Introduction
Connexin 43 (Cx43) is the predominant connexin in
the ventricles is a phosphoprotein, the properties of which are
affected by its phosphorylation state (1). Cx43 is primarily located in the
sarcolemma of cardiomyocytes, and sarcolemmal Cx43 is important in
electrical cell coupling through the formation of a gap junction
(1). A previous study demonstrated
that Cx43 was also present in the mitochondria of cardiomyocytes,
with phosphorylation of >80% of the mitochondrial Cx43 (mtCx43)
(2). In addition, a normal mtCx43
content is essential for ischemic preconditioning-induced
cardioprotection (3). It has been
reported that cardiomyocytes of heterozygous Cx43-deficient mice
have a functional deficit in diazoxide-induced cardioprotection
(4). By contrast, the
mitochondria-specific transgenic overexpression of Cx43 triggers
cytoprotection, induced by preconditioning (5). Increasing evidence has indicated that
mtCx43 is an important sensor of cardioprotective signals and may
be involved in cardiomyocyte pathophysiology. However, whether
mtCx43 is involved in the pathogenesis of dilated cardiomyopathy
(DCM) and its correlation with mitochondrial dysfunction in
cardiomyocytes remain to be fully elucidated.
DCM is the most prevalent type of cardiomyopathy
worldwide and mitochondrial dysfunction is reported to be the major
mechanism responsible for the development of this disease (6). In order to investigate the mechanism
underlying mitochondrial dysfunction in DCM, a rat model of DCM was
established in a previous study by daily oral administration of
furazolidone (FZD) for 30 weeks. This procedure resulted in
apparent mitochondrial dysfunction in the myocardium, accompanied
by increasing left ventricle dimensions and reduced systolic and
diastolic functions (7).
The aim of the present study was to investigate the
role of mtCx43 in mitochondrial dysfunction during the pathogenesis
of DCM. Therefore, the present study examined the expression and
phosphorylation state of mtCx43 in the FZD-treated rat heart and
cardiomyocytes, the role of protein kinase C (PKC)ε in the
phosphorylation of mtCx43 in rat cardiomyocytes and the impact of
mtCx43 suppression on PKCε activator-induced mitochondrial
protection in the FZD-treated cardiomyocytes.
Materials and methods
Animal model
The present study was approved by the Institutional
Animal Research and Ethics Committee of Xi’an Jiaotong University
School of Medicine (Xi’an, China). A total of 36 Sprague-Dawley
rats (3 weeks), weighing between 40 and 60 g, were provided by the
Animal Center of Xi’an Jiaotong University School of Medicine and
were randomly divided into two groups. The rats were housed
individually and provided with ad libitum access to food and
water in a temperature-controlled room (22±2°C), under a 12-h
light/dark cycle (lights on at 08:00 a.m.). The 24 rats in the FZD
group received 700 ppm orally administered FZD solution (Yunpeng
Pharmacy Co., Ltd, Shaanxi, China) dissolved in water daily for 30
weeks and the 12 rats in the control group received tap water only.
Echocardiographic investigations (iE33; Philips, Amsterdam,
Netherlands) were performed in order to identify whether the DCM
model was successfully established, as previously described
(7).
Cell culture and treatment
Primary culture of neonatal rat cardiomyocytes were
prepared and characterized, as previously described (7). The cardiomyocytes were sparsely
plated without cell-cell contact and were cultured in Dulbecco’s
modified Eagle’s medium (DMEM)/F12 (HyClone Laboratories, Inc.,
Waltham, MA, USA) containing 10% heat-inactivated fetal bovine
serum (FBS; Gibco-BRL, Carlsbad, CA, USA), 100 U/ml penicillin
(HyClone; Thermo Fisher Scientific, Waltham, MA, USA) and 100
μg/ml streptomycin sulfate (HyClone) at 37°C with 5%
CO2. Prior to treatment, the cells were starved for 24 h
in DMEM/F12 medium containing 0.5% FBS. Subsequently, the cells
were divided into four groups, as follows: Control group, incubated
with vehicle only; FZD group, incubated with 100 μmol/l FZD
(Sigma-Aldrich, St. Louis, MO, USA) for 48 h; PMA group, incubated
with 100 mmol/l phorbol-12-myristate-13-acetate (PMA;
Sigma-Aldrich), a specific PKC activator, for 60 min following 48 h
treatment with 100 μmol/l FZD; 18β-glycerrhetinic acid (GA)
group, successively treated with 100 μmol/l FZD for 48 h, 50
μmol/l GA (Sigma-Aldrich), a connexin channel inhibitor, for
4 h and 100 nmol/l PMA for 60 min.
