Introduction
Protein tyrosine kinases (PTK) and protein tyrosine
phosphatases (PTP) regulate tyrosine phosphorylation, which is an
important cellular mechanism that regulates intracellular signaling
pathways to control a number of cellular activities, including
proliferation, growth and differentiation (1). Focal adhesion kinase (FAK), a
non-receptor binding to tyrosine kinase, functions as a scaffold
and coordinating center, which may regulate focal adhesion
formation and disassembly. This has been demonstrated in a previous
study, which investigated actin fiber stress and cytoskeletal
organization (2). The most
important site for the regulation of FAK phosphorylation is Tyr397.
This mediates one key intramolecular regulatory interaction between
the protein 4.1, ezrin, radixin, moesin (FERM) and kinase domains
of FAK (3,4). In the integrin signaling pathway,
FAK-Tyr397, accompanied by auto-phosphorylation reconstructs the
Src family kinases (SFK)/FAK composite, which leads to further
phosphorylation and maximal activation of FAK (2).
The important role of SFK in activating FAK has been
demonstrated by the decreasing levels of FAK auto-phosphorylation
following knock out of the Src, Fyn and Yes gene of SFKs in
fibroblasts or by shielding the PTP gene in response to integrin
engagement (5,6). In terms of the molecular mechanisms,
evidence suggests that the FERM domain is responsible for FAK
auto-inhibition by interacting with the kinase domain. The FERM
domain was observed to switch from binding to the kinase domain to
binding with heterologous protein or lipid partners, which led to
FAK-Tyr397 auto-phosphorylation (3,6,7).
PTPα may also regulate the levels of FAK
phosphorylation and a previous study suggested that PTPα and other
PTP family members may catalyze the dephosphorylation of Src-Tyr527
by its own activity (8–10). During the mitotic process,
phosphorylated Src-Tyr527 is able to activate the expression of
Src, the regulation of which is mediated by PTPα (11). However, it has also been suggested
that PTPα-mediated Src activation during mitosis was dependent on
PP2A-mediated dephosphorylation of Ser204 in the juxtamembrane
region of PTPα rather than Tyr789 (12). Therefore, the role of Src-Try789
phosphorylated by PTPα requires further investigation.
In our previous study, a novel splicing mutant,
FAK-Del33, was identified in breast and thyroid cancer by
sequencing (13). The mutation
frequency of The FAK-De133 gene was found to be 14.2% (3/21) in
breast carcinoma; in addition, when this gene was introduced into
normal breast cell lines, it was reported to promote cell migration
and invasion (13). This may be
caused by FAK-Tyr397 hyperphosphorylation and enhancement of
FAK-Src in downstream signaling pathways.
The present study investigated changes in the levels
of amino acid phosphorylation in the signaling pathway of breast
cancer cells and the activity of Src by western blot analysis. A
novel pathway, in which the FAK-Del33 protein may activate c-Src
through PTPα phosphorylation at the Tyr789 site, was identified.
This was the first study, to the best of our knowledge, to
demonstrate the effect of PTPα phosphorylation in a FAK mutant.
Materials and methods
Cell lines
The MDA-MB-468 and MDA-MB-435s cell lines were
isolated from a breast cancer patient and melanoma derived cells
(MDA-MB-435s) were obtained from American Type Culture Collection
(Manassas, VA, USA) and cultured according to the manufacturer’s
instructions. The HEK293T cell line was also obtained from American
Type Culture Collection. The FAK-Del33 mutant cells were obtained
from breast cancer mutation cells lacking exon 33 for FAK and were
stored at the Department of Clinical Laboratory of Shanghai Ruijin
Hospital (Shanghai, China).
