Introduction
Previous studies have demonstrated that
O6-methylguanine DNA methyltransferase (MGMT) can
eliminate the temozolomide (TMZ)-induced
O6-methylguanine (O6MeG) DNA adduct and leads
to TMZ resistance (1–3). However, the MGMT promoter is
methylated in almost 50% of glioblastoma specimens and the
methylation status of the MGMT promoter in the primary tumor is
retained at recurrence (4,5). This suggests other mechanisms are
involved in TMZ resistance, which remain to be elucidated.
TMZ-induced autophagy functions as a cytoprotective
mechanism against TMZ toxicity and contributes to TMZ resistance.
The inhibition of autophagy augments TMZ cytotoxicity in glioma
(6–8). Following nutrient restriction or
stress, adenosine monophosphate-activated protein kinase (AMPK) is
involved in the formation of the autophagosome by interacting with
unc-51-like autophagy activating kinase 1 (ULK1) and induces
autophagy (9,10). Ataxia-telangiectasia mutated (ATM),
a serine/threonine protein kinase, leads to the phosphorylation of
AMPK following ionizing radiation or nutrient restriction, which
suggests that ATM is the upstream kinase of AMPK (11,12).
However, the role of ATM-AMPK in TMZ-induced autophagy remains to
be elucidated.
DNA mismatch repair (MMR), a genetic repair pathway,
is composed of MutS and MutL heterodimers (3,5). The
O6MeG/T mismatches are immediately recognized by MutS
(3) and the mutL homolog 1 (MLH1)
MMR protein then migrates from the nucleus to the cytoplasm via
nuclear export sequences, possibly signaling DNA damage to
downstream pathways (13).
However, whether MLH1, a DNA damage messenger, is involved in
TMZ-induced autophagy remains to be elucidated.
The present study hypothesized that MLH1 signaled
DNA damage to ATM or AMPK and induced autophagy in glioma following
treatment with TMZ. To investigate this hypothesis, the activation
of AMPK-ULK1 and the levels of autophagy following treatment with
MLH1 small interfering (si)RNA and KU-55933 were assessed. The
association between MLH1 and ATM was investigated and the effects
of the inhibition of autophagy, by knock down of MLH1 and
inhibition of ATM-AMPK, on the cytotoxicity of TMZ were
evaluated.
Materials and methods
Cell culture and reagents
The U87MG, U251 and SHG-44 glioma cell lines were
obtained from the Cell Bank of Chinese Academy of Sciences
(Shanghai, China). All cells were plated at a density of
1×106 cells/well in a 6-well plate and maintained in
Dulbecco’s modified Eagle’s medium (Gibco Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (PAA
Laboratories, Pasching, Austria) at 37°C in a 5%
CO2-humidified atmosphere. The U87MG cells were cultured
and passaged in the presence of increasing concentrations of TMZ
(30, 60, 90, 120, 150, 180, 210, 240, 270 and 300 μM) to
generate TMZ-resistant cell lines, termed U87TMZ. The glioma cells
were divided into the following four groups: i) control; ii) TMZ
(100 μM, 72 h); iii) KU-55933 (10 μM, 72 h)/compound
C (5 μM); iv) TMZ (100 μM, 72 h) + KU-55933 (10
μM, 72 h)/compound C (5 μM, 72 h) The TMZ was
supplied by Tasly Pharmaceutical Co., Ltd. (Tianjin, China).
KU-55933 (cat. no. sc-202963) and compound C (cat. no. P5499) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) and
Sigma-Aldrich (St. Louis, MO, USA), respectively. The TMZ (100
μM), KU-55933 (10 μM) and compound C (5 μM)
were dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich). The
final concentration of DMSO in the culture medium did not exceed
0.01%, and therefore did not effect cell viability or protein
expression.
siRNA transfection
For siRNA transfection, the MLH1,
5′-GCCAUGUGGCUCAUGUUACdTdT-3′ siRNA oligo was used and
5′-GCUCAGAUCAAUACGGAGAdTdT-3′ was used as a non-specific negative
control. The siRNAs were purchased from Genepharma Co., Ltd.
(Shanghai, China). The cells were transfected with 100 nM siRNA
using Lipofectamine RNAiMAX transfection reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA). At 48 h post-transfection, the
DMEM culture media was refreshed and the cells were exposed to 100
μM TMZ ro 72 h, followed by western blot analysis or the
detection of autophagosomes. Cells without siRNA transfection were
used as a negative control.
Western blot analysis
Briefly, cells were dissolved and boiled in Laemmli
buffer (Beyotime Institute of Biotechnology, Shanghai, China) for 5
min, separated with 10% SDS-PAGE (Beyotime Institute of
Biotechnology) and transferred to polyvinylidene fluoride membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 80 mA for 2 h.
