedgehog
(Hh) signaling pathway is known to participate in EMT, however the
precise role of this pathway in EMT remains unclear (5). The Hh signaling pathway has been
reported to be activated in a number of human tumors, including
NSCLC and metastatic disease (13)
and ultimately activates the transcription factor human
glioma-associated oncogene homolog 1 (Gli1). Gli1 is also activated
by other cancer-associated signaling pathways, such as the receptor
tyrosine kinase and phosphoinositide 3-kinase (PI3K) pathways
(14).
Despite its association with Hh signaling, the
specific function of Gli1 in EMT remains to be fully elucidated. In
the current study, the role of Gli1 in TGF-β-induced EMT was
investigated in NSCLC cell lines. Gli1 levels in NSCLC cells that
underwent TGF-β1-induced EMT were measured, and the effect of small
interfering RNA (siRNA)- or pharmacological agent-mediated
inhibition of Gli1 activity on TGF-β1-induced EMT was analyzed. To
investigate this, alterations in morphology, phenotypic markers,
invasion and migratory capability were measured.
Materials and methods
Cell lines and reagents
The lung cancer cell lines A549, H460 and SK-MES-1
were purchased from the American Type Culture Collection (Manassas,
VA, USA) and cultured in RPMI-1640 medium containing 10% fetal
bovine serum (FBS) (Gibco Life Technologies, Carlsbad, CA, USA) at
37°C in a humidified atmosphere with 5% CO2. Recombinant
human TGF-β1 and GANT 61 were purchased from PeproTech, Inc. (Rocky
Hill, NJ, USA). Phase contrast images of A549 cells were acquired
using an inverted phase contrast microscope (IX53; Olympus
Corporation, Tokyo, Japan) subsequent to incubation of the cells
with 0, 1, 5 or 10 ng/ml TGF-β1 for 48 h. For western blot and
immunofluorescent analysis, polyclonal rabbit anti-human Gli1
(ab49314), polyclonal rabbit anti-human E-cadherin (ab15148),
monoclonal rabbit anti-human vimentin (ab16700), polyclonal rabbit
anti-human β-actin (ab1801) and horseradish peroxidase
(HRP)-conjugated anti-rabbit secondary antibodies were purchased
from Abcam (Cambridge, MA, USA).
siRNA transfection and drug
treatments
GFP-siRNA specific for Gli1 and nonspecific
GFP-siRNA were diluted in diethylpyrocarbonate (DEPC)-treated water
(all from Life Technologies, Grand Island, NY, USA). The siRNA was
used to deplete Gli1 mRNA and protein levels in the A549 cells.
A549 cells were transfected with DEPC-treated water (control
group), a nonspecific control siRNA (si-VE group) or Gli1-specific
siRNA (si-Gli1 group). A549 cells were cultured until 60–70%
confluence was reached and were then transfected with siRNA using
X-tremeGENE siRNA (Roche Diagnostics, Basel, Switzerland) and
OptiMEM medium (Life Technologies) according to the manufacturer’s
instructions. Following 6-h transfection, the cells were incubated
in RPMI-1640 with 10% FBS and 5 ng/ml TGF-β1. At 24 h subsequent to
transfection, the cells were imaged using the inverted phase
contrast microscope to assess cell morphology, while transfection
efficiency was assessed by detecting green fluorescent protein
(GFP) fluorescence using a confocal laser-scanning microscope
(OLS3100; Olympus, Corporation). Following a 48-h resting period,
cells were collected for Transwell invasion assays, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analyses. For treatment with GANT 61, the H460 and
SK-MES-1 cells were incubated with 10% FBS for 24 h and stimulated
with 5 ng/ml TGF-β1 for 48 h in the presence or absence of 10 μM
GANT 61.
RNA isolation and RT-qPCR
Total RNA from NSCLC cells was isolated using RNAiso
Plus extraction reagent and cDNA was produced using PrimeScript
Reverse Transcriptase (Takara Bio, Inc., Otsu, Japan) according to
the manufacturer’s instructions. RT-qPCR reactions were then
conducted with SYBR Premix Ex Taq II (Takara Bio, Inc.) in 25 μl
reactions with 2 μl cDNA and 0.4 μM each of the forward and reverse
primers using the following thermocycling conditions: 95°C for 30
sec, 95°C for 5 sec and 60°C for 30 sec. Data was collected over 40
cycles and all expression levels were normalized to β-actin. The
following primers were used: Gli1, F 5′-CTG GAC CTG CAG ACG GTT
ATC-3′ and R 5′-AGC CTC CTG GAG ATG TGC AT-3′; β-actin, F 5′-TGA
CGT GGA CAT CCG CAA AG-3′ and R 5′-CTG GAA GGT GGA CAG CGA GG-3′
(Sangon Biotech Co., Ltd., Shanghai, China).
Western blot analysis
For biochemical analysis, cells were washed with
ice-cold phosphate-buffered saline (PBS; Beyotime Institute of
Biotechnology, Jiangsu, China) and lysed in
radioimmunoprecipitation assay lysis buffer [50mM Tris, pH 7.4;
150mM NaCl; 1% Triton X-100; 1% sodium deoxycholate; 0.1% sodium
dodecyl sulfate (SDS); Beyotime Institute of Biotechnology]. The
lysates were kept on ice for 30 min and clarified by centrifugation
at 12,000 × g for 25 min at 4°C. The clarified lysate was then
collected and the vimentin, β-actin, E-cadherin and Gli1 proteins
were separated by 12% SDS-polyacrylamide gel electrophoresis
(30–100 μg protein/lane) and transferred to a polyvinylidine
fluoride membrane (all from Beyotime Institute of Biotechnology).
