Introduction
Cellular proteins are continually renewed by
synthesis and degradation. Macroautophagy, a process by which cells
autodigest parts of their cytoplasm, is a fundamental cellular
mechanism of protein degradation (1). Autophagy is an evolutionarily
conserved pathway that leads to stress-induced degradation of
intracellular proteins and organelles (2). This type of degradation has been
reported to be mediated by interactions with microtubule-associated
protein 1 light chain 3 (LC3), a mammalian homologue of
autophagy-related gene 8, which was demonstrated to proceed via
recruitment to the phagophore/isolation membrane, where it remained
associated with the completed autophagosome (3).
The present study investigated the involvement of
autophagy in the degradation of gap junction proteins following
traumatic brain injury (TBI). Gap junctions are components of the
plasma membrane that contain clusters of oligomeric assemblies of
proteins called connexins (Cxs), which form channels to allow the
passage of ions and small molecules between adjacent cells.
Previous studies have shown that several Cxs are expressed by
numerous types of cells in the central nervous system (4). Cx43 was shown to be expressed
exclusively in astrocytes (5),
demonstrated using immunogold labeling for Cx43 (6). Phosphorylated Cx43 (p-Cx43), via the
extracellular regulated protein kinase signaling pathway, reduces
intercellular permeability via gap junctions, correlating with
tissue homeostasis (7). Using HeLa
and normal rat kidney epithelial cell models, Lichtenstein et
al (8) reported that
autophagy-regulated connexin degradation may be induced by
starvation. However, the mechanism of neuronic autophagy, which
contributed to astrocytic p-Cx43 degradation in the hippocampus
following TBI in vivo, remains to be elucidated. In the
present study, the involvement of autophagy in the degradation of
p-Cx43 was examined in the rat hippocampus following experimental
TBI.
Materials and methods
Animals and TBI model
The study was approved by the ethics committee of
the Hebei United University (Tangshan, China). All experimental
procedures were performed in accordance with the guidelines of the
Chinese Council on Animal Protection and were approved by Hebei
United University Committee for the Use of Animals in Research
(Tangshan, China). A total of 60 male Sprague-Dawley rats (age,
12–16 weeks; weight, 350–375 g; Vital River Laboratory Animal
Technology Co., Ltd., Beijing, China) were used in the present
study. All animals were housed using the standard 12-h light/dark
cycle and access to water and food ad libitum prior to and
following surgery or sham operation. The rat model of TBI was
induced using a modified weight-drop device, as described
previously by Marmarou et al (9). In brief, rats were anaesthetized
using sodium pentobarbital (Nembutal, 60 mg/kg). A midline incision
was made that exposed the skull between the bregma and lambda
suture lines, and a steel disc (10 mm in diameter and 3 mm in
thickness; Tangshan Railway Vehicle Co., Ltd., Tangshan, China) was
then adhered to the skull using acrylic polymers. Animals were
subsequently placed onto a foam mattress (Tangshan Railway Vehicle
Co., Ltd.) positioned underneath a weight-drop device where a 450-g
weight fell freely through a vertical tube from 1.5 m onto the
steel disc. Sham-operated animals underwent an identical surgical
procedure without weight-drop impact. Following surgery, rats were
housed in individual cages and placed on heat pads (37°C) for 24 h
to maintain a normal body temperature during the recovery
period.
Group determination and administration of
drugs
The rats were randomly divided into three groups:
Sham, TBI, and TBI treated with 3-methyladenine (3-MA;
Sigma-Aldrich, St. Louis, MO, USA) (n=5 for each group). The rats
were sacrificed at time-points of 3, 6, 24 and 48 h following TBI.
1 μl 3-MA (sc-205596, 400 nmol; Santa Cruz Biotechnology,
Santa Cruz, CA, USA) was administered through right ventricle
injection 10 min prior to the induction of TBI. For
intracerebroventricular injections, animals were fixed in a
stereotaxic apparatus (RWD68025; RWD Life Science Co., Ltd.,
Shenzhen, China), a midline incision was made in the skin, followed
by the induction of a small hole in the cranial region. A Hamilton
syringe (RWD68025; RWD Life Science Co., Ltd.) was used to inject
3-MA or 1 μl saline into the right cerebral ventricle
according to the following coordinates: Bregma, advanced placement
−0.8 mm, right +1.6 mm (midline) and deep 3.4 mm from dura
(10). Sham-operated rats received
intracerebroventricular injection of 1 μl normal saline.