Following blocking with bovine serum albumin (Thermo Fisher
Scientific, Waltham, MA, USA), the membranes were incubated with
the primary antibodies in Tris-buffered saline with Tween-20 (TBST;
Amresco LLC, Solon, OH, USA) at 37°C for 2 h. Subsequently,
secondary antibodies in TBST were added and incubated for 2 h at
room temperature. Bound secondary antibodies were detected using an
enhanced chemiluminescence detection system (Beyotime Institute of
Biotechnology). Antibodies specific for MLH1 (cat. no. 3515),
phosphorylated (p)ATM (Ser1981; cat. no. 5883), total ATM (cat. no.
2873), total AMPKα (cat. no. 2532), pAMPKα (Thr172; cat. no. 2535),
pULK1 (Ser467; cat. no. 4634) and microtubule-associated protein 1
light chain 3 β (LC3B; cat. no. 2775), with β-actin (cat. no. 4970)
as a loading control, were purchased from Cell Signaling
Technology, Inc (Danvers, MA, USA). Polyclonal anti-γH2AX antibody
(cat. no. ab2893) was obtained from Abcam (Cambridge, MA, USA).
Histone subtypes (γH2AX) provide a sensitive marker of DNA
double-strand breaks (DSBs) (14)
and LC3B, which is cleaved into LC3B- and LC3B-II during autophagy,
was used as an autophagy marker (15,16).
Detection of the autophagosome/acidic
vesicular organelles (AVOs)
Following TMZ and/or KU-55933/Compound C treatment,
acridine orange (cat. no. sc-358795, Santa Cruz Biotechnology,
Inc.) was added at a final concentration of 1 μg/ml for 15
min. The cells were collected, excited (488 nm) and analyzed by
flow cytometric analysis using a Guava Easycyte 5HT flow cytometer
(EMD Millipore, Billerica, MA, USA). The nucleoli of the acridine
orange-stained cells fluoresced bright green and the acidic
vesicles emitted a bright red fluorescence. The cells containing
AVOs were identified as double positive cells.
Detection of apoptosis
The apoptotic cells were detected using an annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit (cat. no. C1063; Beyotime Institute of Biotechnology,
Jiangsu, China) according to the manufacturer’s instructions, using
flow cytometric analysis on a Guava Easycyte 5HT flow cytometer
(EMD Millipore). The data were analyzed using Guava Soft 2.2
software (EMD Millipore).
Cell viability analysis
MTT assays were performed to assess the sensitivity
of the cells to the drug treatments. Briefly, the cells were seeded
at a density of 3,000 cells/well in 96-well microplates. Following
drug treatment, 20 μl MTT (5 mg/ml) was added to each well
and the plates were incubated at 37°C for 4 h. The culture media
was subsequently replaced with 100 μl DMSO to lyse the
cells, followed by the measurement of absorbance at 570 nm using a
Thermo Fisher Scientific Multiskan GO microplate reader (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
Statistical analysis
All the experiments were performed in triplicate and
data are expressed as the mean ± standard deviation. Statistical
analysis of the data was performed using Student’s t-test for two
groups or a one-way analysis of variance for three or more groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TMZ-induced autophagy is
MLH1-dependent
Western blot analysis revealed that TMZ failed to
induce the phosphorylation of AMPK or ULK1 following MLH1 knock
down in the U87MG, U251 or SHG-44 cells (Fig. 1A and B). In the TMZ+MLH1 siRNA
group, the cleavage of LC3B was decreased and the number of AVOs
was reduced, including under cytotoxic stress, compared with the
TMZ group (10.85±1.36%, vs. 34.02±1.77%, respectively; P<0.001).
Additionally, the MLH1 siRNA group had fewer AVOs compared with the
control group (5.60±0.64%, vs. 12.29±0.91%, respectively;
P<0.001; Fig. 1C), consistent
with the LC3B cleavage results (Fig.
1B). This suggested that MLH1 was important in the activation
of TMZ-induced autophagy.