Subsequent to transfer, the membranes were incubated in 5% milk for
1 h, and then with the Gli1 (2.5 μg/ml), E-cadherin (1:500),
vimentin (1:500) or β-actin (1:1000) antibodies diluted in non-fat
milk. The membranes were then washed with Tris-buffered saline with
Tween-20 (Beyotime Institute of Biotechnology) and incubated with
the HRP-conjugated secondary antibodies. Immunoreactive proteins
were visualized using a BeyoECL Plus kit (Beyotime Institute of
Biotechnology).
Immunofluorescence
For the immunofluorescence analysis, NSCLC cells
were seeded onto glass slides 24 h prior to treatment and were
transfected with TGF-β1 and siRNA. Following treatment for 24 h,
cells were fixed with 4% paraformaldehyde (Beyotime Institute of
Biotechnology) for 15 min, washed with PBS, and permeabilized with
0.1% Triton X-100 (Beyotime Institute of Biotechnology) for 30 sec.
To examine cell-surface expression of E-cadherin, the cells were
not permeabilized following fixation. Cells were incubated with 5%
sheep serum albumin (Beyotime Institute of Biotechnology) for 1 h
to block non-specific proteins. Subsequent to blocking, cells were
incubated with the Gli1, E-cadherin, vimentin or β-actin
antibodies, washed with PBS three times, and then incubated with
fluorescein isothiocyanate-conjugated goat anti-rabbit and
tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit
secondary antibodies (ZF-0311; Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) diluted in distilled water
for 1 h at 37°C. The stained cells were placed on a coverslip in
mounting medium (Beyotime Institute of Biotechnology) with
4′,6-diamidino-2-phenylindole (Beyotime Institute of Biotechnology)
to label the nuclei. Cells were imaged using the OLS3100
fluorescence microscope (magnification, ×400).
Transwell cell migration and invasion
assay
The invasive and migratory ability of NSCLC cells
was assessed using Matrigel-coated or uncoated chambers (Corning
Incorporated, New York, NY, USA). At 48 h subsequent to
transfection or GANT 61 (PeproTech, Inc.) treatment,
1×105 cells were seeded into the upper Transwell
chamber, while the lower chamber was filled with RPMI-1640
supplemented with 20% FBS and 5 ng/ml TGF-β1. The chambers were
incubated at 37°C with 5% CO2 for 24 h. Cells on the
upper surface of the filter were removed using a cotton swab, while
cells that invaded/migrated to the lower surface of the filter were
fixed with 4% paraformaldehyde for 15 min at room temperature, and
stained with 0.1% crystal violet (Beyotime Institute of
Biotechnology) for 30 min. Crystal violet-stained cells were
observed and images were captured under the phase contrast
microscope, and counted in five randomly selected fields
(magnification, ×100). The experiments were performed in triplicate
and each experiment was performed three times.
Wound healing cell migration assay
NSCLC cells were seeded into six-well plates and
allowed to form confluent monolayers. The cell monolayers were
scratched using a 200 μl pipette tip to create a wound, washed
three times with PBS and incubated in RPMI-1640 supplemented with
0, 1, 5 and 10 ng/ml TGF-β1. Wound width was measured using phase
contrast microscopy and images were captured immediately subsequent
to the addition of TGF-β1 (0 h) and at 48 h of incubation.
Statistical analysis
Statistical analysis was conducted using SPSS,
version 18.0 (SPSS, Inc., Chicago, IL, USA). All results are
presented as the mean ± standard deviation. Statistical analysis
was performed using analysis of variance with the Tukey-Kramer
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
TGF-β1 induces EMT in NSCLC cells
Epithelial cancer cells undergo EMT early in the
process of metastasis (15). A549
cells undergo EMT in response to TGF-β1 exposure (16,17),
hence these cells can be used as a model to study EMT in NSCLC. To
establish the role of TGF-β1 in EMT, A549 cells were treated with
0, 1, 5 or 10 ng/ml TGF-β1 for 48 h and the morphological
alterations occurring were assessed by phase contrast microscopy.
The untreated A549 cells were observed to exhibit classic
epithelial morphology, whereas subsequent to 48-h TGF-β1 treatment,
cell morphology was clearly altered. The cells acquired a
mesenchymal phenotype, becoming elongated, spindle-shaped cells
with reduced cell-cell contacts (Fig.
1A). The extent of these morphological alterations was
dose-dependent for TGF-β1, suggesting that the effects observed
were a result of TGF-β1 treatment.
The phenotypic alterations occuring suggested that
A549 cells had undergone EMT, thus the expression of specific EMT
markers, including the epithelial marker E-cadherin and the
mesenchymal marker vimentin were measured (7,11).
Following the treatment of A549 cells with a range of
concentrations of TGF-β1 for 48 h, E-cadherin and vimentin
expression was assessed by western blot analysis. TGF-β1 treatment
led to reduced expression of the E-cadherin and increased
expression of the vimentin compared with the control, in a
concentration-dependent manner (Fig.
1B). Similar results were obtained in the other NSCLC cell
lines, with 5 ng/ml TGF-β1-treatment of H460 and SK-MES-1 cells for
48 h resulting in increased expression levels of vimentin but a
reduction in E-cadherin expression, compared with untreated cells
(Fig. 1C and D). These data
suggest that TGF-β1 induces EMT in NSCLC cells.