Hippocampal tissue (the entire ipsilateral hippocampus, including
the impact site and surroundings) from each injured rat was
dissected and used for numerous assays.
Western blot analysis
Western blot analysis was performed as previously
described (11). In brief, rats
were deeply anesthetized and underwent intracardiac perfusion with
0.1 mol/l phosphate-buffered saline (PBS; pH 7.4; Wuhan Boster
Biological Engineering Co., Ltd., Wuhan, China). The hippocampus
was rapidly isolated; total proteins were extracted and protein
concentration was determined using the bicinchoninic acid reagent
(Solarbio, Beijing, China) method. Samples were subjected to
SDS-PAGE. Separated proteins on the gel were transferred onto
polyvinylidene fluoride membranes (Roche Diagnostics, Mannheim,
Germany). Blots were blocked using 5% fat-free milk powder (Wuhan
Boster Biological Engineering Co., Ltd.,) for 1 h at room
temperature. Subsequently, blots were incubated overnight at 4°C
with indicated primary antibodies, including rabbit anti-p-Cx43
polyclonal antibodies, rabbit anti-LC3 polyclonal antibody and
mouse anti-β-actin monoclonal antibody (dilution, 1:500; Santa Cruz
Biotechnology; Santa Cruz, CA, USA). The blots were then incubated
with horseradish peroxidase-conjugated anti-rabbit and anti-mouse
immunoglobulin G (dilution, 1:5,000; Cell Signaling Technology,
Danvers, MA, USA) for 2 h at room temperature. Following incubation
with titrated secondary antibodies, the immunoblot on the
polyvinylidene fluoride membrane was visible once developed using
an enhanced chemiluminescence detection system (ChemiDoc XRS;
Bio-Rad Laboratories, Hercules, CA, USA), and the densitometric
signals were quantified using imaging software (Image Lab 4.1; Bio
Rad Laboratories). Immunoreactive bands of the protein expression
levels were normalized to the intensity of corresponding β-actin
bands. The western blot results were analyzed Image J software
(Bethesda, MD, USA).
Immunofluorescence analyses
The brain tissues were fixed in 4% paraformaldehyde
(Wuhan Boster Biological Engineering Co., Ltd.) for 24 h, followed
by immersion in 30% sucrose solution until sinking to the bottom.
Sections were made 200 μm apart from the anterior to
posterior hippocampus (bregma −1.90 to −3.00 mm) of the TBI animals
and then embedded in optimal cutting temperature compound (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China).
15-μm frozen sections were sliced with a cryostat (CM 1950,
Heidelberger, Germany) treated with 0.4% Triton-100 for 10 min and
then blocked in normal donkey serum for 1 h (both Beijing Solarbio
Science & Technology Co., Ltd.). For triple labeling, frozen
sections were incubated with a mixture of goat anti-p-Cx43
polyclonal antibody, rabbit anti-LC3 polyclonal antibody and mouse
anti-glial fibrillary acidic protein (GFAP) monoclonal antibody
(dilution, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA)
overnight at 4°C. The next day, the sections were incubated with
Alexa Fluor 647-conjugated, 594-conjugated and 488-conjugated
secondary antibodies (dilution, 1:500; Invitrogen, Carlsbad, CA,
USA) against the appropriate species for 2 h at 37°C in the dark.
Sections were mounted with DAPI (Vector Laboratories, Burlingame,
CA, USA). Images were captured using a laser scanning confocal
microscope (Olympus FV1000; Olympus, Tokyo, Japan). Primary
antibodies were replaced with PBS in the negative control
group.
Electron microscopy
Freshly dissected tissues were fixed in 3%
glutaraldehyde and 1% osmic acid for 2 h, followed by
propionaldehyde (Beijing Zhongshan Jinqiao Biology & Technology
Co., Ltd., Beijing, China) dehydration and epoxy resin embedding.