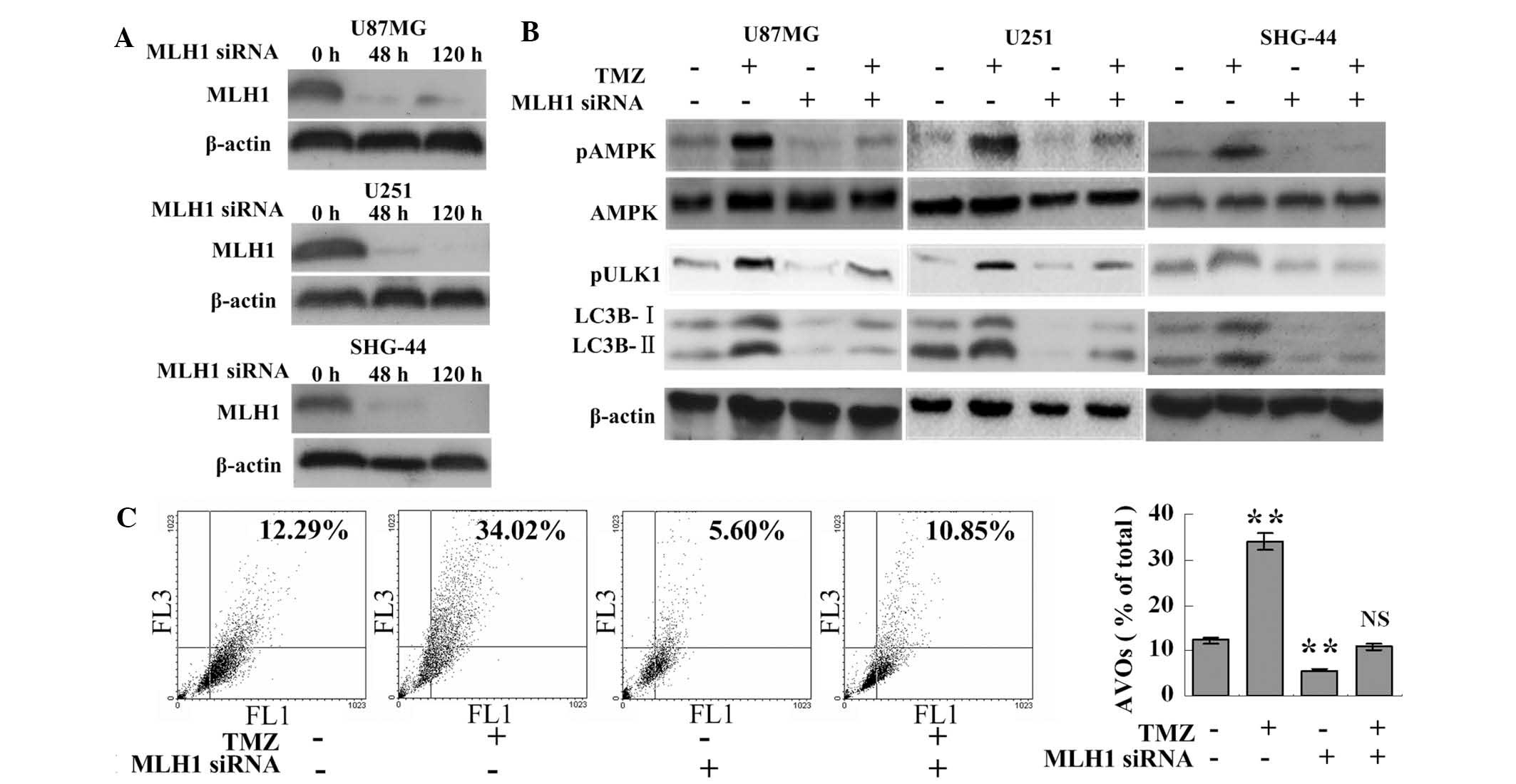 | Figure 1TMZ induces autophagy via MLH1. (A)
Glioma cells were transfected with 100 nM MLH1 siRNA and harvested
at the indicated time points for western blot analysis. (B) Glioma
cells and the MLH1-knockdown glioma cells (100 nM MLH1 siRNA for 48
h) were incubated with 100 μM TMZ or vehicle (DMSO) for 72
h. Following treatment, the cells were collected for western blot
analysis. (C) U87MG cells and the MLH1-knockdown U87MG cells were
incubated with 100 μM TMZ or vehicle for 72 h. Following
treatment, the cells were collected for AVO detection using flow
cytometric analysis. The data are expressed as the mean ± standard
deviation (**P<0.01, vs. control group; n=3 for each
group). NS, not statistically significant compared with the control
group; MLH1, mutL homolog 1; TMZ, temozolomide; siRNA, small
interfering RNA; AMPK, adenosine monophosphate-activated protein
kinase; ULK, unc-51-like autophagy activating kinase 1; LC3B,
microtubule-associated protein 1 light chain 3 β; AVO, acidic
vesicular organelle, p, phosphorylated; FL1, green fluorescence;
FL3, red fluorescence. |
TMZ induces autophagy via pATM, which is
MLH1-dependent
TMZ failed to induce the phosphorylation of AMPK or
ULK1 when used in combination with 10 μM KU-55933. The
TMZ+KU-55933 group had lower expression levels of LC3B-I and
LC3B-II compared with the TMZ or the control group (Fig. 2A). Consistently, the inhibition of
ATM resulted in fewer AVOs in the TMZ+KU-55933 group compared with
the TMZ group (9.24±0.38%, vs. 29.90±2.14%, respectively;
P<0.001; Fig. 2B). The levels
of LC3B cleavage and AVOs were also reduced in the KU-55933 group
compared with the control group (Fig.