The development of metastatic disease is the most
frequent cause of cancer-associated mortality (18). EMT is often associated with
metastasis, as the invasive and migratory capacity of cells
increases following the induction of EMT. To analyze the migratory
capacity of A549 cells following TGF-β1 treatment, wound healing
assays were conducted. In this assay, it was observed that TGF-β1
treatment led to increased migration after 48 h compared with
untreated cells (Fig. 1E). To
assess the effect of TGF-β1 on the invasive capacity of NSCLC
cells, Transwell invasion assays were conducted using
TGF-β1-treated H460 and SK-MES-1 cells, which exhibited increased
invasion compared with the control (Fig. 1F and G). These results indicate
that TGF-β1 induces enhanced migration and invasion of NSCLC cells,
which is consistent with the observed phenotypic alterations. These
data support an involvement of TGF-β1 in the induction of EMT in
NSCLC cells.
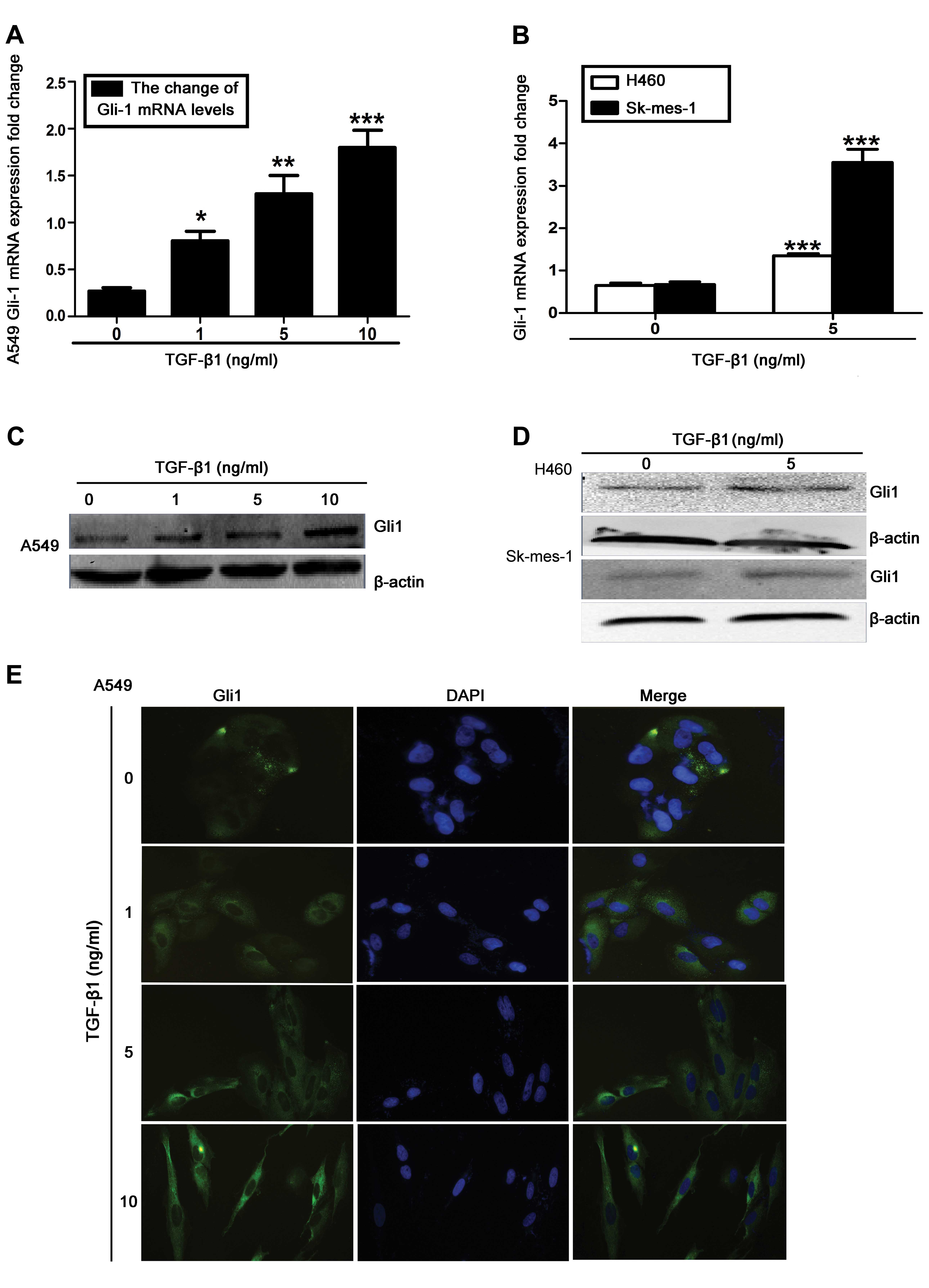
Upregulation of Gli1 expression levels in
TGF-β1-stimulated NSCLC cells
Gli1 is commonly observed to be overexpressed in
NSCLC, and has been implicated in the induction of EMT and
metastasis (13,19–21).
Therefore, the role of Gli1 was investigated in TGF-β1-induced EMT.
Using RT-qPCR and western blot analysis, it was observed that
TGF-β1 stimulation significantly increased Gli1 mRNA and protein
levels in A549, H460 and SK-MES-1 cells, compared with untreated
cells (Fig. 2A–D). A similar
induction of Gli1 was observed in the immunofluorescence analysis
of TGF-β1-treated A549 cells (Fig.
2E). These results indicate that Gli1 upregulation occurs
during TGF-β1-induced EMT in NSCLC cells.