Construction and identification of the
expression system
The FAK-Del33 mutant cell suspension was harvested
after 48 h in culture and the nucleic acids were then extracted
using phenol-chloroform extraction technology. In brief, 500
μl phenol:chloroform:isoamyl alcohol (25:24:1) was added to
the cell suspension and it was vortexed for ~20 sec, then
centrifuged at 16,000 x g for 5 min at room temperature. The
supernatant was transferred to a fresh tube and ethanol
precipitation was performed by adding cold 95% ethanol, incubating
for 1 h at −20°C, centrifuging at 16,000 × g for 15 min, decanting
the supernatant, adding 80% ethanol and repeating the
centrifugation. Polymerase chain reaction (PCR) was performed to
amplify the FAK-Del33 gene, using the following primers: forward
5′-TCA GAA ACA GAT GAT TTT GCT GAG AT T ATA GAT GA-3′ and reverse
5′-AGC AAA ATC ATC TGT TTC TGA CAC AGA GA-3′. Furthermore,
BamHI and NotI restriction sites were introduced at
the 5′ and 3′ ends, respectively. The purified PCR fragment was
inserted into multiple cloning sites in pCDNA3.1 (+). Construction
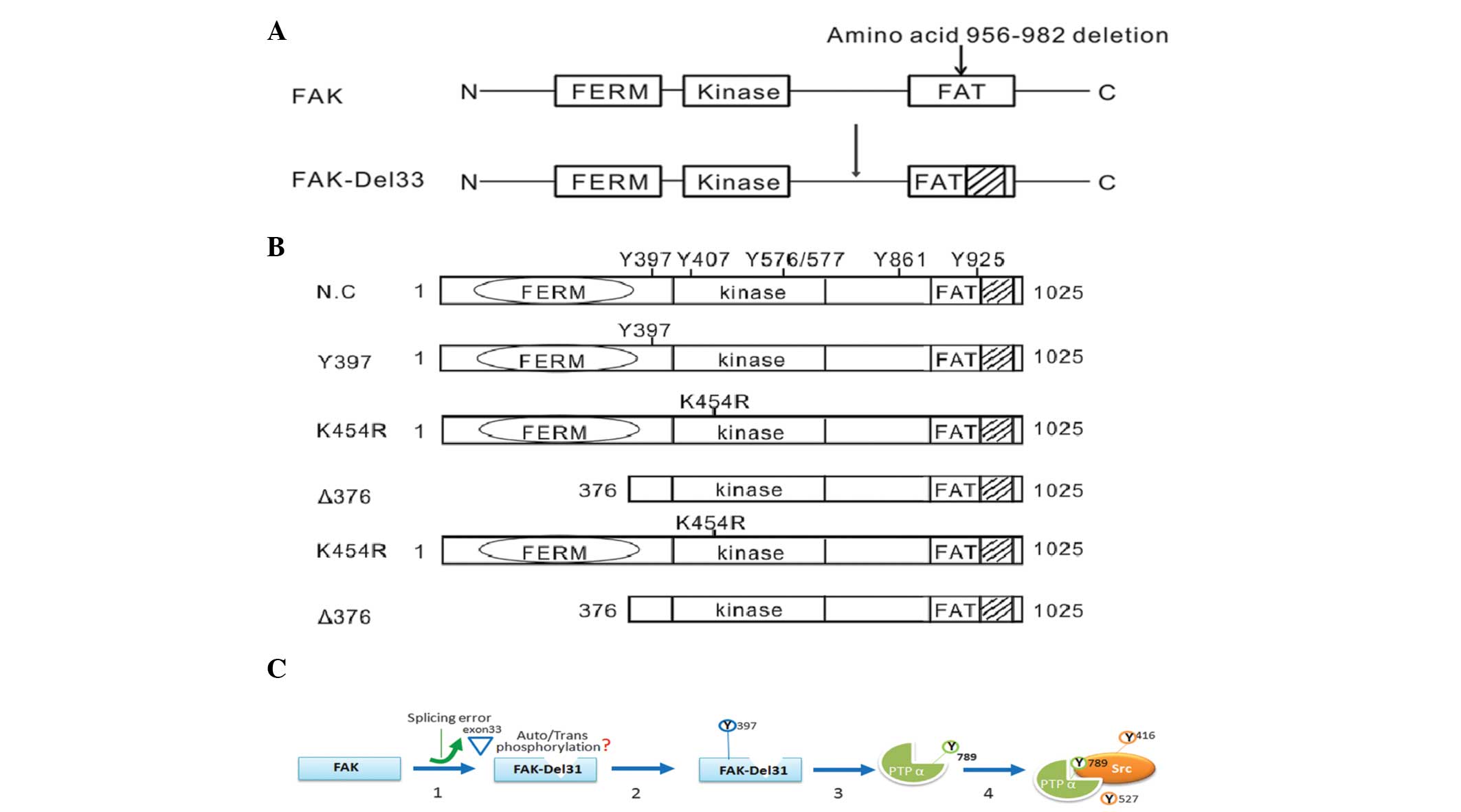
of the expression plasmids used in the present study are shown in
Fig. 1A.
Retroviral expression system
The pGIPZ lentiviral vector-based expression system
(Open Biosystems, Inc., Victoria, Australia) was used to clone and
express the FAT-wild-type (Wt) and FAK-del33 (exon 33 of FAK
deletion) genes in the cells, according to the manufacturers
instructions, which packaged the lentivirus and infected the
HEK293T cells to culture and amplify the virus.