Tissue was sliced using a cryostat and stained using 2% uranyl
acetate and lead citrate (Beijing Zhongshan Jinqiao Biology &
Technology Co., Ltd.). Images were observed and recorded using
transmission electron microscopy (Hitachi, Tokyo, Japan).
Statistical analysis
All experiments were repeated three times and
similar results were obtained. Statistical analysis was performed
using the SPSS 16.0 statistics software (IBM, Armonk, NY, USA).
Data are expressed as the mean ± standard deviation. Statistical
analysis was performed using analysis of variance, followed by the
Dunnett’s post-hoc test. P<0.05 was considered to indicate a
statistically significant difference between values.
Results
General
No significant differences were observed in body
weight or temperature between the TBI and sham-injured groups.
Furthermore, no differences were found in injury levels among the
3-, 6-, 24-, or 48-h TBI groups.
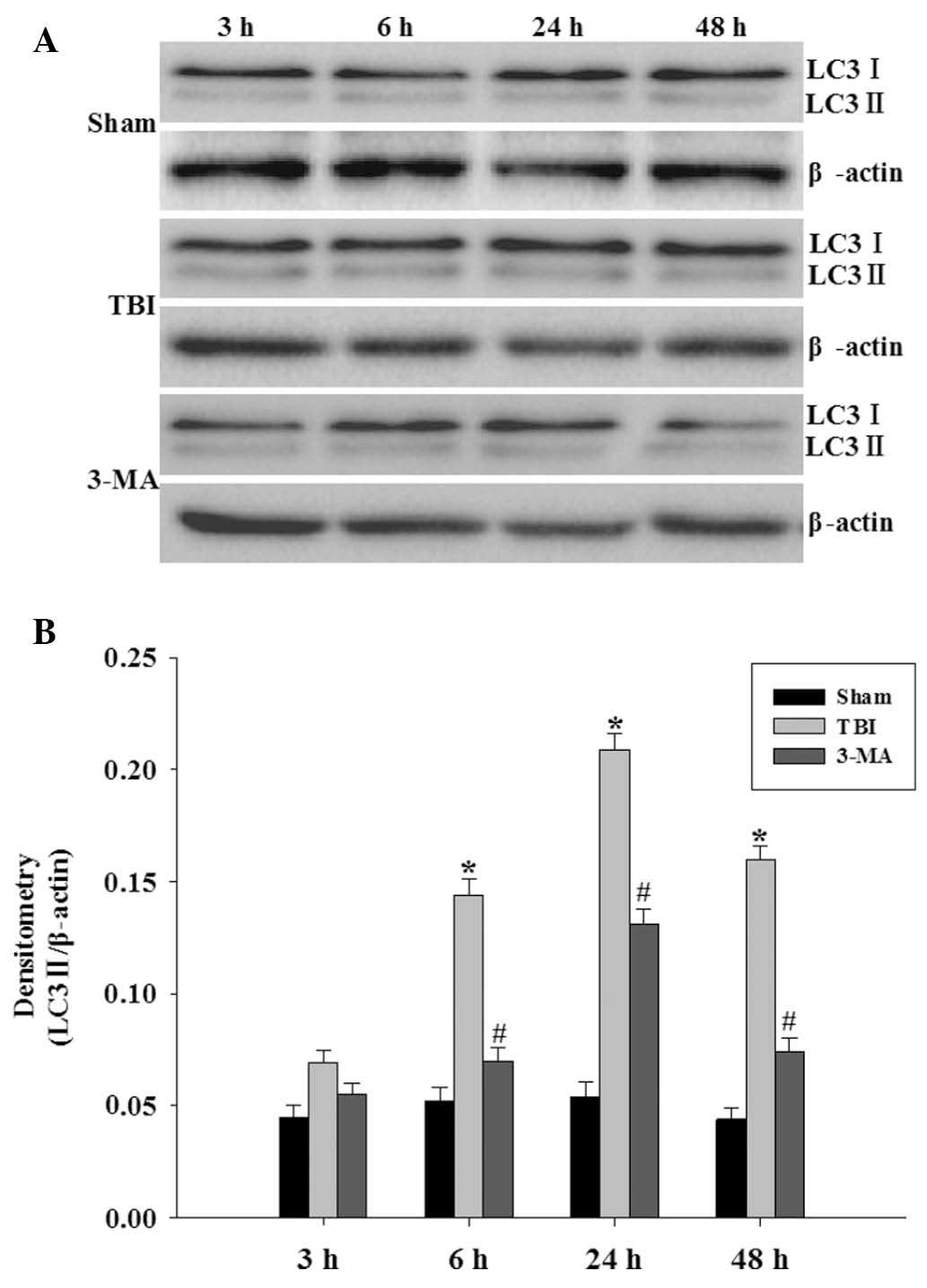
3-MA treatment depresses LC3-II protein
levels
LC3-II protein levels were analyzed using western
blot analysis (Fig. 1A). LC3-II