Effect of Gli1-siRNA and GANT 61
treatment on Gli1 induction by TGF-β1 in NSCLC cells
To determine the function of Gli1 in TGF-β1-induced
EMT, siRNA was used to deplete Gli1 mRNA and protein levels in A549
cells. The cells were transfected with DEPC-treated water
(control), Gli1-specific siRNA (si-Gli) or nonspecific control
siRNA (si-VE) for 24 h. Fluorescence microscopy analysis indicated
that the siRNAs were efficiently transfected, as demonstrated by
the presence of GFP expression (Fig.
3A). Subsequent to a 48-h resting period, Gli1 mRNA expression
levels were significantly suppressed by Gli1-specific siRNA
compared with control, as measured by RT-qPCR (P<0.001; Fig. 3B). This siRNA-mediated depletion of
Gli1 was further confirmed by western blotting (Fig. 3C) and immunofluorescence analysis
(Fig. 3D). As an alternative to
siRNA-mediated depletion of Gli1, the effects of pharmacological
inhibition of Gli1 were investigated using the Gli1-specific
inhibitor GANT 61 in TGF-β1-treated H460 and SK-MES-1 cells. Cells
were treated with 10 μM GANT 61 for 48 h prior to stimulation with
5 ng/ml TGF-β1 for an additional 48 h. This was demonstrated by
RT-qPCR and western blot analysis to downregulate Gli1 mRNA and
protein levels in the H460 (P<0.001) and SK-MES-1 (P<0.05)
cell lines, compared with controls (Fig. 3E and F).
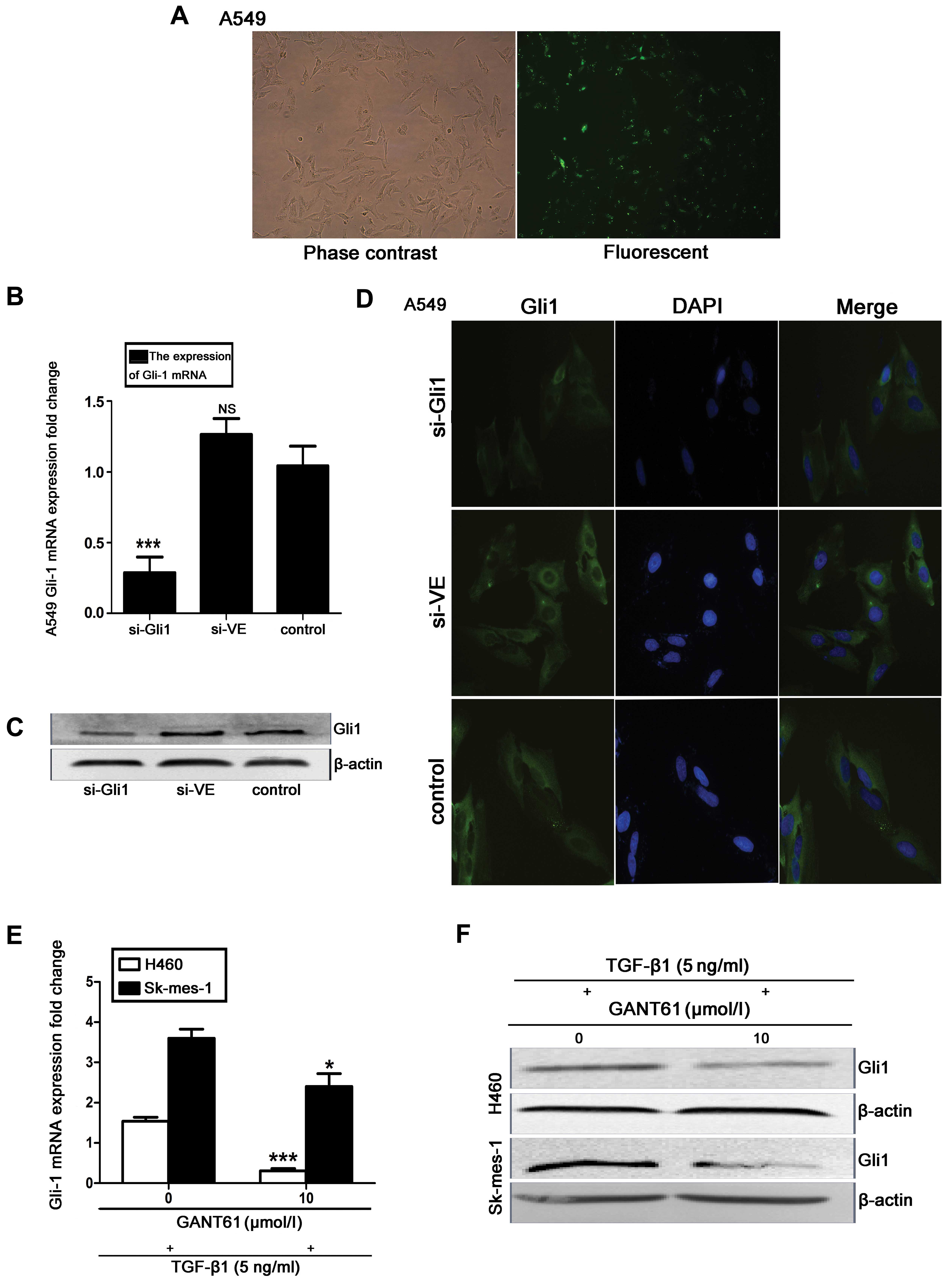 | Figure 3Effect of Gli1-siRNA and GANT 61 on
Gli1 expression in TGF-β1-induced NSCLC cells. (A) Confocal laser
scanning microscope images to assess transfection efficiency. (B)
Gli1 mRNA expression levels were assessed by RT-qPCR. (C) Western
blot analysis of Gli1 protein expression in the si-Gli1, si-VE and
control groups, with β-actin as the loading control. (D)
Immunofluorescence staining (magnification, ×400). All experiments
were performed 3 times. (E) RT-qPCR assessment and (F) western blot
analysis of H460 and SK-MES-1 cells treated with the Gli1 inhibitor
GANT 61 (10 μM) for 48 h, followed by 48 h stimulation with TGF-β1.