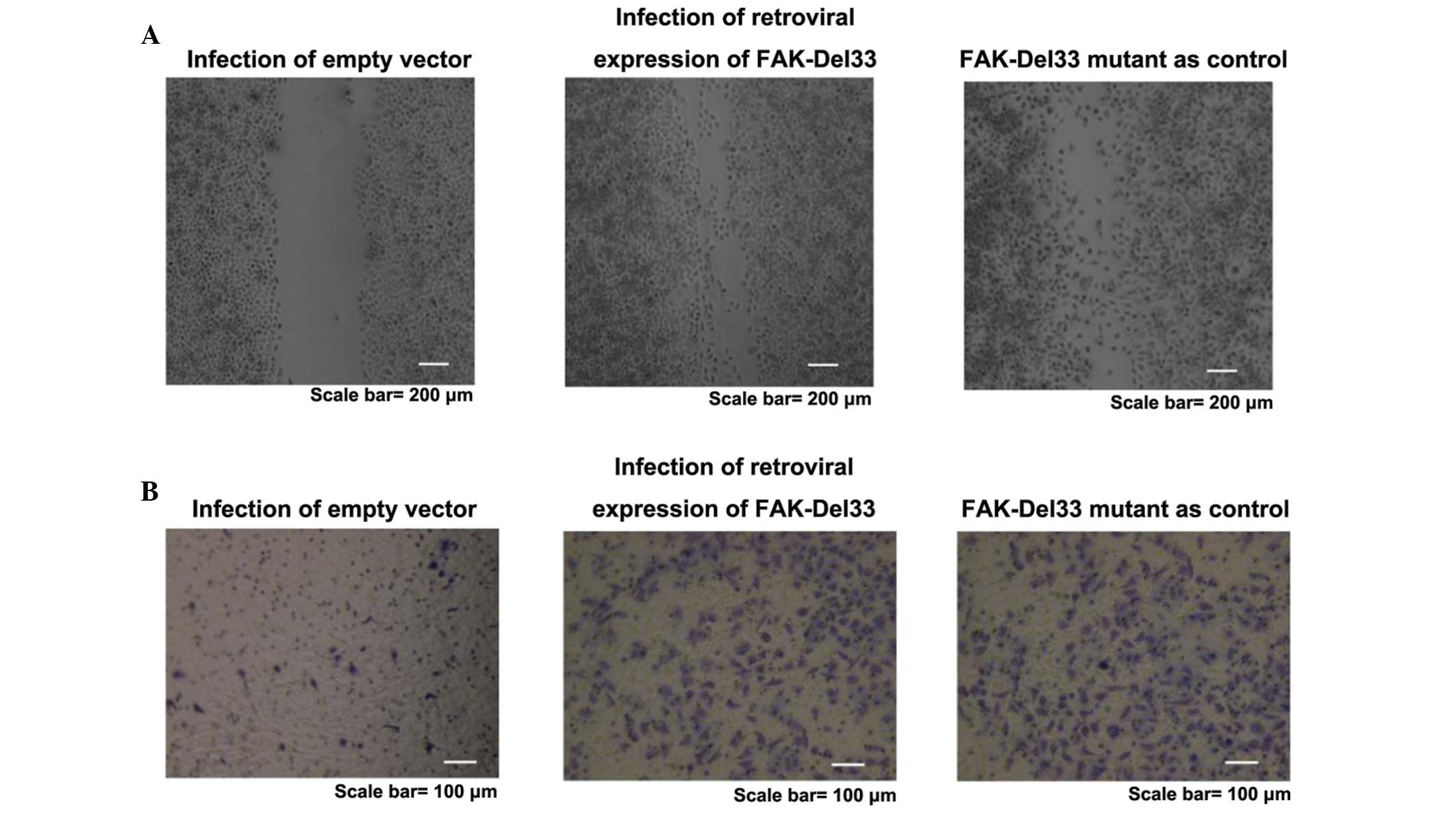
Wound-healing assay
A wound-healing assay was performed to detect
changes in the motility and migration of the MDA-MB-468 and
MDA-MB-435s cell lines. The FAK-Del33 mutant was used as a positive
control. Cells, which were infected with the recombinant virus and
empty vector were used as a negative control.
The cells were seeded (3×105) into 60
mm2 plates and cultured for 12 h at 37°C. A vertical
scratch was made using a p20 pipette tip in the middle of the plate
to remove cells from this area. Following this, the cells were
washed three times using phosphate-buffered saline (PBS) and
cultured in serum-free medium. Images of the wound-healing distance
were captured for each cell line at 0, 6, 12 and 24 h using an
inverted microscope (IX73; Olympus Corp., Tokyo, Japan).
Migration assay
The cells (3×105) were cultured in 60
mm2 plates containing Transwell filters with an 8
μm pore size (Corning, Inc., New York, NY, USA) coated with
10 μg/ml fibronectin for 12 h. The culture medium
(Dulbecco’s modified Eagle’s medium) was designed with a serum-free
upper layer containing 0.1% bovine serum albumin (BSA) and a lower
layer containing 20% fetal bovine serum (Gibco-BRL, Gaithersburg,
MD, USA) in different chambers. After 24 h, the bottom of the
filter was stained with Giemsa (Sangon Biotech Co., Ltd, Shanghai,
China) to calculate the efficiency of the cell migration.
Sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and western blot analysis
The FAK-Del33 mutant and FAK-Wt cell suspensions
were harvested and centrifuged at 1,000 × g for 10 min (Thermo
Fisher Scientific, Waltham, MA, USA). The supernatant was discarded
and the pellet was resuspended in lysis buffer [20 mM Tris (pH
7.5), 150 mM NaCl, 1% Triton X-100; Beyotime Institiute of
Technology, Jiangsu, China], containing 1% protease inhibitor
cocktail (Sigma-Aldrich, St Louis, MO, USA), 25 mM NaF
(Sigma-Aldrich) and 1 mM Na3VO4
(Sigma-Aldrich).
