Introduction
Chronic obstructive pulmonary disease (COPD) is
characterized by persistent airflow limitation, and is a major
public health problem (1). By the
year 2030, COPD is predicted to have become the seventh most common
disease and the fourth leading cause of mortality worldwide
(2). Cigarette smoking is the most
common risk factor for developing COPD, and the disease prevalence
in numerous countries is associated with the rate of smoking
(3). COPD also has a proven
association with persistent systemic inflammation, excessive
oxidative stress and abnormal immune function (4). These numerous effects highlight its
multidimensional nature; although COPD damage is primarily observed
in the lungs, it is often accompanied by disruptions to
cardiovascular, musculoskeletal, renal and intestinal function
(5).
Small intestinal integrity is maintained by
intercellular tight junctions (TJs) between luminal cells. TJs
regulate solute and ion transport through the paracellular pathway
(6), which is the dominant pathway
for passive transepithelial solute flow in the small intestine
(7). A number of compounds
regulate TJs, including transmembrane proteins such as occludin and
claudins, and peripheral membrane proteins such as zonula occludens
(ZOs). Claudins are integral TJ proteins regulating the size
selectivity of the barrier, occludin is considered to provide the
primary intercellular seal between epithelial cells, and ZOs
provide the critical connection between transmembrane TJ components
and the intracellular actin cytoskeleton (8).
TJ protein expression is regulated by extracellular
stimuli including oxidative stress responses and pro-inflammatory
cytokines (9,10). Hypoxia-inducible factor 1 (HIF-1),
a transcription factor regulating oxidative stress and apoptotic
genes (11), is a notable
modulator of TJ protein expression (12,13).
HIF-1 is selectively stabilized and activated during local hypoxia,
and coordinates the tissue’s adaptive response to hypoxia by
facilitating oxygen supply, glucose transport and angiogenesis
(14). Functionally, HIF-1 is a
heterodimer comprising an inducible HIF-1α subunit and a
constitutively expressed HIF-1β subunit. HIF-1α is upregulated in
response to hypoxia and accounts for the disruption of the
intestinal luminal structure (15).
Significantly, the respiratory and intestinal tracts
have a similar luminal structure; the two organs have an extensive
luminal surface area, selective epithelial barrier and overlying
mucus-gel layer (16). The
absorptive and barrier function of the intestinal epithelium is
sensitive to tissue hypoxia and reduced perfusion, and similarly,
tissue hypoxia is one of the most notable characteristics in COPD
pathology (17). Clinical findings
indicate a combination of disturbed intestinal integrity,
intestinal hyperpermeability and enhanced enterocyte loss in COPD
patients (18,19). However, the underlying molecular
mechanisms of this small intestinal damage remain unknown.
In the present study, we exposed rats to cigarette
smoke (CS) to induce COPD, which is a well-established model
(20). Gene expression analysis of
intestinal samples by reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) was used to evaluate the effect of CS on
oxidative stress and inflammation. The present study may provide an
essential molecular mechanism for intestinal barrier dysfunction
and hyperpermeability in patients with COPD.
Materials and methods
Ethics
Rats were used in strict accordance with the
protocol approved by the Animal Care Committee of Tianjin Medical
University, China.
Exposure of rats to cigarette smoke
Male Wistar rats weighing 180±20 g and aged 6 weeks
were purchased from the Model Animal Center of the Radiological
Medicine Research Institute, Chinese Academy of Medical Science
(Tianjin, China). Rats were housed in standard laboratory cages
(five per cage) and had free access to food and water. Rats were
randomly divided into two groups (n=15 per group) matched by body
weight; a CS-exposed group and an unexposed control group. As
described previously (21), the CS
group received whole-body exposure to the smoke of five unfiltered
cigarettes (Daqianmen™, tar ≤15 mg, nicotine ≤1.1 mg and CO ≤13 mg)
for 30 min twice daily (before 9 am and after 5 pm), every day for
14 weeks inside a 0.6 m3 custom plexiglass chamber (made
in-house), which included five ventilation holes. Naïve animals
underwent an identical protocol but without CS exposure. Smoke
contamination between the study groups was prevented by using
different chambers for each.