All values are presented as the mean, error bars represent the
standard deviation; NS, not significant, *P<0.05,
**P<0.005 and ***P<0.001 vs. control.
Gli1, glioma-associated oncogene homolog 1; si-Gli1, Gli1 siRNA
group; si-VE, nonspecific siRNA group; TGF-β1, transforming growth
factor-β1; NSCLC, non-small cell lung cancer; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
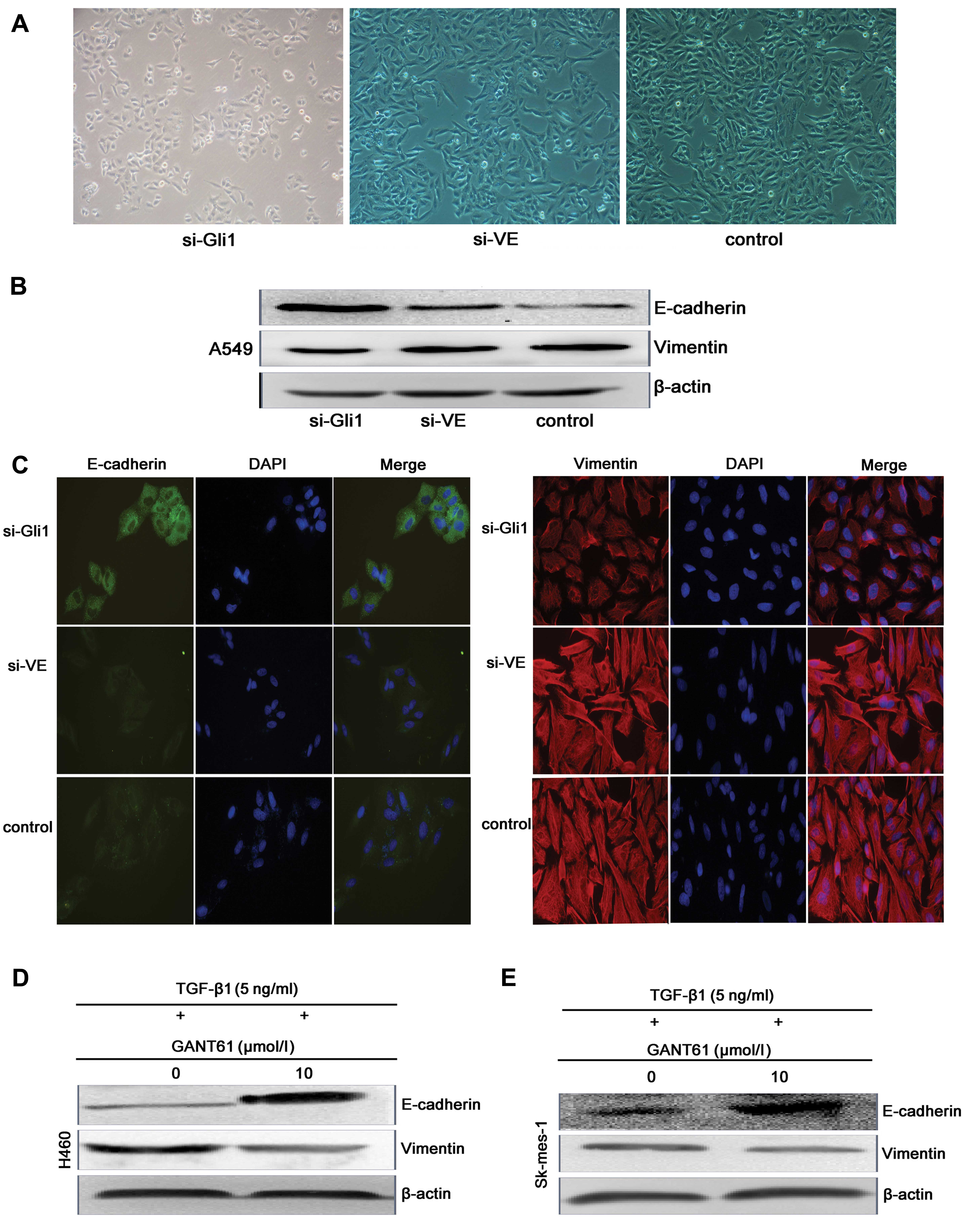
Effects of Gli1 inhibition on
TGF-β1-induced EMT of NSCLC cells
To determine if Gli1 participated in the induction
of EMT in NSCLC cells, phase contrast microscopy was used to assess
the morphology of A549 cells 24 h subsequent to si-Gli1, si-VE or
blank transfection, and 18 h subsequent to TGF-β1 stimulation
(Fig. 4A). The expression of EMT
markers in TGF-β1 following siRNA-mediated depletion of Gli1 was
also assessed. Western blot and immunofluorescence analysis
demonstrated that, compared with cells transfected with control
siRNA, E-cadherin expression was elevated in cells transfected with
Gli1 siRNA, while vimentin protein levels were reduced (Fig. 4B and C). The effects of
pharmacological inhibition of Gli1 on TGF-β1-induced EMT in H460
and SK-MES-1 cells were also investigated. An elevation of
E-cadherin protein levels and a reduction of vimentin expression
was observed following 48-h GANT 61 treatment in H460 and SK-MES-1
cells, compared with untreated cells (Fig. 4D and E). These results suggest that
the downregulation of Gli1 inhibits TGF-β1-induced EMT of NSCLC
cells.