Isolation of mitochondria
Mitochondria were isolated from the left ventricles
of the rats and the cultured cardiomyocytes through differential
centrifugation, as previously described (7). Briefly, the rats were narcotized with
30 mg/kg nembutal (Sigma-Aldrich) and the thorax was opened, in
order to excise the heart. The arterial and vessel tissues were
removed, and the ventricular tissues were retained and cut into 1
mm sections. The homogenates were centrifuged at 1,000 × g for 10
min and the supernatant was then centrifuged at 10,000 × g for 10
min. Subsequently, the sediment was suspended in isolation buffer
(300 mmol/l sucrose, 2 mmol/l HEPES, 0.1 mmol/l EGTA, pH 7.4;
Shanghai Genmed Gene Pharmaceutical Technology Co., Ltd., Shanghai,
China) and added to the top of 30% percoll solution (Sigma-Aldrich)
for centrifugation at 35,000 × g for 30 min. The mitochondrial
fraction was collected and washed twice using isolation buffer
through centrifugation at 8,000 × g for 5 min.
Mitochondrial membrane potential
assay
JC-1 dye (Invitrogen Life Technologies, Carlsbad,
CA, USA) was used to assess the mitochondrial membrane potential
(MMP) level through the measurement of quantitative fluorescence.
The freshly isolated rat myocardial mitochondria or cultured
cardiomyocytes were incubated at 37°C for 10 min in DMEM/F12 medium
containing 10 μg/ml JC-1. Subsequently, the mitochondria or
cardiomyocytes were rinsed twice with phosphate-buffered saline and
then scanned in a fluorescence microplate reader (Infinite M200;
Tecan Inc., Maennedorf, Switzerland) at 488 nm excitation and 535
nm or 590 nm emission, respectively.
Enzyme activities measurement
The activities of cytochrome c oxidase (COX)
and succinate dehydrogenase (SDH) were determined using COX and SDH
quantitative colorimetric assay kits (Genemed Co., Ltd, Shanghai
and Jiancheng Chemical Industrial Co., Ltd, Nanjing, China,
respectively) according to the manufacturer’s instructions. The
activity of COX was calculated according to changes in absorbance
at 550 nm, measured using a spectrophotometer. The activity of SDH
was determined by the speed of the 2,6-dichlorophenol-indophenol
reduction reaction (the change in absorbance value at the same
time), accompanied by the reduction of flavin adenine dinucleotide
(FAD) to FADH.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the rat left ventricle
tissues and cardiomyocytes using TRIzol reagent (Invitrogen Life
Technologies) and then 1 μg total RNA was reverse
transcribed into cDNA using a cDNA Synthesis kit (Fermentas; Thermo
Fisher Scientific), according to the manufacturer’s instructions.
The cDNA was amplified using the appropriate primers and the SYBR
Premix Ex Taq™ II (Takara Biotechnology Co., Ltd., Dalian, China).
qPCR reactions were carried out with an initial denaturation at
95°C for 30 sec, followed by 40 cycles consisting of: Denaturing at
95°C for 5 sec, annealing at 60°C for 30 sec, and extension at 72°C
for 30 sec, followed by a melting curve analysis, on an iCycler iQ™
Real Time PCR Detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The primers were as follows: Cx43, forward
5′CATGGGTGACTGGAG-3′ and reverse 5′-AGGACCCAGAAGCGCA-3′;
glyceraldahyde-3-phosphate dehydrogenase (GADPH), forward
5′-TTGTGATGGGTGTGAACC-3′ and reverse 5′-TTCTGAGTGGCAGTGATG-3′
(Takara Biotechnology Co., Ltd.). The amount of relative gene
expression was calculated by 2-ΔΔCt.