The mixture was freeze-thawed four times at −80°C
prior to centrifuging at 10,000 × g for 30 min at 4°C (Thermo
Fisher Scientific). The supernatant containing FAK-Del33 protein
was collected and analyzed using 8% SDS-PAGE. The FAK-Wt breast
cancer cells were used for comparison. Following SDS-PAGE, the
proteins were transferred onto a polyvinylidene difluoride membrane
(Millipore, Billerica, MA, USA).
The membrane was blocked using 5% BSA in Tris-HCl
buffered saline containing 0.05% Tween-20 for 2 h at 25°C. The
membrane was then incubated with the following primary monoclonal
antibodies at 4°C overnight (1:1,000): Mouse anti-PTPα, rabbit
anti-phospho-PTPα (Tyr789), rabbit anti-Src, rabbit
anti-phospho-Src (Tyr527); rabbit anti-non-phospho-Src (Tyr527);
rabbit anti-phospho-Src (tyr416) and rabbit anti-PTP1B, followed by
incubation at room temperature for 2 h with anti-rabbit or
anti-mouse immunoglobulin G secondary antibodies. All antibodies
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). GADPH levels were used as an internal standard. The protein
bands were detected using enhanced chemiluminescence (Bestbio,
Shanghai, China). The Wt breast cancer cells were used as a
negative control.
siRNA-induced apoptosis
The following siRNA sequences were used to target:
PTPα forward 5′-GCU GGG AGC CAU UCC AAU UdTdT-3′ and reverse
primers PTPα 5′GCU AGG AGC UAU UCC GAU U dTdT-3′, PTP1B 5′-AAU ACA
GUG CGA CAG CUA GAA dTdT-3′ and control siRNA 5′-AAU UAA CCU UAC
CGA GGG UCG dTdT-3′ (14). All the
siRNA duplexes were synthesized by GenePharma, Inc. (Shanghai,