Downregulation of Gli1 reduces the
invasive and migratory capacity of the TGF-β1-stimulated NSCLC
cells
Given the effects of Gli1 inhibition on the
expression of EMT markers, the effect of Gli1 siRNA on the invasive
and migratory abilities of TGF-β1-induced A549 cells was
investigated. Compared with cells transfected with non-specific
control siRNA and untransfected cells, cells with Gli1 siRNA
exhibited significantly reduced invasion and migration in a
Transwell assay (P<0.001; Fig.
5A–C), suggesting that Gli1 is required for migration in
response to TGF-β1. The effects of Gli1 inhibition on the invasive
capacity of H460 and SK-MES-1 cells were also investigated.
Suppression of Gli1 using GANT 61 significantly inhibited the
ability of H460 and SK-MES-1 cells to invade through Matrigel
compared with untreated cells (P<0.05; Fig. 5D and E). These data suggest that
Gli1 contributes to the enhanced migration and invasion of
TGF-β1-stimulated NSCLC cells.
 | Figure 5Inhibition of Gli1 reduced the
invasive and migratory capacity of TGF-β1-induced NSCLC cells. (A)
Cells were seeded 48 h subsequent to si-Gli1, si-VE, or blank
transfection into the upper transwell chambers, which were either
pre-coated with 20 mg Matrigel (invasion assay), or uncoated
(migration assay). Following a 24-h resting period, cells which had
migrated (upper panels) or invaded (lower panels) were stained with
crystal violet and imaged with a phase contrast microscope
(magnification, ×100). Five fields were randomly chosen from each
filter, and values representing the mean numbers of cells which had
(B) invaded or (C) migrated were obtained. Each experiment was
performed in triplicate. (D and E) Transwell Matrigel invasion
assay results from TGF-β1-stimulated H460 and SK-MES-1 cells
treated with 10 μM GANT 61. All values represent the mean number of
cells, error bars represent the standard deviation. NS, not
significant; *P<0.05, **P<0.005 and
***P<0.001 vs. control. Gli1, glioma-associated
oncogene homolog 1; TGF-β1, transforming growth factor-β1; NSCLC,
non-small cell lung cancer; si-Gli1, Gli1 siRNA group; si-VE,
nonspecific siRNA group. |
Discussion
EMT is an important biological process that allows
cancer cells to develop metastatic characteristics. One of the
major alterations that occurs during EMT is the loss of the
cell-cell adhesion molecule E-cadherin and overexpression of
vimentin, N-cadherin and MMP-9 (6). In the current study, A549 NSCLC cells
were demonstrated to undergo morphological alterations associated
with EMT, following stimulation with TGF-β1, in a dose-dependent
manner. Following 48-h TGF-β1 stimulation, the A549, H460 and
SK-MES-1 cells were observed to exhibit a mesenchymal phenotype,
with elongated, spindle-shaped cells and a reduction in cell-cell
contacts. These morphological alterations were accompanied by the
downregulation of E-cadherin and upregulation of vimentin, which is
consistent with classical EMT.
In addition to these morphological alterations, EMT
is associated with increased motility and invasive ability.
Consistent with this, the current study observed that TGF-β1
treatment led to an increase in the migratory behavior of A549
cells compared with untreated cells in a wound healing assay.
Furthermore, TGF-β1 enhanced the invasive ability of H460 and
SK-MES-1 cells in a Transwell Matrigel invasion assay. Taken
together, these data indicate that TGF-β1 promotes EMT and its
associated metastatic behavior in NSCLC cells.
To investigate the molecular mechanism of
TGF-β1-induced EMT in NSCLC, the current study focused on Gli1.
Notably, TGF-β1 stimulation was observed to upregulate Gli1
expression in A549 cells in a dose-dependent manner, as assessed by
western blotting, RT-qPCR and immunofluorescence analysis,
suggesting that Gli1 may serve an important function in
TGF-β1-induced EMT. Inhibition of Gli1 using siRNA or the Gli1
inhibitor GANT 61 prevented morphological alterations in NSCLC
cells following TGF-β1 stimulation. Furthermore, inhibition of Gli1
attenuated the induction of the mesenchymal marker vimentin, and
upregulated the epithelial marker E-cadherin following TGF-β1
treatment. In addition, inhibition of Gli1 reduced the migratory
and invasive capacity of NSCLC cells. These results suggest that
Gli1 is important in the induction of EMT by TGF-β1 and that
inhibition of Gli1 may reverse the EMT phenotype in NSCLC cells,
thus potentially reducing metastasis.
TGF-β1 is a multifunctional cytokine that is closely
associated with cell growth, fibrosis and tumorigenesis. It is also
able to promote EMT and tumor cell metastasis (11,22),
and this has been demonstrated in several cancer cell lines
(23–26). The Gli1 protein is a zinc finger
transcription factor that is activated by the Hh signaling pathway,
which involves Hh proteins, the Patched protein, the Smoothened
(Smo) protein and the five-zinc finger Gli transcription factor
family (Gli1, Gli2 and Gli3) (27,28).
Gli1 and Gli2 act as the main activators of Hh-target genes, while
Gli3 acts as a repressor of Hh target genes (29,30).