2A and B). Therefore, TMZ-induced autophagy and the
phosphorylation of AMPK were ATM-dependent. Subsequently, the
association between MLH1 and ATM was investigated. Compared with
the significant phosphorylation of ATM in the TMZ group, the knock
down of MLH1 inhibited the phosphorylation of ATM in the MLH1 siRNA
and the TMZ+MLH1 siRNA groups (Fig.
2C), which indicated that the TMZ-induced phosphorylation of
ATM was MLH1-dependent.
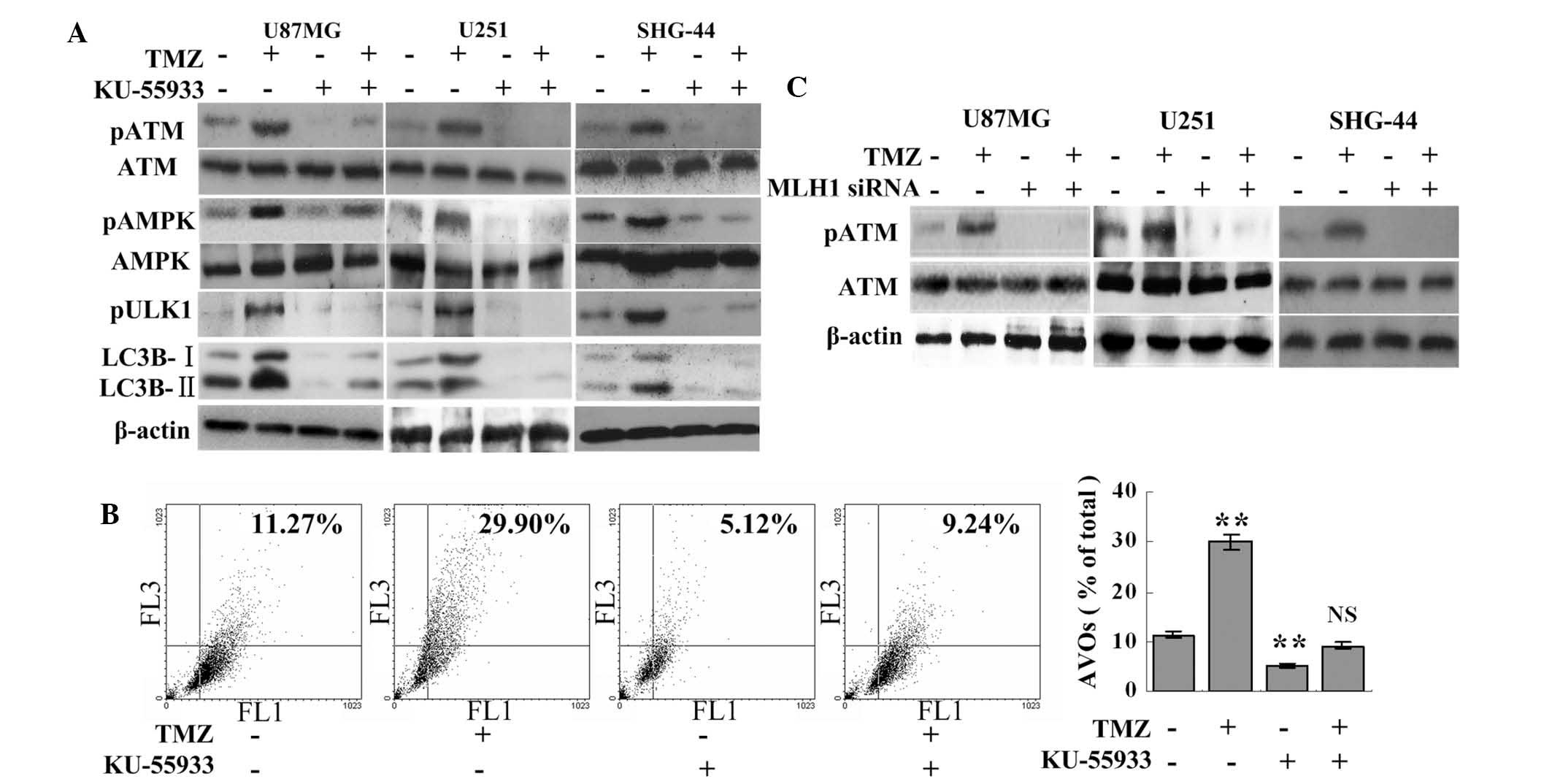 | Figure 2TMZ induces autophagy via pATM, which
is MLH1-dependent. (A) U87MG, U251 and SHG-44 cells were treated
with 100 μM TMZ and/or 10 μM KU-55933, as indicated,
for 72 h. Subsequently, the cells were collected for western blot
analysis. dimethyl sulfoxide was used as a negative control. (B)
U87MG cells were treated with 100 μM TMZ and/or 10 μM
KU-55933, as indicated, for 72 h. Following treatments, the cells
were collected and the number of AVOs were detected using flow
cytometric analysis. (C) Glioma cells and MLH1-knockdown glioma
cells (100 μM MLH1 siRNA for 48 h) were incubated with 100
μM TMZ or vehicle (DMSO) for 72 h. Subsequently, the levels
of pATM were assessed using western blot analysis. The data are
expressed as the mean ± standard deviation (**P<0.01,
vs. control group; n=3 for each group). NS, not statistically
significant compared with the control group; MLH1, mutL homolog 1;
ATM, ataxia telangiectsia; p, phosphorylated; TMZ, temozolomide;
siRNA, small interfering RNA; AMPK, adenosine
monophosphate-activated protein kinase; ULK, unc-51-like autophagy
activating kinase 1; LC3B, microtubule-associated protein 1 light
chain 3 β; AVO, acidic vesicular organelle; FL1, green