Western blot analysis
The proteins (20 μg) were electrophoresed on
8 or 10% SDS-PAGE (Xi’an Wolsen Bio-technology, Co., Ltd., Xi’an,
China) and transferred onto a polyvinylidene difluoride membrane
(Millipore, Bedford, MA, USA). The membrane was incubated with 5%
non-fat dry milk in Tris-buffered saline solution for 2 h, followed
by the addition of the following antibodies: Rabbit polyclonal Cx43
(1:500; cat. no. sc-9059, Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), rabbit polyclonal serine 368-phosphorylated Cx43 (p-S368
Cx43; 1:200; cat. no. sc-25165-R, Santa Cruz Biotechnology, Inc.),
rabbit monoclonal PKCε (1:1,000; cat. no. 2683, Cell Signaling
Technology, Inc., Danvers, MA, USA), rabbit monoclonal p-
myristoylated alanine-rich C kinase substrate (p-MARCKS; 1:1,000;
cat. no. 11992, Cell Signaling Technology, Inc.), mouse monoclonal
voltage-dependent anion channels (VDAC; 1:3,000; cat. no.
sc-390996, Santa Cruz Biotechnology, Inc.) or mouse monoclonal
β-actin (1:2,000; cat. no. sc-47778, Santa Cruz Biotechnology,
Inc.) at 4°C overnight. The membranes were then incubated with
peroxidase-conjugated secondary antibodies (1:4,000–1:10,000; cat.