Total RNA isolation
Following treatment, rats were anesthetized and
sacrificed. The abdominal cavity was opened, and the duodenum was
excised, rinsed in ice-cold phosphate-buffered saline (pH 7.4;
Sangon Biotech, Shanghai, China), frozen in liquid nitrogen and
stored at −80°C until analysis. RNA was extracted from duodenum
tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA).
Extract yield and quality were determined by measuring absorbance
at 260 and 280 nm with the MaestroNano Micro-Volume
spectrophotometer (Maestrogen, Inc., Las Vegas, NV, USA). The
absorbance ratio at 260:280 was between 1.8 and 2.0. The RNA was
subsequently reverse transcribed into complementary DNA (cDNA).
RT-qPCR
Messenger RNA (mRNA; 3 μg) was reverse
transcribed with oligo (dT) primer for 1 h at 50°C using a
TIANScript RT kit (Tiangen Biotech Co., Ltd, Beijing, China)
according to the manufacturer’s instructions. The cDNA served as
templates for RT-qPCR, which was performed using SYBR-Green PCR
core reagents (Bio-Rad Laboratories, Hercules, CA, USA). Specific
gene primers were designed using Primer-Quest SM software available
at http://www.idtdna.com/Scitools/Applications/PrimerQuest/
(Integrated DNA Technologies, Inc., Coralville, IA, USA), then
commercially produced (BGI Tech, Shenzhen, Guangdong, China;
Table I). DNA amplifications were
performed on a CFX96 real-time system (Bio-Rad Laboratories) under
the following reaction conditions: an initial heating cycle of 95°C
for 2 min; 40 cycles alternating between denaturation at 95°C for
25 sec and primer annealing at 60°C for 25 sec; and extension at
72°C for 20 sec. Melt curves clarified the identity of amplicons,
and the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase
(GAPDH), served as an internal control. The relative mRNA
expression of targeted genes was calculated by the comparative
threshold cycle (CT) method normalized to GAPDH mRNA in the same
sample. Briefly, specific ΔCt was calculated as follows: ΔCt=
(Cttarget)-(CtGAPDH); relative expression was defined
as: 2−ΔCt.
 | Table IDNA primer sequences for quantitative
reverse transcription-quantitative polymerase chain reaction. |
Table I
DNA primer sequences for quantitative
reverse transcription-quantitative polymerase chain reaction.
| Gene | Forward primer | Reverse primer |
|---|
| GAPDH |
5′-TGGAGTCTACTGGCGTCTTC-3′ |
5′-TTCACACCCATCACAAACATG-3′ |
| claudin-1 |
5′-TGTCCACCATTGGCATGAAG-3′ |
5′-GCCACTAATGTCGCCAGACC-3′ |
| claudin-2 |
5′-ACAGCACTGGCATCACCCA-3′ |
5′-GCGAGGACATTGCACTGGAT-3′ |
| claudin-4 |
5′-AAGGCCAAGGTCATGATCACAG-3′ |
5′-GAAGTCGCGGATGACGTTGT-3′ |
| occludin |
5′-CTACTCCTCCAACGGCAAAG-3′ |
5′-AGTCATCCACGGACAAGGTC-3′ |
| zo-1 |
5′-ATTCAGTTCGCTCCCATGAC-3′ |
5′-GCTGTGGAGACTGTGTGGAA-3′ |
| HIF-1α |
5′-AAGAAACCGCCTATGACGTG-3′ |
5′-CCACCTCTTTTTGCAAGCAT-3′ |
| bax |
5′-CCAGGACGCATCCACCAAGAAGC-3′ |
5′-TGCCACACGGAAGAAGACCTCTCG-3′ |
| bcl-2 |
5′-GGATGACTTCTCTCGTCGCTACCGT-3′ |
5′-CGAGTGAGGATGTGCATGAA-3′ |
| SOD |
5′-GCAGAAGGCAAGCGGTGAAC-3′ |
5′-TCACACCACAAGCCAAGCGG-3′ |
|
p22phox |
5′-AAGTACCTGACCGCTGTGG-3′ |
5′-AGGTAGATCACACTGGCAATG-3′ |
| nox2 |
5′-GGCTGTGAATGAGGGACTC-3′ |
5′-CCAGTGCTGACCCAAGAAG-3′ |
| TNF-α |
5′-CGTCGTAGCAAACCACCAAG-3′ |
5′-CACAGAGCAATGACTCCAAAG-3′ |
| NF-κB |
5′-AGCCCTATGCCTTTTCAACAT-3′ |
5′-CACTCCTGGGTCTGTGTTGTT-3′ |
Statistical analysis
Results are presented as the means ± SEM.