fluorescence; FL3, red fluorescence. |
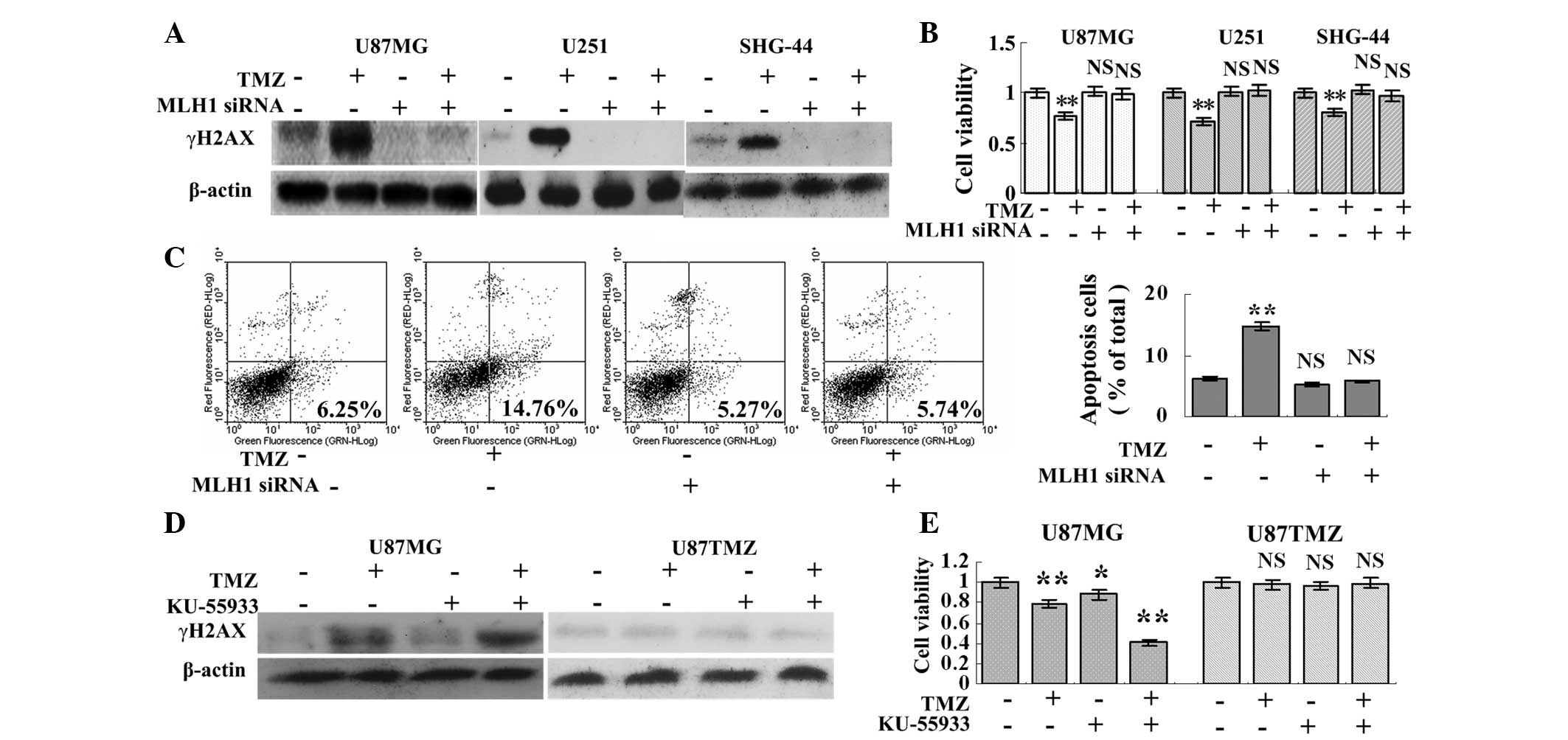
Knock down of MLH1 leads to TMZ
resistance
Since autophagy is critical for tumor survival, the
effect of MLH1 knock down on TMZ cytotoxicity was evaluated. The
knock down of MLH1 resulted in a significant decrease in the
expression of γH2AX (Fig. 3A). The
knock down of MLH1 also favored U87MG, U251 and SHG-44 cell growth
following treatment with TMZ. The MLH1 siRNA and TMZ+MLH1 siRNA
groups exhibited normal cell growth, while the cells in the TMZ
group exhibited the slowest rate of cell growth (Fig. 3B). Additionally, the levels of
apoptosis in the MLH1 siRNA and TMZ+MLH1 siRNA groups were
5.27±0.67% and 5.74±1.24%, respectively, which were not
significantly different compared with the control group
(6.25±1.15%), however, levels were lower compared with the TMZ
group (14.76±1.59%; Fig. 3C). This
indicated that knock down of MLH1 resulted in TMZ resistance,
despite inhibition of autophagy.
ATM inhibition augments TMZ cytotoxicity
in inherently TMZ-sensitive glioma cells
In the TMZ+KU-55933 group, increased expression of
γH2AX and reduced cell viability were observed compared with the
other groups in the U87MG cells. By contrast, TMZ and KU-55933 had
no effect on the expression of γH2AX or on the viability of the
U87TMZ cells, suggesting that the inhibition of ATM augmented TMZ
cytotoxicity in the inherently TMZ-sensitive cells (Fig. 3D and E).
AMPK inhibition disrupts autophagy and
augments TMZ cytotoxicity in inherently TMZ-sensitive glioma
cells
The inhibition of AMPK resulted in lower expression
levels of LC3B- and LC3B-II in the TMZ+compound C group compared
with the TMZ group, suggesting that the inhibition of AMPK