nos. sc-2370/1, Santa Cruz Biotechnology) at room temperature for 2
h. The blots were visualized using an enhanced chemiluminescence
detection system (Pierce Biotechnology, Inc., Shaanxi, China).
β-actin and VDAC were used as loading control for the whole
cellular and mitochondrial proteins, respectively.
Statistical analysis
Data from three independent experiments are
expressed as the mean ± standard deviation and statistical analyses
were performed using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). Differences between the groups were evaluated using one-way
analysis of variance and a least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Mitochondrial dysfunction in the
myocardium
Freshly isolated mitochondria from the left
ventricles were stained with JC-1 to assess the MMP levels.
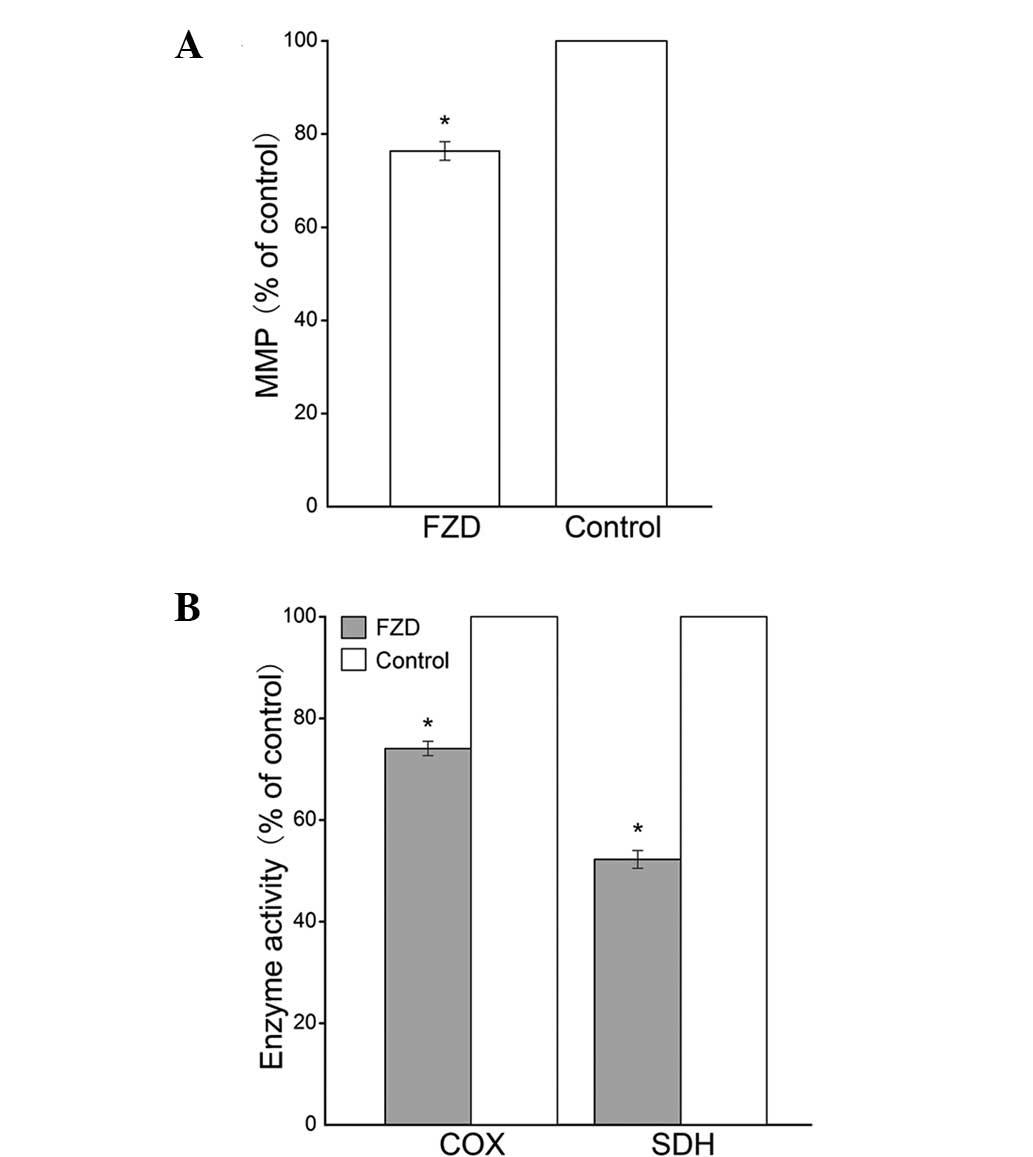
Compared with those in the control group, the MMP levels were
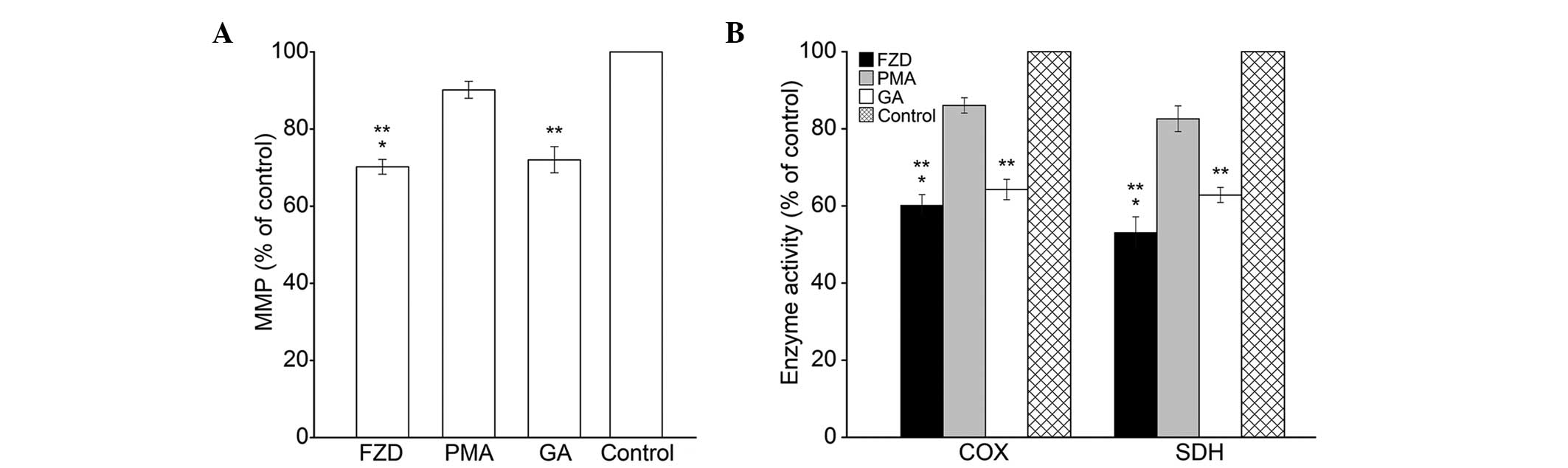
significantly decreased in the FZD group (P<0.05; Fig. 1A). In addition, the activities of
COX and SDH in the FZD group were significantly reduced (P<0.0;
Fig. 1B).