protein was identified at low levels in the hippocampus of rats in
the sham group. Activation of LC3-II in the hippocampus was
significantly increased at 6 h following injury, peaked at 24 h,
and by 48 h activation of LC3-II had slightly decreased but was
still significant (P<0.05 vs. sham group). The results shown in
Fig 1B demonstrated that 3-MA
pretreatment significantly inhibited the upregulation of LC3-II
protein levels compared with those of the TBI groups at 6, 24 and
48 h (P<0.05 vs. TBI group).
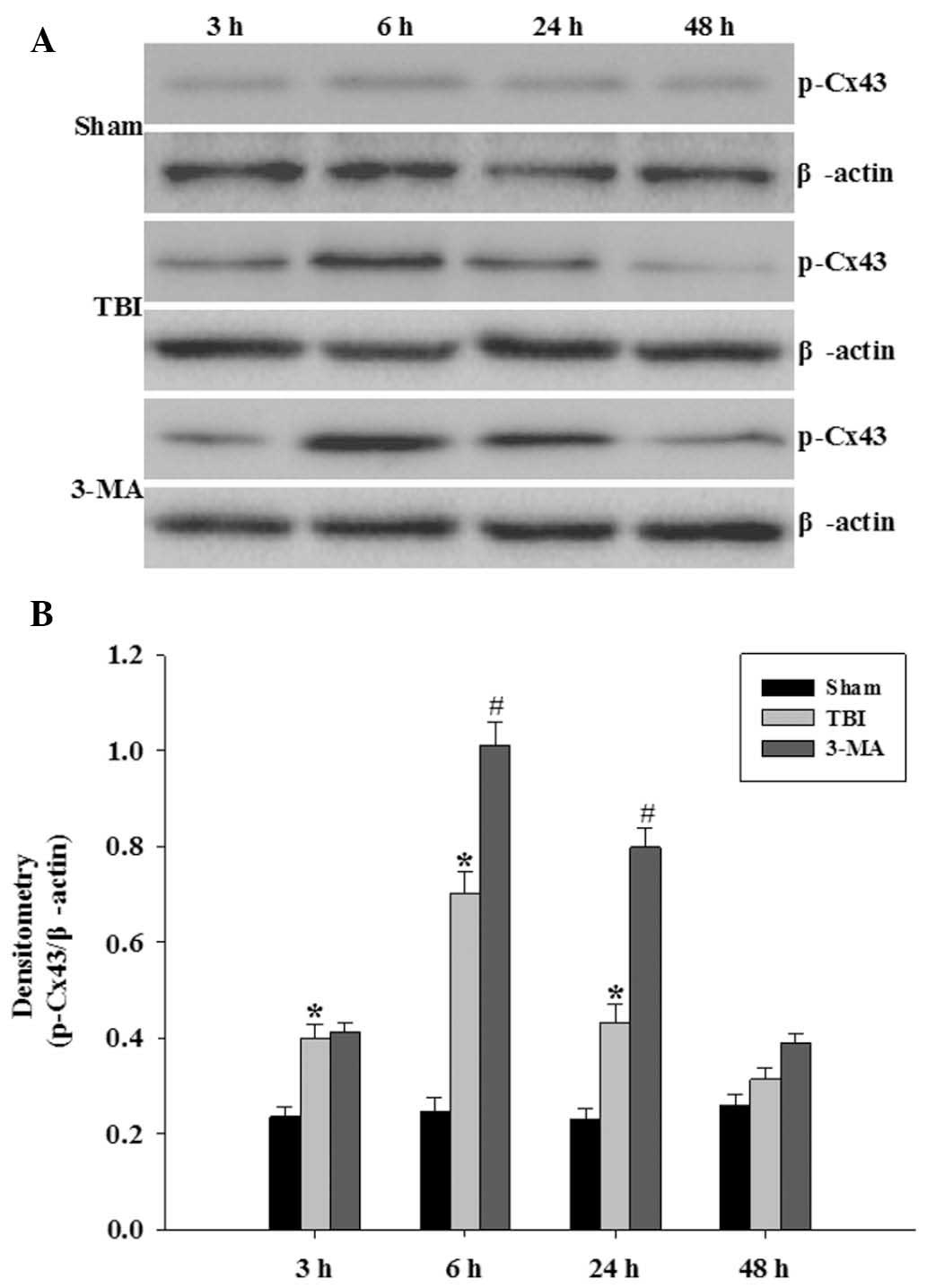
p-Cx43 is increased following TBI in the
rat hippocampus
Protein levels of p-Cx43 were determined in order to
observe altered p-Cx43 activity following TBI and confirm the
ability of 3-MA to regulate p-Cx43 (Fig. 2A). Levels of p-Cx43 protein in the
hippocampus were significantly upregulated at 3 h following TBI,
which persisted at high levels until 24 h following injury
(P<0.05 vs. sham group). As demonstrated in Fig. 2B, pretreatment with 3-MA
significantly increased the relative protein abundance of p-Cx43 in
the hippocampus at 6 and 24 h following TBI (P<0.05 vs. TBI
group).
Neuronic autophagy regulates