The Hh signaling pathway is implicated in developmental processes
and tumor malignancies (31,32,34),
and the Gli family of transcription factors mediate a number of
important cellular processes, including EMT, migration, metastasis
and tumorigenesis (21). In NSCLC
specifically, Gli1 has been demonstrated to be elevated in tumor
tissue samples and NSCLC cells lines (13,33).
In addition, upregulation of Hh signaling has been indicated to
contribute to TGF-β1-induced EMT in NSCLC cells (19). These data are consistent with the
observations of the current study, demonstrating that Gli1 levels
are elevated in TGF-β1-stimulated NSCLC cells that have undergone
EMT, thus it is suggested that Gli1 is important in EMT downstream
of TGF-β1.
The current study suggests that Hh signaling, and
Gli1 in particular, may provide a novel therapeutic target in
NSCLC. As the Gli1 transcription factor mediates the terminal
effects of the Hh pathway, the upstream positive regulator of Gli1
(Smo) has been investigated as a therapeutic target, with several
Smo-targeted small molecule inhibitors currently undergoing
clinical trials. One Smo-targeted small molecule inhibitor,
vismodegib (GDC-0449), has been demonstrated in phase I clinical
trials to be suitable for the treatment of multiple types of cancer
(35). In addition to Smo, other
signaling pathways, such as oncogenic epidermal growth factor
receptor-RAS-protein kinase B (AKT) signaling have been
demonstrated to activate Gli1 (36). In particular, the PI3K/AKT and
mitogen-activated protein kinase/extracellular-signal-regulated
kinase (ERK)1/2 signaling pathways also regulate TGF-β1-induced EMT
in A549 cells (37). However, the
precise role Gli1 in PI3K and ERK signaling remains to be fully
elucidated.
In conclusion, the results of the current study
indicate that the loss of Gli1 in NSCLC cells dramatically
attenuates TGF-β1-induced EMT. Gli1 appears to be required for the
phenotypic alterations, in addition to the enhancement of motility
and invasion associated with TGF-β1-induced EMT. The results of the
present study suggest that inhibition of Gli1 may serve as a useful
strategy to target metastatic disease in patients with NSCLC.
Additionally, as EMT affects the sensitivity of NSCLC cell lines to
common therapeutic agents including erlotinib, cisplatin and
paclitaxel (38–40), targeting Gli1 may improve the
efficacy of these therapies. EMT also affects the response to
radiotherapy, including promoting radiation-induced fibrosis and
post-radiotherapy-associated metastasis (40), which is suggested to occur through
the loss of E-cadherin (41,42).
Therefore, reversal of EMT via Gli1 inhibition may potentially
resensitize NSCLC cells to chemotherapy and radiation, which would
contribute to improved prognosis for patients.
Acknowledgements
The authors thank Ms. Zhu Yang and Mr. Tao Yu for
their academic assistance. The current study was supported by the
College of Life Science and Biological Engineering of Chongqing
Medical University (Chongqing, China).
References
|
1
|
Cagle PT, Allen TC, Dacic S, et al:
Revolution in lung cancer: new challenges for the surgical
pathologist. Arch Pathol Lab Med. 135:110–116. 2011.PubMed/NCBI
|
|
2
|
Juergens R and Brahmer J: Targeting the
epidermal growth factor receptor in non-small-cell lung cancer:
who, which, when, and how? Curr Oncol Rep. 9:255–264. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alberg AJ, Ford JG and Samet JM; American
College of Chest Physicians. Epidemiology of lung cancer: ACCP
evidence-based clinical practice guidelines (2nd edition). Chest.
132(Suppl): 29S–55S. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gavert N and Ben-Ze’ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prudkin L, Liu DD, Ozburn NC, et al:
Epithelial-to-mesenchymal transition in the development and
progression of adenocarcinoma and squamous cell carcinoma of the
lung. Mod Pathol. 22:668–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li LP, Lu CH, Chen ZP, et al: Subcellular
proteomics revealed the epithelial-mesenchymal transition phenotype
in lung cancer. Proteomics. 11:429–439. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang G, Dong W, Shen H, et al: A
comparison of Twist and E-cadherin protein expression in primary
non-small-cell lung carcinoma and corresponding metastases. Eur J
Cardiothorac Surg. 39:1028–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pirozzi G, Tirino V, Camerlingo R, et al:
Epithelial to mesenchymal transition by TGFβ-1 induction increases
stemness characteristics in primary non small cell lung cancer cell
line. PLoS One. 6:e215482011. View Article : Google Scholar
|
|
10
|
Soltermann A, Tischler V, Arbogast S, et
al: Prognostic significance of epithelial-mesenchymal and
mesenchymal-epithelial transition protein expression in non-small
cell lung cancer. Clin Cancer Res. 14:7430–7437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang HJ, Wang HY, Zhang HT, et al:
Transforming growth factor-β1 promotes lung adenocarcinoma invasion
and metastasis by epithelial-to-mesenchymal transition. Mol Cell
Biochem. 355:309–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gialmanidis IP, Bravou V, Amanetopoulou
SG, Varakis J, Kourea H and Papadaki H: Overexpression of hedgehog
pathway molecules and FOXM1 in non-small cell lung carcinomas. Lung
Cancer. 66:64–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stecca B and Ruiz I Altaba A:
Context-dependent regulation of the GLI code in cancer by HEDGEHOG
and non-HEDGEHOG signals. J Mol Cell Biol. 2:84–95. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aokage K, Ishii G, Ohtaki Y, et al:
Dynamic molecular changes associated with epithelial-mesenchymal
transition and subsequent mesenchymal-epithelial transition in the
early phase of metastatic tumor formation. Int J Cancer.