China) using 2′-ACE protection chemistry. The Stemfect™ RNA
Transfection kit (Stemgent, Cambridge, MA, USA) was used to
transfect the siRNA into the cells. Fig. 1B shows the amino acid positions in
the FAK protein.
Statistical analysis
Statistical analysis was performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Differences between the
two groups were evaluated using a one-way analysis of variance. A
χ2 test was used to calculate the significance of
detection rates between the two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Wound-healing and migration assay
The healing rate of the FAK-Del33 mutant was faster
than that of MDA-MB-468 and MDA-MB-435s cells. Following infection
with a lentivirus contaning the FAK-Del33 gene, the healing
efficiency of the MDA-MB-468 and MDA-MB-435s cells was higher
compared with those observed in the cells containing the empty
vector. The FAK-Del33 protein may regulate cell growth. The results
obtained from the migration assays supported the hypothesis that
this protein also regulates cell motility and migration (Fig. 2).
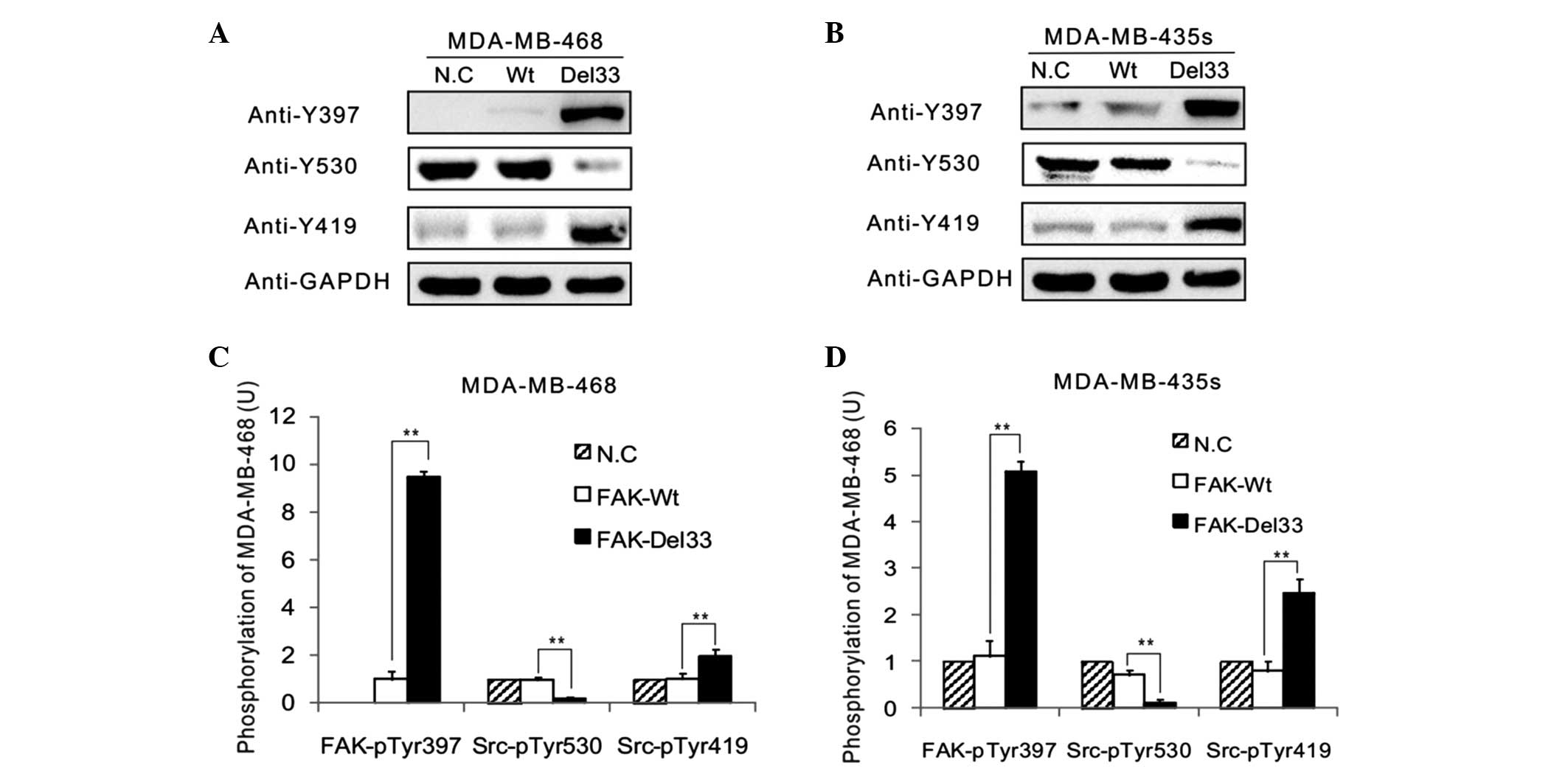
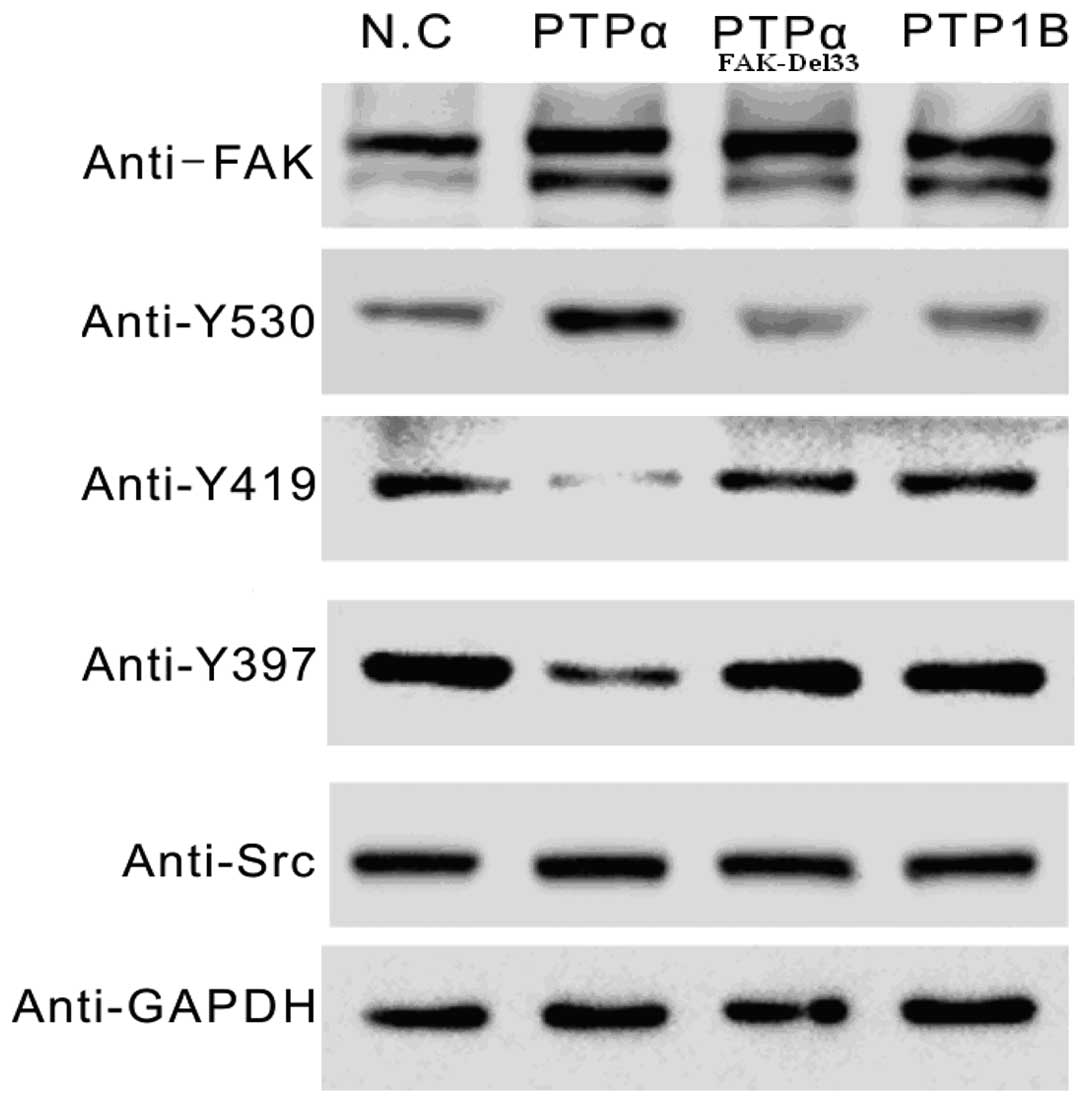
Western blot analysis
The main autophosphorylation of FAK, Src-Tyr397, was
overexpressed in the FAK-Del33 mutant compared with the FAK-Wt
cells. The results revealed ~60–80% of the Src-Tyr530 was
dephosphorylated in the MDA-MB-468 and MDA-MB-435s cells compared
with the FAK-Del mutant cells. The phosphorylation levels of
Src-Tyr419 were 1.90-3.07-fold higher in the FAK-Del33 mutant
compared with the FAK-Wt cells (Fig.