immunofluorescence intensity of astrocytic p-Cx43 in the
hippocampus following TBI
An overlap was observed between GFAP and p-Cx43 in
hippocampal astrocytes, indicating that p-Cx43 strongly colocalized
with the hippocampal astrocytes (Fig.
3; arrows). DAPI analysis revealed that LC3 specifically
colocalized within neurons of the hippocampus. The
immunofluorescence intensity of LC3 and p-Cx43 in the hippocampus
was significantly increased at 24 h following TBI. It was also
found that pretreatment with the autophagy inhibitor 3-MA further
enhanced the TBI-induced increase in p-Cx43 immunofluorescence
intensity in hippocampal astrocytes.
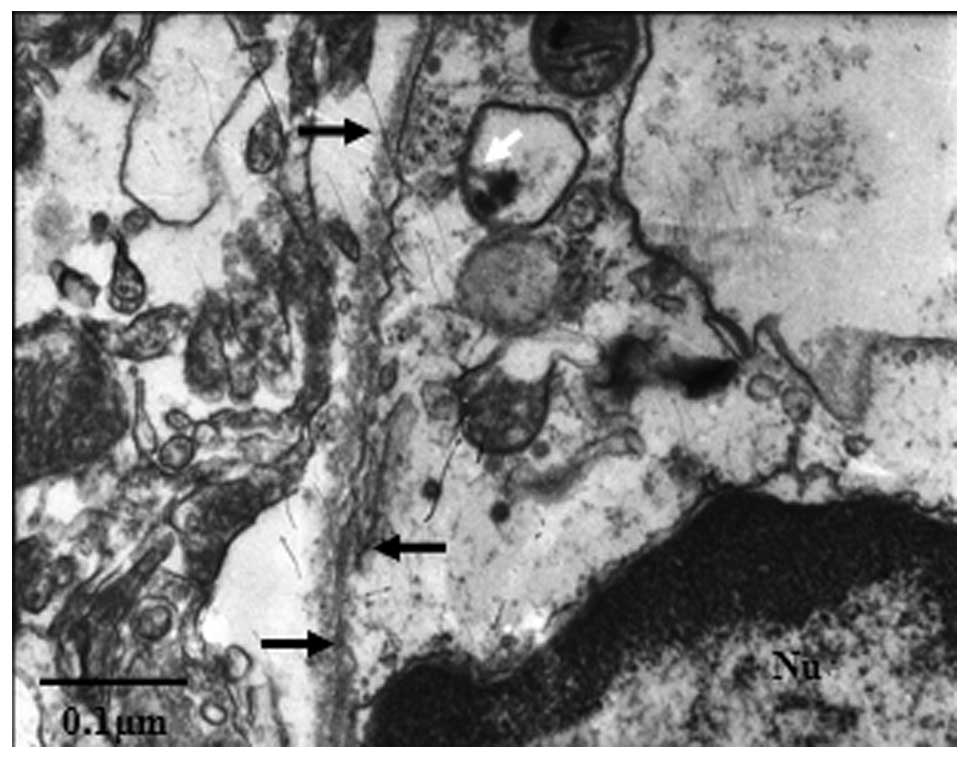
Internalized gap junctions are present in
lysosomes in the hippocampal neuron
Connexin and gap junction degradation was further
analyzed by examining the ultrastructure of rat hippocampi using
transmission electron microscopy 24 h following TBI. Gap junction
structures were found between astrocytes and neurons (Fig. 4; black arrows). Profiles of the
internalized gap junctions were observed in the neuronic cytoplasm.