Statistical comparisons were made using the one-tailed Student’s
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Exposure to CS affects oxidative
stress-related genes
Both patients with COPD and animals exposed to CS
demonstrate oxidative stress and inflammation systemically
(4,22). In this study, we exposed rats to CS
and then evaluated the effects of CS exposure on the expression of
genes that are associated with oxidative stress and inflammation in
the small intestine of rats by RT-qPCR. There was a significant
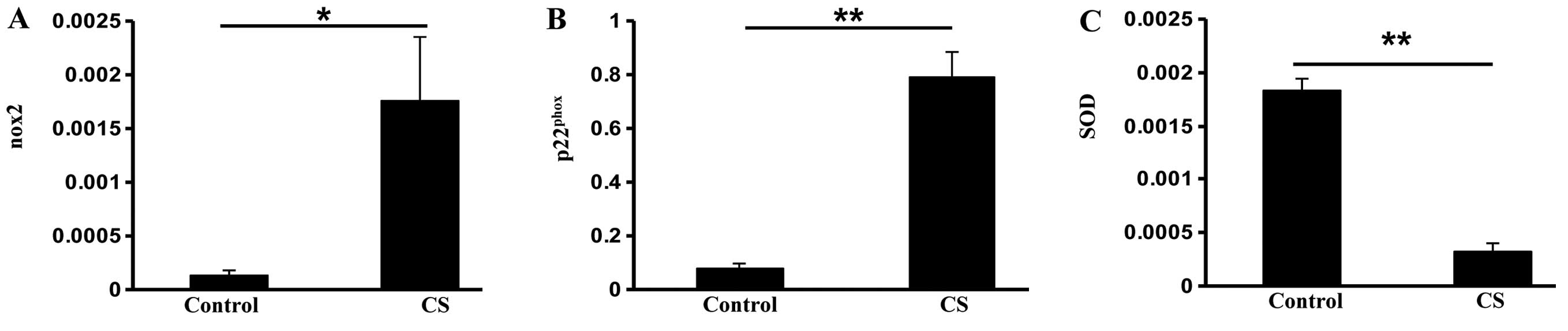
increase in the expression of the nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase subunits nox2 (P<0.05) and
p22phox (P<0.01) in the CS group (Fig. 1A and B). However, the antioxidant
enzyme super-oxide dismutase (SOD) was reduced in the CS group
compared with the control group (P<0.01; Fig. 1C), indicating that CS exposure
increases oxidative stress in the small intestine.
To investigate the correlation between CS-associated
oxidative stress and intestinal apoptosis, we then measured
pro-apoptotic bax and anti-apoptotic bcl-2 gene expression. The bax
mRNA expression was over five times greater in the CS group
compared with the control group (P<0.01; Fig. 2A). In addition, bcl-2 expression
was significantly reduced in the CS group (P<0.01; Fig. 2B). These data suggested that CS
exposure induces apoptosis in intestinal cells.
The transcription factor NF-κB was evaluated to
assess small intestinal inflammation in the study groups. NF-κB
expression was higher in the CS group compared with the control
group, but this change was not significant (P=0.09; Fig. 3A). There was no significant
difference in the expression of inflammatory cytokine tumor
necrosis factor α (TNF-α), which is controlled by NF-κB (23), between the two groups (Fig. 3B). These data together suggested
that CS exposure induces oxidative stress and apoptosis in the
small intestine.