interrupted TMZ-induced autophagy (Fig. 4A and C). Since autophagy favors
cell survival, the present study investigated whether the
inhibition of AMPK augmented TMZ cytotoxicity. As expected,
compound C augmented TMZ cytotoxicity, which was indicated by an
increase in the expression of γH2AX and in the number of apoptotic
cells in the TMZ+compound C group compared with the other groups
(Fig. 4C and D). The cell
viability analysis also revealed that the slowest rate of cell
growth was observed in the TMZ+compound C group (Fig. 4B).
 | Figure 4AMPK inhibition decreases levels of
autophagy and augments TMZ cytotoxicity in inherently TMZ-sensitive
glioma cells. (A) U87MG, U251 and SHG-44 cells were treated with 5
μM compound C or vehicle (DMSO) for 72 h. The cells were
then harvested for western blot analysis to determine the levels of
pAMPKα (Thr172). (B–D) U87MG, U251 and SHG-44 cells were treated
with 100 μM TMZ and/or 5 μM compound C for 72 h. (B)
MTT assays were performed to assess the cell viability (n=6 for
each group) and (C) western blot analysis was performed to detect
the expression of LC3B and γH2AX. (D) U87MG cells were treated with
100 μM TMZ and/or 5 μM compound C for 72 h. Apoptosis
was detected using annexin-V-fluorescin isothiocyanate/propidium
iodide double staining (n=3 for each group). U87TMZ cells were
treated with 100 μM TMZ and/or 5 μMm compound C for
72 h. (E) Western blot analysis was performed to detect the
expression levels of pULK1 and LC3B. (F) MTT assays were performed
to assess the cell viability (n=6 for each group). The data are
expressed as the mean ± standard deviation (*P<0.05
and **P<0.01, vs. control group or indicated group).