Certain profiles of partially disrupted internalized gap junctions
were observed inside membranous structures; these structures are
likely to represent secondary lysosomes (Fig. 4; white arrow).
Discussion
Autophagy is a proteolytic process which, under
normal conditions, demonstrates low levels of activity; however,
under pathological conditions, the activity of this pathway is
significantly altered (12). The
ability of cells to increase their autophagic proteolytic activity
enables them to adapt to changes in their environment, such as TBI
(13). The results of the present
study showed that protein levels of p-Cx43 in the hippocampus were
significantly upregulated at 3, 6 and 24 h following TBI, whereas
other studies indicated that no significant changes in the total
amount of Cx43 in the hippocampus were present following injury
(5). The results of the present
study suggested that p-Cx43 in the hippocampus is associated with
molecular sequelae of TBI. Previous studies have demonstrated using
biochemical and colocalization studies that astrocytic p-Cx43
regulated neuronal autophagy in the hippocampus following TBI in
rats (14). Autophagy was found to
be involved in TBI-induced cell membrane breakdown, cell loss as
well as motor and cognitive outcome deficits (15). However, studies have revealed that
reduction of cell-cell permeability through gap junctions may lead
to partial pathological hippocampal dysfunction, including
post-traumatic amnesia and post-traumatic stress disorders
(16,17). This therefore highlights the
importance of elucidating the cellular mechanisms of the
involvement of neuronic autophagy in astrocytic connexin
degradation.
Ultrastructural studies in rat liver, incisor and
fetal epidermis (18) as well as
equine hoof wall (19) have
revealed the internalization of gap junctions enclosed within
double membrane structures, therefore indicating the involvement of
autophagic activity in the degradation of gap junctions or
connexins. Gap junctions between neurons and astrocytes function as
an interdependent network with bidirectional communication
(20). In the present study,
autophagic double membrane structures were observed at the 1-h
time-point post-TBI in neurons, and three days later in astrocytes.
Western blot analysis revealed that 3-MA significantly increased
protein levels of p-Cx43 following TBI-induction in the
hippocampus, therefore indicating the involvement of autophagy in
the TBI-induced degradation of p-Cx43 in the hippocampus.
Immunofluorescence assays also demonstrated increased
immunofluorescence activity of astrocytic p-Cx43 in the hippocampus
following treatment with 3-MA compared with that of the TBI group
without 3-MA-treatment. This therefore demonstrated the involvement
of neuronic autophagy in the degradation of astrocytic p-Cx43 in
the hippocampus. Internalized gap junctions surrounded by lysosomal
structures were observed in hippocampal neurons using
ultrastructural studies, implicating the involvement of neuronic
autophagy in the degradation of gap junction plaques, which may be
formed of astrocytic connexin. It was therefore suggested that
astrocytic p-Cx43 targeted for subsequent degradation and gap
junction plaques between astrocyte and neuron are sequestered by an
LC3-containing membrane following stimulation of neuronic autophagy
by TBI in the hippocampus.
In conclusion, the present study suggested that
autophagy in neurons was involved in the degradation pathway for
p-Cx43 in hippocampal astrocytes following TBI.