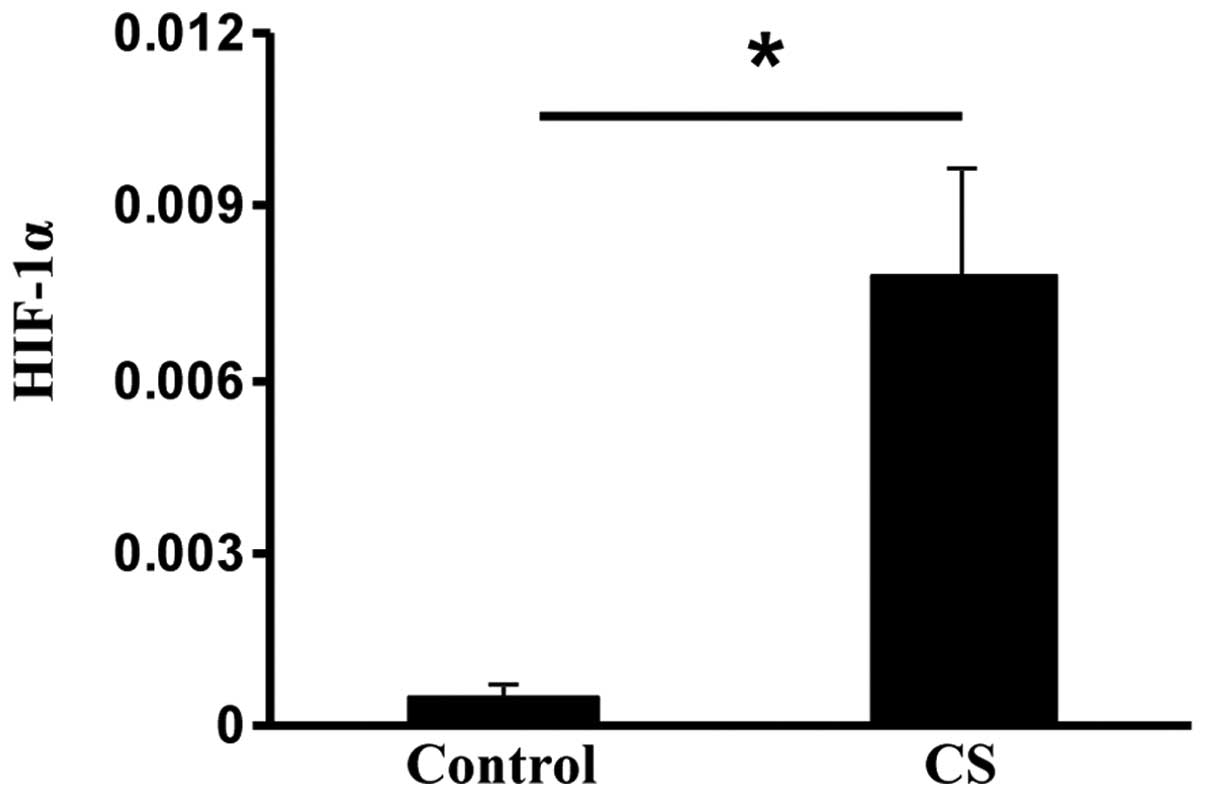
Exposure to CS upregulates HIF-1α
HIF-1α is usually elevated during hypoxic stress,
but it may also be induced by oxidative stress (24). The HIF-1α subunit is selectively
stabilized and primarily determines HIF-1 activity (14). Compared with the control rats,
HIF-1α expression was significantly increased in the CS rats
(P<0.05; Fig. 4). These data
suggested that CS exposure may induce oxidative stress by
upregulating HIF-1 in the small intestine.