NS, not statistically significant, MLH1, mutL homolog 1; TMZ,
temozolomide; siRNA, small interfering RNA; AMPK, adenosine
monophosphate-activated protein kinase; ULK, unc-51-like autophagy
activating kinase 1; LC3B, microtubule-associated protein 1 light
chain 3 β; p, phosphorylated. |
In the U87TMZ cells, the expression levels of pULK1
and LC3B cleavage in the TMZ group were indistinguishable from
those in the control group, suggesting that TMZ failed to increase
the levels of autophagy in the TMZ-resistant glioma cells. Although
compound C decreased LC3B cleavage and reduced cell viability in
the compound C and the TMZ+compound C groups, no significant
differences were observed in the levels of pULK1, LC3B cleavage or
cell viability between these two groups, suggesting that the
reduction in autophagy in the U87TMZ cells was not associated with
TMZ (Fig. 4E and F). These results
indicated that a reduction in autophagy using AMPK inhibitors only
augmented TMZ cytotoxicity in inherently TMZ-sensitive tumor
lines.
Discussion
Following 6-thioguanine or 5-fluorouracil treatment,
MLH1 is involved in G2/M cell cycle arrest for damage repair and,
at a later stage, in the induction of autophagy for irreparable DNA
damage (17–19). The present study indicated that
MLH1 is also important in TMZ-induced autophagy.
AMPK is a conserved sensor of intracellular energy,
which is activated in response to low nutrient availability and
cellular stresses and is involved in the initiation of
autophagosome formation by interacting with the ULK1
autophagy-initiating kinase (9,10).
Following DNA damage or nutrient restriction, pATM leads to the
phosphorylation of AMPK, indicating that ATM is an upstream kinase
of AMPK (11,12). The results from the present study
demonstrated that TMZ-induced AMPK phosphorylation was regulated by
ATM. Following ATM inhibition, TMZ failed to induce the
phosphorylation of AMPK or ULK1, which led to decreased levels of
LC3B cleavage and AVO formation. The activation of AMPK-ULK1 was
also under MLH1 regulation, however the association between MLH1
and ATM remained to be determined.
Following the detection of DNA mismatches by MutS,
MLH1 is recruited to the foci, undergoes conformational changes
through cycles of ATP binding and hydrolysis and recruits and/or
activates downstream effector proteins (20–22).
The present study hypothesized that ATM was a downstream protein of
MLH1 and the results supported this hypothesis, as TMZ induced
autophagy via the phosphorylation of ATM in an MLH1-dependent
manner.
Notably, knock down of MLH1 and inhibition of
ATM-AMPK decreased autophagy, however, the opposite effect was
observed on TMZ cytotoxicity. TMZ-induced O6MeG/T
mismatches are firstly bound and recognized by MutS, which
subsequently recruits MutL to the foci (21,23).
The MutS-MutL complex excises the mispaired thymine, while
O6MeG persists in the template strand (3). Such failed replication cycles with
the repeated reinsertion and excision of thymine results in
replication fork collapse and DSBs (3). MLH1 is a major component of the MMR
pathway and MLH1 deficiency leads to failure of the MMR pathway to
recognize the O6MeG/T mismatches, leading to TMZ
resistance (3,24,25).
By contrast, ATM-AMPK had no effect on the failed cell cycles or on
the formation of DSB, however, it was involved in autophagy.
Therefore, the inhibition of ATM-AMPK decreased autophagy and
increased the therapeutic efficacy of TMZ in the inherently
TMZ-sensitive cells. Kanzawa et al demonstrated that
inhibiting autophagy at an early stage using 3-methyladenine
suppresses the characteristic recruitment of LC3 to autophagosome
membranes and rescues tumor cells from cell death (26). Whether MLH1 is involved in the
localization of LC3 requires further investigation.
The present study demonstrated for the first time,
to the best of our knowledge, that TMZ induced autophagy through
the ATM-AMPK pathways, which occured in an MLH1-dependent manner.
Although the knock down of MLH1 and inhibition of ATM-AMPK
decreased TMZ-induced autophagy, MLH1 knock down led to
TMZ-resistance in the glioma cells, while ATM-AMPK inhibition
augmented TMZ cytotoxicity.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81302201), the China Postdoctoral
Science Foundation (no. 2012M512182) and the Guangdong Natural
Science Foundation (no. S2012040006588).
Abbreviations:
|
TMZ
|
temozolomide
|
|
MMR
|
DNA mismatch repair
|
|
MLH1
|
mutL homolog 1
|
|
ATM
|
ataxia-telangiectasia mutated
|
|
AMPK
|
adenosine monophosphate-activated
protein kinase
|
|
ULK1
|
unc-51-like autophagy activating
kinase 1
|
References
|
1
|
Cen L, Carlson BL, Pokorny JL, et al:
Efficacy of protracted temozolomide dosing is limited in MGMT
unmethylated GBM xenograft models. Neuro Oncol. 15:735–746. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang G, Li LT, Xin Y, Zhang L, Liu YQ and
Zheng JN: Strategies to improve the killing of tumors using
temozolomide: targeting the DNA repair protein MGMT. Curr Med Chem.