Acknowledgments
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (no. H2012401071).
References
|
1
|
Reggiori F and Klionsky DJ:
Autophagosomes: biogenesis from scratch? Curr Opin Cell Biol.
17:415–422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pozuelo-Rubio M: Regulation of autophagic
activity by 14-3-3ζ proteins associated with class III
phosphatidylinositol-3-kinase. Cell Death Differ. 18:479–492. 2011.
View Article : Google Scholar :
|
|
3
|
Zheng YT, Shahnazari S, Brech A, Lamark T,
Johansen T and Brumell JH: The adaptor protein p62/SQSTM1 targets
invading bacteria to the autophagy pathway. J Immunol.
183:5909–5916. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Bock M, Kerrebrouck M, Wang N and
Leybaert L: Neurological manifestations of oculodentodigital
dysplasia: a Cx43 channelopathy of the central nervous system?
Front Pharmacol. 4:1202013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Maio A, Vega VL and Contreras JE: Gap
junctions, homeostasis, and injury. J Cell Physiol. 191:269–82.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rash JE, Yasumura T, Dudek FE and Nagy JI:
Cell-specific expression of connexins and evidence of restricted
gap junctional coupling between glial cells and between neurons. J
Neurosci. 21:1983–2000. 2001.PubMed/NCBI
|
|
7
|
Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF
and Burt JM: Mechanism of v-Src-and mitogen-activated protein
kinase-induced reduction of gap junction communication. Am J
Physiol Cell Physiol. 284:C511–C520. 2003. View Article : Google Scholar
|
|
8
|
Lichtenstein A, Minogue PJ, Beyer EC and
Berthoud VM: Autophagy: a pathway that contributes to connexin
degradation. J Cell Sci. 124:910–920. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats. Part I: Pathophysiology and biomechanics. J
Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Milad M, Vidal-Gonzalez I and Quirk G:
Electrical stimulation of medial prefrontal cortex reduces
conditioned fear in a temporally specific manner. Behav Neurosci.
118:389–394. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song SX, Gao JL, Wang KJ, et al:
Attenuation of brain edema and spatial learning deficits by the
inhibition of NADPH oxidase activity using apocynin following
diffuse traumatic brain injury in rats. Mol Med Rep. 7:327–331.
2013.
|
|
12
|
Meijer AJ and Codogno P: Autophagy:
regulation and role in disease. Critical Rev Clin Lab Sci.
46:210–240. 2009. View Article : Google Scholar
|
|
13
|
Zhang YB, Li SX, Chen XP, et al: Autophagy
is activated and might protect neurons from degeneration after
traumatic brain injury. Neurosci Bull. 24:143–149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun LQ, Gao JL, Cui CM, et al: Astrocytic
p-connexin 43 regulates neuronal autophagy in the hippocampus
following traumatic brain injury in rats. Mol Med Rep. 9:77–82.
2014.
|
|
15
|
Luo CL, Li BX, Li QQ, et al: Autophagy is
involved in traumatic brain injury-induced cell death and
contributes to functional outcome deficits in mice. Neuroscience.
184:54–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rami A, Volkmann T and Winckler J:
Effective reduction of neuronal death by inhibiting gap junctional
intercellular communication in a rodent model of global transient
cerebral ischemia. Exp Neurol. 170:297–304. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ohsumi A, Nawashiro H, Otani N, Ooigawa H,
Toyooka T and Shima K: Temporal and spatial profile of
phosphorylated connexin43 after traumatic brain injury in rats. J
Neurotrauma. 27:1255–1263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sasaki T and Garant PR: Fate of annular
gap junctions in the papillary cells of the enamel organ in the rat
incisor. Cell Tissue Res. 246:523–530. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leach DH and Oliphant LW: Degradation of
annular gap junctions of the equine hoof wall. Acta Anat (Basel).
120:214–219. 1984. View Article : Google Scholar
|
|
20
|
Zhang YB, Li SX, Chen XP, et al: Autophagy
is activated and might protect neurons from degeneration after
traumatic brain injury. Neurosci Bull. 24:143–149. 2008. View Article : Google Scholar : PubMed/NCBI
|