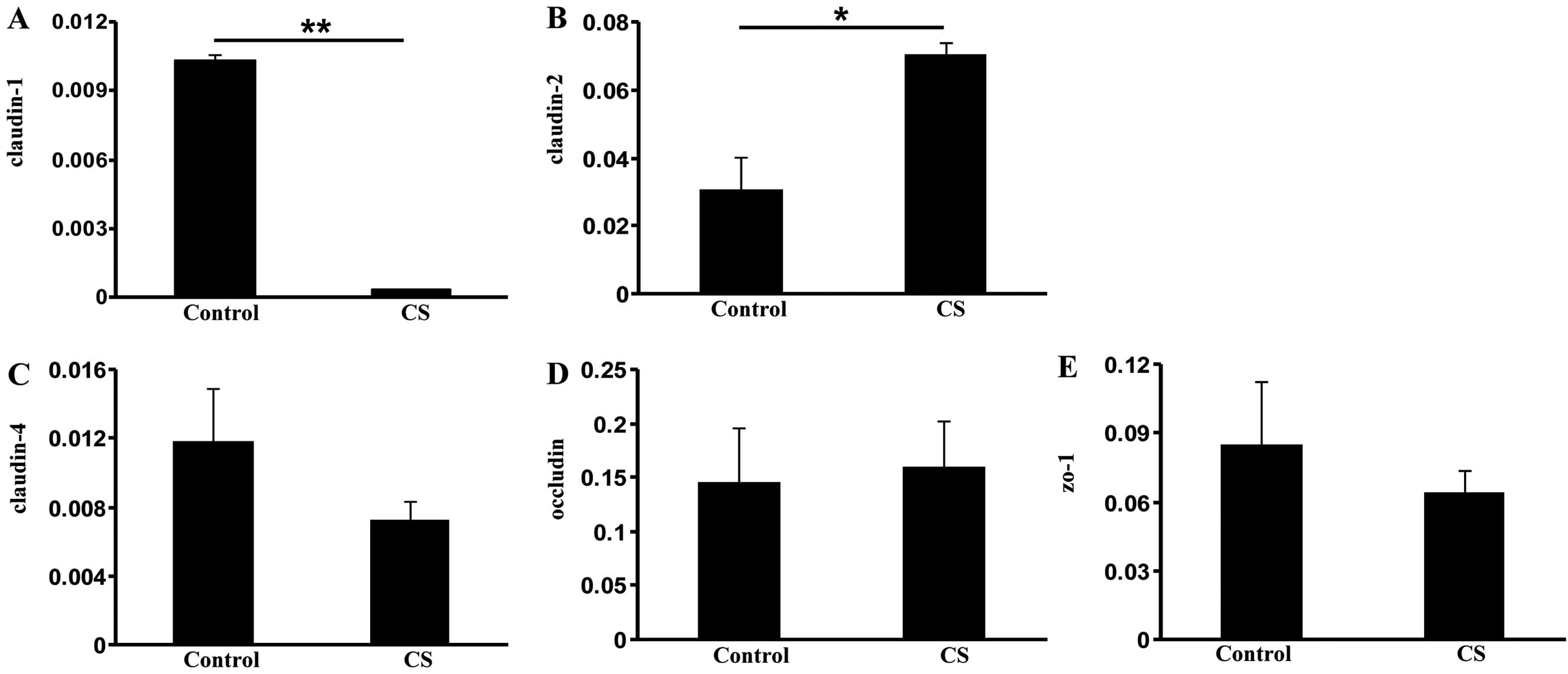
Exposure to CS alters expression of TJ
protein genes
The activation of HIF-1 is associated with the
disruption of TJs (25). To gain
insight into the mechanism of increased intestinal permeability in
COPD, we measured the expression of several TJ components:
claudin-1, claudin-2, claudin-4, occludin and zo-1. Compared with
the control group, the CS group demonstrated significantly reduced
claudin-1 expression (P<0.01; Fig.
5A), indicating that CS exposure loosens the TJ of intestinal
luminal cells (26). By contrast,
elevated expression of claudin-2 (P<0.05), a leaky protein
playing an opposing role to claudin-1 (27), was observed in the CS group
compared with the control group (Fig.
5B). However, no significant changes in claudin-4, occludin and
zo-1 expression were observed (Fig.
5C–E). These data suggest that CS exposure increases intestinal
permeability through the dysregulation of TJ components.
Discussion
Patients with COPD exhibit disrupted intestinal
integrity. To highlight the mechanisms underlying this disorder, we
used a CS model, which revealed that CS exposure increases
oxidative stress and induces apoptosis in the rat small intestine,
while the inflammatory cytokine TNF-α levels remained unchanged.
Our data suggested that the disrupted intestinal barrier in COPD
may be associated with HIF-1α elevation, which causes dysregulation
of TJ components.