3).
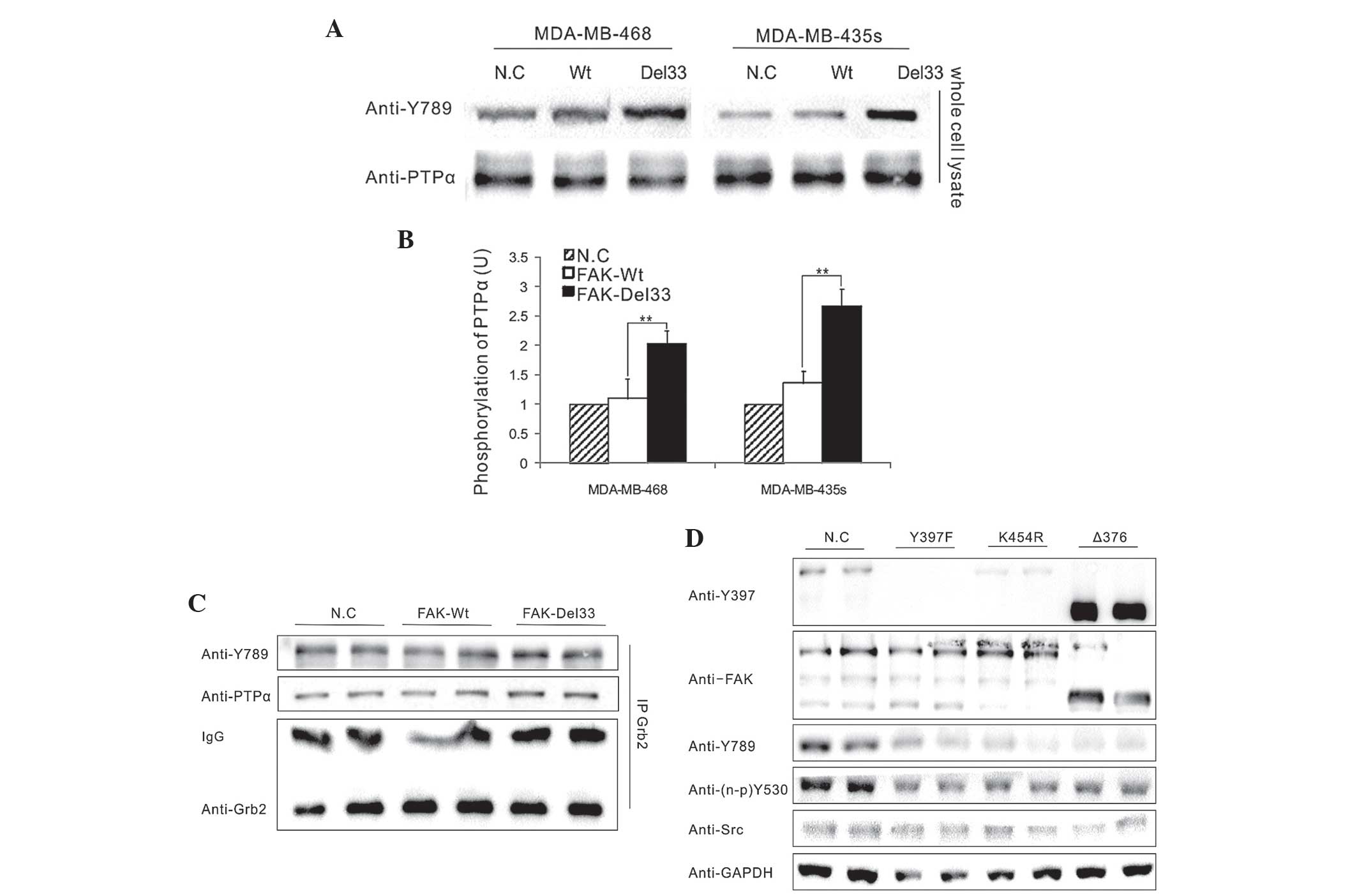
Significant differences were observed in the
phosphorylation levels of PTPα-Tyr789 between the FAK-Del33 mutant
and the FAK-Wt cells (Fig. 4A) and
an association between phosphorylated PTPα-Tyr789 and
dephosphorylated Src-Tyr530 was identified in the FAK-Del33 mutant
cells (Fig. 4B).