19:3886–3892. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
4
|
Skiriute D, Vaitkiene P, Saferis V, et al:
MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in
glioblastoma. BMC Cancer. 12:2182012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Felsberg J, Thon N, Eigenbrod S, et al:
Promoter methylation and expression of MGMT and the DNA mismatch
repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and
recurrent glioblastomas. Int J Cancer. 129:659–670. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin CJ, Lee CC, Shih YL, et al:
Resveratrol enhances the therapeutic effect of temozolomide against
malignant glioma in vitro and in vivo by inhibiting autophagy. Free
Radic Biol Med. 52:377–391. 2012. View Article : Google Scholar
|
|
7
|
Katayama M, Kawaguchi T, Berger MS and
Pieper RO: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar
|
|
8
|
Natsumeda M, Aoki H, Miyahara H, et al:
Induction of autophagy in temozolomide treated malignant gliomas.
Neuropathology. 31:486–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong PM, Puente C, Ganley IG and Jiang X:
The ULK1 complex: sensing nutrient signals for autophagy
activation. Autophagy. 9:124–137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
She C, Zhu LQ, Zhen YF, Wang XD and Dong
QR: Activation of AMPK protects against hydrogen peroxide-induced
osteoblast apoptosis through autophagy induction and NADPH
maintenance: New implications for osteonecrosis treatment? Cell
Signal. 26:1–8. 2014. View Article : Google Scholar
|
|
11
|
Singh K, Matsuyama S, Drazba JA and
Almasan A: Autophagy-dependent senescence in response to DNA damage
and chronic apoptotic stress. Autophagy. 8:236–251. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Duan X, Ponomareva L, Veeranki S and
Choubey D: IFI16 induction by glucose restriction in human
fibroblasts contributes to autophagy through activation of the
ATM/AMPK/p53 pathway. PLoS One. 6:e195322011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brieger A, Adam R, Passmann S, Plotz G,
Zeuzem S and Trojan J: A CRM1-dependent nuclear export pathway is
involved in the regulation of MutLalpha subcellular localization.
Genes Chromosomes Cancer. 50:59–70. 2011. View Article : Google Scholar
|
|
14
|
Takahashi A and Ohnishi T: Does gammaH2AX
foci formation depend on the presence of DNA double strand breaks?
Cancer Lett. 229:171–179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
16
|
Wu J, Dang Y, Su W, et al: Molecular
cloning and characterization of rat LC3A and LC3B - two novel
markers of autophagosome. Biochem Biophys Res Commun. 339:437–442.
2006. View Article : Google Scholar
|
|
17
|
Zeng X and Kinsella TJ: BNIP3 is essential
for mediating 6-thio-guanine- and 5-fluorouracil-induced autophagy
following DNA mismatch repair processing. Cell Res. 20:665–675.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng X and Kinsella TJ: Mammalian target
of rapamycin and S6 kinase 1 positively regulate
6-thioguanine-induced autophagy. Cancer Res. 68:2384–2390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zeng X and Kinsella TJ: A novel role for
DNA mismatch repair and the autophagic processing of chemotherapy
drugs in human tumor cells. Autophagy. 3:368–370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson JR, Erdeniz N, Nguyen M, Dudley S
and Liskay RM: Conservation of functional asymmetry in the
mammalian MutLalpha ATPase. DNA Repair (Amst). 9:1209–1213. 2010.
View Article : Google Scholar
|
|
21
|
Polosina YY and Cupples CG: MutL:
conducting the cell’s response to mismatched and misaligned DNA.
Bioessays. 32:51–59. 2010. View Article : Google Scholar
|
|
22
|
Chahwan R, van Oers JM, Avdievich E, et
al: The ATPase activity of MLH1 is required to orchestrate DNA
double-strand breaks and end processing during class switch
recombination. J Exp Med. 209:671–678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quiros S, Roos WP and Kaina B: Processing
of O6-methylguanine into DNA double-strand breaks requires two
rounds of replication whereas apoptosis is also induced in
subsequent cell cycles. Cell Cycle. 9:168–178. 2010. View Article : Google Scholar
|
|
24
|
Sarkaria JN, Kitange GJ, James CD, et al:
Mechanisms of chemoresistance to alkylating agents in malignant
glioma. Clin Cancer Res. 14:2900–2908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taverna P, Liu L, Hanson AJ, Monks A and
Gerson SL: Characterization of MLH1 and MSH2 DNA mismatch repair
proteins in cell lines of the NCI anticancer drug screen. Cancer
Chemother Pharmacol. 46:507–516. 2000. View Article : Google Scholar
|
|
26
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|