128:1585–1595. 2011. View Article : Google Scholar
|
|
16
|
Kasai H, Allen JT, Mason RM, Kamimura T
and Zhang Z: TGF-beta1 induces human alveolar epithelial to
mesenchymal cell transition (EMT). Respir Res. 6:562005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JH, Jang YS, Eom KS, et al:
Transforming growth factor beta1 induces epithelial-to-mesenchymal
transition of A549 cells. J Korean Med Sci. 22:898–904. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sporn MB: The war on cancer. Lancet.
347:1377–1381. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maitah MY, Ali S, Ahmad A, Gadgeel S and
Sarkar FH: Up-regulation of sonic hedgehog contributes to
TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells.
PLoS One. 6:e160682011. View Article : Google Scholar
|
|
20
|
Zhu H and Lo HW: The human
glioma-associated oncogene homolog 1 (GLI1) family of transcription
factors in gene regulation and diseases. Curr Genomics. 11:238–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng X, Vittar NB, Gai X, et al: The
transcription factor GLI1 mediates TGFβ1 driven EMT in
hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One.
7:e495812012. View Article : Google Scholar
|
|
22
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ko H, So Y, Jeon H, Jeong MH, et al:
TGF-β1-induced epithelial-mesenchymal transition and acetylation of
Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung
cancer cells. Cancer Lett. 10:205–213. 2013. View Article : Google Scholar
|
|
24
|
Gunaratne A, Thai BL and Di Guglielmo GM:
Atypical protein kinase C phosphorylates Par6 and facilitates
transforming growth factor β-induced epithelial-to-mesenchymal
transition. Mol Cell Biol. 33:874–886. 2013. View Article : Google Scholar :
|
|
25
|
Liu LC, Tsao TC, Hsu SR, Wang HC, Tsai TC,
Kao JY and Way TD: EGCG inhibits transforming growth
factor-β-mediated epithelial-to-mesenchymal transition via the
inhibition of Smad2 and Erk1/2 signaling pathways in nonsmall cell
lung cancer cells. J Agric Food Chem. 60:9863–9873. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao M, Seike M, Soeno C, et al: MiR-23a
regulates TGF-β-induced epithelial-mesenchymal transition by
targeting E-cadherin in lung cancer cells. Int J Oncol. 41:869–875.
2012.PubMed/NCBI
|
|
27
|
Pasca di Magliano M and Hebrok M: Hedgehog
signalling in cancer formation and maintenance. Nat Rev Cancer.
3:903–911. 2003. View
Article : Google Scholar
|
|
28
|
Ruel L, Rodriguez R, Gallet A,
Lavenant-Staccini L and Thérond PP: Stability and association of
Smoothened, Costal2 and Fused with Cubitus interruptus are
regulated by Hedgehog. Nat Cell Biol. 5:907–913. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang B, Fallon JF and Beachy PA:
Hedgehog-regulated processing of Gli3 produces an
anterior/posterior repressor gradient in the developing vertebrate
limb. Cell. 100:423–434. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bai CB, Stephen D and Joyner AL: All mouse
ventral spinal cord patterning by hedgehog is Gli dependent and
involves an activator function of Gli3. Dev Cell. 6:103–115. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teglund S and Toftgård R: Hedgehog beyond
medulloblastoma and basal cell carcinoma. Biochim Biophys Acta.
1805.181–208. 2010.
|
|
32
|
Katoh Y and Katoh M: Hedgehog target
genes: mechanisms of carcinogenesis induced by aberrant hedgehog
signaling activation. Curr Mol Med. 9:873–886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yuan Z, Goetz JA, Singh S, et al: Frequent
requirement of hedgehog signaling in non-small cell lung carcinoma.
Oncogene. 26:1046–1055. 2007. View Article : Google Scholar
|
|
34
|
Yoo YA, Kang MH, Lee HJ, et al: Sonic
hedgehog pathway promotes metastasis and lymphangiogenesis via
activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer
Res. 71:7061–7070. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
LoRusso PM, Rudin CM, Reddy JC, et al:
Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449)
in patients with refractory, locally advanced or metastatic solid
tumors. Clin Cancer Res. 17:2502–2511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruiz I Altaba A, Mas C and Stecca B: The
Gli code: an information nexus regulating cell fate, stemness and
cancer. Trends Cell Biol. 17:438–447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen XF, Zhang HJ, Wang HB, et al:
Transforming growth factor-β1 induces epithelial-to mesenchymal
transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2
signaling pathways. Mol Biol Rep. 39:3549–3556. 2012. View Article : Google Scholar
|
|
38
|
Yauch RL, Januario T, Eberhard DA, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shintani Y, Okimura A, Sato K, et al:
Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jung JW, Hwang SY, Hwang JS, Oh ES, Park S
and Han IO: Ionising radiation induces changes associated with
epithelial-mesenchymal transdifferentiation and increased cell
motility of A549 lung epithelial cells. Eur J Cancer. 43:1214–1224.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou YC, Liu JY, Li J, et al: Ionizing
radiation promotes migration and invasion of cancer cells through
transforming growth factor-beta-mediated epithelial-mesenchymal
transition. Int J Radiat Oncol Biol Phys. 81:1530–1537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Theys J, Jutten B, Habets R, et al:
E-Cadherin loss associated with EMT promotes radioresistance in
human tumor cells. Radiother Oncol. 99:392–397. 2011. View Article : Google Scholar : PubMed/NCBI
|