To assess the interference of Grb2 in the
phosphorylation of PTPα-Tyr789, which was bound to the PTPα
carboxyl terminus, western blot analysis was performed. The
endogenous Grb2 antibody was obtained from lysates of the
MDA-MB-468 cells by immunoprecipitation and its effects in the
FAK-Del33 mutant cells were examined (Fig. 4C). The results demonstrated that
PTPα-Tyr789 exhibited the same level of phosphorylation and was not
associated with the Grb2 antibody, suggesting that Grb2 is not
involved in the dephosphorylation of Src-Tyr530.
Mutational analysis of amino acids
A total of three amino acid sites were selected for
mutational analysis; Y397F (autophosphorylation site), K454R
(ATP-binding site) and Δ375 (deletion of FERM domain). As shown in
Fig. 4D, all three FAK mutations
in FAK-Del33 background failed to induce PTPα-Tyr789
phosphorylation; this therefore indicated that the ability of the
FAK-Del33 protein to induce PTPα-Tyr789 phosphorylation was
dependent on Tyr397, K454R and the FERM domain.
RNAi in the FAK-Del33 mutant
The Src gene sites of Tyr530 dephosphorylation and
Tyr419 phosphorylation changed significantly in the FAK-Del33
mutant. Following transfection of cells with PTPα siRNA, the
expression efficiency of these two Src genes were reduced
significantly. In addition, PTPα siRNA reduced the phosphorylation
levels of Src-Tyr397 (Fig. 5),
which was a feedback effect of the regulation of Src activation.
However, these effects were not observed in the PTP1B siRNA. In
addition, wound-healing and migration analyses revealed that the
PTPα siRNA had an effect on the efficiency of cell migration and
growth of the MDA-MB-468 cells Therefore, PTPα siRNA inhibited the
FAK-Del33-mediated action on tumorigenesis.
A schematic diagram of a model of c-Src activation
in cells with the FAK-Del33 mutation is shown in Fig. 1C, in which the deletion of exon 33
may be important in the regulation of Tyr789 phosphorylation. The
identification of differences in protein binding sites and the
exact role of the FAK mutant in PTPα regulation are important areas
for future investigation.
Discussion
According to previous studies, PTPα acts as an
upstream activator in the modulation of FAK phosphorylation
(13,14). The present study demonstrated a
novel pathway, that the FAK-Del33 mutation regulated PTPα-Tyr789
phosphorylation, leading to Src-Tyr530 dephosphorylation. Previous
studies involving PTPα-deficient fibroblasts revealed that the
phosphorylation of FAK, particularly at the Tyr397
auto-phosphorylation site can be identified in PTPα-deficient
fibroblasts and restored by reintroducing active, but not inactive,
PTPα. It was suggested that PTPα is required for maximal
integrin-stimulated Src-PTK activity as an upstream regulator of
FAK-Tyr397 phosphorylation (15,16).
The overexpression of PTPα has also been observed by increasing the
tyrosine phosphorylation of FAK (17).
However, previous studies have demonstrated that FAK
is required for PTPα phosphorylation and that the phos-phorylation
of PTPα may be a consequence of, rather than a prerequisite for,
FAK-Src-PTK activation (16,18).
In mouse embryonic fibroblasts, it has been observed that PTPα was
phosphorylated in response to integrin stimulation and required
SFKs (Src or Fyn/Yes), FAK and an intact cytoskeleton. This
suggested that the phosphorylation of PTPα-Tyr789 is mediated by
the active SFK/FAK complex (18,19).
The present study confirmed this hypothesis and, to the best of our
knowledge, was the first to demonstrate the phosphorylation of
PTPα-Tyr789 by an upstream activator, leading to Src activation.
The majority (~20%) of Tyr789 phosphorylation is bound to Grb2 in
NIH3T3 cells (19), however, this
was not observed in >20% of the endogenous PTPα-Tyr789 in the
present study, suggesting that free PTPα-Tyr789 displaces the
pTyr530 binding to the Src SH2 domain for Tyr530 dephosphorylation.
However, other studies have demonstrated that the dephosphorylation
of pTyr530 also occurs in the Tyr789-independent activation of Src
under certain circumstances and in other cell types (18,20).
Therefore, the role of tyrosine phosphorylation of PTPα in Src
activation remains to be elucidated.
Previous studies have demonstrated that PTPα
dephosphorylates Tyr419 and Tyr530 at Src (8,11,21,22),
leading to Src kinase activation. This indicates that Tyr789 leads
to substrate specificity, not overall catalytic activity and is
important in overexpressing the FAK-Del33 protein in breast cancer
cells.