Tissue hypoxia is a key player in a number of the
extrapulmonary comorbidities occurring in COPD (17). This hypoxia is primarily driven by
a ventilation/perfusion mismatch resulting from progressive airflow
limitation and emphysematous destruction of the pulmonary capillary
bed. The risk of alveolar hypoxia and subsequent hypoxemia
increases as the disease progresses (1). Hypoxia caused by COPD greatly impacts
the expression of oxygen-dependent genes, including HIF-1α. Aside
from oxygen-dependent regulation, HIF-1α expression is also
regulated by growth factors and free radicals. Studies reveal that
nicotine exposure, a significant component of cigarettes, mimics
hypoxic effects by increasing the generation of mitochondrial
reactive oxidative species (ROS) and thus stimulates HIF-1α
accumulation and activity (28).
An oxidative stress response frequently follows smoke exposure, and
potentially, hypoxic stimulation in the CS exposure model worsened
the oxidative stress response in the small intestine, resulting in
HIF-1α upregulation.
The roles of HIF-1α in the regulation of barrier
function remain controversial. One study suggests that HIF-1α is a
protective factor for intestinal barrier function, as demonstrated
in experimental murine colitis (29). However, another study reveals that
HIF-1α injures the barrier function and TJs (12). Prolonged intestinal HIF-1α exposure
is associated with potential ischemia/reperfusion-induced gut
mucosal injury, which contributes to gut barrier function loss,
bacterial translocation, apoptosis, gut-derived inflammatory
response and resulting villous injury (30). The adverse effects of HIF-1α are
apparently mediated by intestinal bacteria or bacterial products
(31). Hence, HIF-1α appears to be
involved intestinal barrier regulation, but its role, whether
protective or injurious, may depend on the exact physiological
conditions. Reportedly, HIF-1α upregulation is coupled to a
reduction in zo-1 protein concentration and dislocation of zo-1
from the TJ, which enables redistribution of other TJ proteins,
including claudin-1 (13). HIF-1α
suppression is correlated with attenuated intestinal barrier
dysfunction and morphologic redistribution of TJ proteins zo-1,
occludin and claudin-1, triggered by pro-inflammatory cytokines
(32). Therefore, HIF-1α may cause
small intestinal epithelial barrier damage and hyperpermeability
through TJ protein regulation.
Only claudin-1 and claudin-2 were affected by CS
exposure in the present study, suggesting that CS selectively
affects different TJ components. This selective effect has been
previously observed (33). In a
side-stream smoking study, increased TJ protein expression and
reduced inflammation were observed in the large intestine (34). The divergence between these studies
may result from the different smoke simulation methods and variable
effect on the different intestinal segments evaluated by the
studies.
Increased small intestine apoptosis following CS
exposure has also been observed by other groups (35). HIF-1α induces apoptosis through an
overexpression of pro-apoptotic proteins at the transcriptional
level (36), which bind and
inhibit the anti-apoptotic protein bcl-2 (37). Bcl-2 expression is correlated with
intestinal damage and TJ breakdown (38). However, it is unclear whether
intestinal cell apoptosis is the primary event associated with CS
or whether it is induced indirectly by HIF-1α. Cell apoptosis can
be induced by oxidative stress, which may impair intestinal
epithelial integrity and TJs. ROS-induced damage represents an
essential mechanism contributing to the TJ barrier defect (9). The antioxidant enzyme SOD is the
first line of tissue defense against ROS. SOD expression increased
in the rat small intestine following CS exposure in the present
study, in line with a previous study (39). This may indicate a decreased
radical-scavenging ability of the intestine following smoke
exposure, increasing gut susceptibility to oxidative damage. In
addition, the NADPH oxidase subunits, nox2 and p22phox,
are elevated in the CS intestine; NADPH oxidase is potentially a
source of ROS in CS exposure. Notably, COPD development is
associated with NADPH oxidase increase (40), and this same redox status may occur
in the small intestine in patients with COPD. Future analysis of
intestinal protein expression using western blotting or
immunohistochemistry is required to confirm this hypothesis.
In conclusion, CS exposure elevates oxidative
stress, apoptosis and TJ component dysregulation, potentially
mediated by HIF-1α. These data provide new insight into the
mechanisms behind cigarette smoke and associated intestinal mucosal
disruption in COPD patients.
Acknowledgments
This study was supported by the Natural Science
Foundation of Tianjin City (13JCYBJC22400 and 13JCYBJC40000) and
the National Natural Science Foundation of China (81270144).
References
|
1
|
Vestbo J, Hurd SS, Agusti AG, et al:
Global strategy for the diagnosis, management, and prevention of
chronic obstructive pulmonary disease: GOLD executive summary. Am J
Respir Crit Care Med. 187:347–365. 2013. View Article : Google Scholar
|
|
2
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4422006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pauwels RA and Rabe KF: Burden and
clinical features of chronic obstructive pulmonary disease (COPD).
Lancet. 364:613–620. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Celli BR: Update on the management of
COPD. Chest. 133:1451–1462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Divo M, Cote C, de Torres JP, et al:
Comorbidities and risk of mortality in patients with chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
186:155–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsukita S, Furuse M and Itoh M:
Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol.
2:285–293. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anderson JM and Van Itallie CM: Tight
junctions and the molecular basis for regulation of paracellular
permeability. Am J Physiol. 269:G467–G475. 1995.PubMed/NCBI
|
|
8
|
Mitic LL, Van Itallie CM and Anderson JM:
Molecular physiology and pathophysiology of tight junctions I.
Tight junction structure and function: lessons from mutant animals
and proteins. Am J Physiol Gastrointest Liver Physiol.
279:G250–G254. 2000.PubMed/NCBI
|
|
9
|
Zhu H and Li YR: Oxidative stress and
redox signaling mechanisms of inflammatory bowel disease: updated
experimental and clinical evidence. Exp Biol Med (Maywood).
237:474–480. 2012. View Article : Google Scholar
|
|
10
|
Bruewer M, Luegering A, Kucharzik T, et
al: Proinflammatory cytokines disrupt epithelial barrier function
by apoptosis-independent mechanisms. J Immunol. 171:6164–6172.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee KA, Roth RA and LaPres JJ: Hypoxia,
drug therapy and toxicity. Pharmacol Ther. 113:229–246. 2007.
View Article : Google Scholar
|
|
12
|
Rosenberger P, Khoury J, Kong T,
Weissmuller T, Robinson AM and Colgan SP: Identification of
vasodilator-stimulated phosphoprotein (VASP) as an HIF-regulated
tissue permeability factor during hypoxia. FASEB J. 21:2613–2621.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nico B, Mangieri D, Crivellato E, et al:
HIF activation and VEGF overexpression are coupled with ZO-1
up-phosphorylation in the brain of dystrophic mdx mouse. Brain
Pathol. 17:399–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)alpha: its protein stability
and biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Semenza GL: Regulation of oxygen
homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda).
24:97–106. 2009. View Article : Google Scholar
|
|
16
|
Keely S, Talley NJ and Hansbro PM:
Pulmonary-intestinal cross-talk in mucosal inflammatory disease.
Mucosal Immunol. 5:7–18. 2012. View Article : Google Scholar
|
|
17
|
Kent BD, Mitchell PD and McNicholas WT:
Hypoxemia in patients with COPD: cause, effects, and disease
progression. Int J Chron Obstruct Pulmon Dis. 6:199–208.
2011.PubMed/NCBI
|
|
18
|
Rutten EP, Lenaerts K, Buurman WA and
Wouters EF: Disturbed intestinal integrity in patients with COPD:
effects of activities of daily living. Chest. 145:245–252. 2014.
View Article : Google Scholar
|
|
19
|
Beloborodova EI, Akimova LA, Kritskaia NG,
Asanova AV, Semenenko EV and Burkovskaia VA: Disturbed absorptive
function of small intestines in patients with chronic obstructive
pulmonary disease. Klin Med (Mosk). 90:54–59. 2012.In Russian.
|
|
20
|
Wright JL, Cosio M and Churg A: Animal
models of chronic obstructive pulmonary disease. Am J Physiol Lung
Cell Mol Physiol. 295:L1–L15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng J, Wang QS, Chiang A and Chen BY: The
effects of sleep hypoxia on coagulant factors and hepatic
inflammation in emphysematous rats. PLoS One. 5:e132012010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barreiro E, Peinado VI, Galdiz JB, et al:
Cigarette smoke-induced oxidative stress: A role in chronic
obstructive pulmonary disease skeletal muscle dysfunction. Am J
Respir Crit Care Med. 182:477–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fukushima H, Matsumoto A, Inuzuka H, et
al: SCF (Fbw7) modulates the NFkB signaling pathway by targeting
NFkB2 for ubiquitination and destruction. Cell Rep. 1:434–443.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang T, Leng YF, Zhang Y, Xue X and Kang
YQ: Oxidative stress and hypoxia-induced factor 1alpha expression
in gastric ischemia. World J Gastroenterol. 17:1915–1922. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Engelhardt S, Al-Ahmad AJ, Gassmann M and
Ogunshola OO: Hypoxia selectively disrupts brain microvascular
endothelial tight junction complexes through a hypoxia-inducible
factor-1 (HIF-1) dependent mechanism. J Cell Physiol.