In conclusion, the present study revealed an
unanticipated role for the FAK-Del33 protein in the regulation of
PTPα phosphorylation, which is important in Src-Tyr530
dephosphorylation and, consequently, in enhancing cell migration
and invasion.
Acknowledgments
This project was funded by Shanghai Committee of
Science and Technology, China (grant no. 15ZR1426800). It was also
funded by the National Natural Science Foundation of China under
the grant 81071745.
References
|
1
|
Stoker AW: Protein tyrosine phosphatases
and signaling. J Endocrinol. 185:19–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parsons JT: Focal adhesion kinase: the
first ten years. J Cell Sci. 116:1409–1416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frame MC, Patel H, Serrels B, et al: The
FERM domain: organizing the structure and function of FAK. Nat Rev
Mol Cell Biol. 11:802–814. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao X and Guan JL: Focal adhesion kinase
and its signaling pathways in cell migration and angiogenesis. Adv
Drug Deliv Rev. 63:610–615. 2011. View Article : Google Scholar :
|
|
5
|
Klinghoffer RA, Sachsenmaier C, Cooper JA,
et al: Src family kinases are required for integrin but not PDGFR
signal transduction. EMBO J. 18:2459–2471. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salazar EP and Rozengurt E: Src family
kinases are required for integrin-mediated but not for G
protein-coupled receptor stimulation of focal adhesion kinase
autophosphorylation at Tyr-397. J Biol Chem. 276:17788–17795. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim ST, Mikolon D, Stupack DG, et al: FERM
control of FAK functions: implications for cancer therapy. Cell
Cycle. 7:2306–2314. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng XM, Wang Y and Pallen CJ: Cell
transformation and activation of pp60c-src by overexpression of a
protein tyrosine phosphatase. Nature. 359:336–339. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ponniah S, Wang DZ, Lim KL, et al:
Targeted disruption of the tyrosine phosphatase PTPalpha leads to
constitutive downregulation of the kinases Src and Fyn. Curr Biol.
9:535–538. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arias-Salgado EG, Haj F, Dubois C, et al:
PTP-1B is an essential positive regulator of platelet integrin
signaling. J Cell Biol. 170:837–845. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng XM, Resnick RJ and Shalloway D: A
phosphotyrosine displacement mechanism for activation of Src by
PTPalpha. EMBO J. 19:964–978. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vacaru AM and den Hertog J: Serine
dephosphorylation of receptor protein tyrosine phosphatase alpha in
mitosis induces Src binding and activation. Mol Cell Biol.
30:2850–2861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang XQ, Liu XF and Yao L: Somatic
mutational analysis of FAK in breast cancer: a novel
gain-of-function mutation due to deletion of exon 33. Biochem
Biophys Res Commun. 443:363–369. 2014. View Article : Google Scholar
|
|
14
|
Zheng XM, Resnick RJ and Shalloway D:
Mitotic activation of protein-tyrosine phosphatase alpha and
regulation of its Src-mediated transforming activity by its sites
of protein kinase C phosphorylation. J Biol Chem. 277:21922–21929.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schlaepfer DD and Hunter T: Evidence for
in vivo phosphorylation of the Grb2 SH2-domain binding site on
focal adhesion kinase by Src-family protein-tyrosine kinases. Mol
Cell Biol. 16:5623–5633. 1996.PubMed/NCBI
|
|
16
|
Zeng L, Si X, Yu WP, et al: PTP alpha
regulates integrin-stimulated FAK autophosphorylation and
cytoskeletal rearrangement in cell spreading and migration. J Cell
Biol. 160:137–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harder KW, Moller NP, Peacock JW, et al:
Protein-tyrosine phosphatase alpha regulates Src family kinases and
alters cell-substratum adhesion. J Biol Chem. 273:31890–31900.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen M, Chen SC and Pallen CJ:
Integrin-induced tyrosine phosphorylation of protein-tyrosine
phosphatase-alpha is required for cytoskeletal reorganization and
cell migration. J Biol Chem. 281:11972–11980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
den Hertog J, Tracy S and Hunter T:
Phosphorylation of receptor protein-tyrosine phosphatase alpha on
Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in
vivo. EMBO J. 13:3020–3032. 1994.PubMed/NCBI
|
|
20
|
Yang LT, Alexandropoulos K and Sap J:
c-SRC mediates neurite outgrowth through recruitment of Crk to the
scaffolding protein Sin/Efs without altering the kinetics of ERK
activation. J Biol Chem. 277:17406–17414. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang J, Yao L, Xu R, et al: Activation of
Src and transformation by an RPTPalpha splice mutant found in human
tumours. EMBO J. 30:3200–3211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Yu L and Zheng X:
PTPalpha-mediated Src activation by EGF in human breast cancer
cells. Acta Biochim Biophys Sin (Shanghai). 45:320–329. 2013.
View Article : Google Scholar
|