229:1096–1105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Assimakopoulos SF, Tsamandas AC,
Tsiaoussis GI, et al: Altered intestinal tight junctions’
expression in patients with liver cirrhosis: a pathogenetic
mechanism of intestinal hyperpermeability. Eur J Clin Invest.
42:439–446. 2012. View Article : Google Scholar
|
|
27
|
Zhang YG, Wu S, Xia Y and Sun J:
Salmonella infection upregulates the leaky protein claudin-2 in
intestinal epithelial cells. PloS One. 8:e586062013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo L, Li L, Wang W, Pan Z, Zhou Q and Wu
Z: Mitochondrial reactive oxygen species mediates nicotine-induced
hypoxia-inducible factor-1alpha expression in human non-small cell
lung cancer cells. Biochim Biophys Acta. 1822:852–861. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robinson A, Keely S, Karhausen J, Gerich
ME, Furuta GT and Colgan SP: Mucosal protection by
hypoxia-inducible factor prolyl hydroxylase inhibition.
Gastroenterology. 134:145–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feinman R, Deitch EA, Watkins AC, et al:
HIF-1 mediates pathogenic inflammatory responses to intestinal
ischemia-reperfusion injury. Am J Physiol Gastrointest Liver
Physiol. 299:G833–G843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koury J, Deitch EA, Homma H, et al:
Persistent HIF-1alpha activation in gut ischemia/reperfusion
injury: potential role of bacteria and lipopolysaccharide. Shock.
22:270–277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao M, Wang P, Sun C, He W and Wang F:
Amelioration of IFN-gamma and TNF-alpha-induced intestinal
epithelial barrier dysfunction by berberine via suppression of
MLCK-MLC phosphorylation signaling pathway. PLoS One. 8:e619442013.
View Article : Google Scholar
|
|
33
|
McGilligan VE, Wallace JM, Heavey PM,
Ridley DL and Rowland IR: The effect of nicotine in vitro on the
integrity of tight junctions in Caco-2 cell monolayers. Food Chem
Toxicol. 45:1593–1598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang H, Zhao JX, Hu N, Ren J, Du M and Zhu
MJ: Side-stream smoking reduces intestinal inflammation and
increases expression of tight junction proteins. World J
Gastroenterol. 18:2180–2187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Verschuere S, Bracke KR, Demoor T, et al:
Cigarette smoking alters epithelial apoptosis and immune
composition in murine GALT. Lab Invest. 91:1056–1067. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sowter HM, Ratcliffe PJ, Watson P,
Greenberg AH and Harris AL: HIF-1-dependent regulation of hypoxic
induction of the cell death factors BNIP3 and NIX in human tumors.
Cancer Res. 61:6669–6673. 2001.PubMed/NCBI
|
|
37
|
Boyd JM, Malstrom S, Subramanian T, et al:
Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set
of cellular proteins. Cell. 79:341–351. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mazzon E and Cuzzocrea S: Role of
TNF-alpha in ileum tight junction alteration in mouse model of
restraint stress. Am J Physiol Gastrointest Liver Physiol.
294:G1268–G1280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Guo X, Ko JK, Mei QB and Cho CH:
Aggravating effect of cigarette smoke exposure on experimental
colitis is associated with leukotriene B(4) and reactive oxygen
metabolites. Digestion. 63:180–187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee IT and Yang CM: Role of NADPH
oxidase/ROS in pro-inflammatory mediators-induced airway and
pulmonary diseases. Biochem Pharmacol. 84:581–590. 2012. View Article : Google Scholar : PubMed/NCBI
|