Expression of Cx43 in the myocardium and
myocardial mitochondria
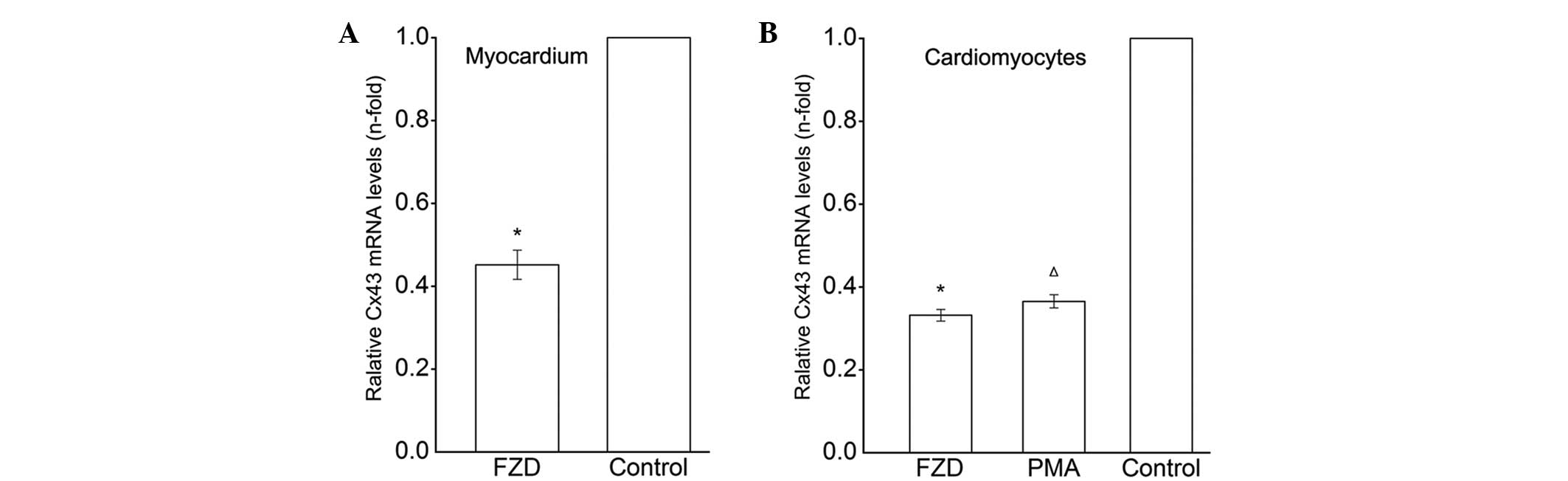
RT-qPCR analysis revealed that the mRNA expression
of Cx43 in the left ventricle myocardium was markedly downregulated
following treatment with FZD compared with that of the untreated
control (P<0.05; Fig. 2A). A
marked reduction in the mRNA expression levels of Cx43 was also
detected in the FZD-treated cardiomyoctes (P<0.05; Fig. 2B). Western blot analysis identified
three typical Cx43-specific bands in the myocardial protein
extracts. The overall expression of Cx43 in the FZD group was
significantly decreased compared with that of the control group
(P<0.05; Fig. 3). In addition,
the phosphorylation status of Cx43 was analyzed using western blot
analysis, which demonstrated that the immunoreactivity of p-S368
Cx43 was reduced by 37.7±4.4% in the myocardium following FZD
treatment compared with that of the untreated control (P<0.05;
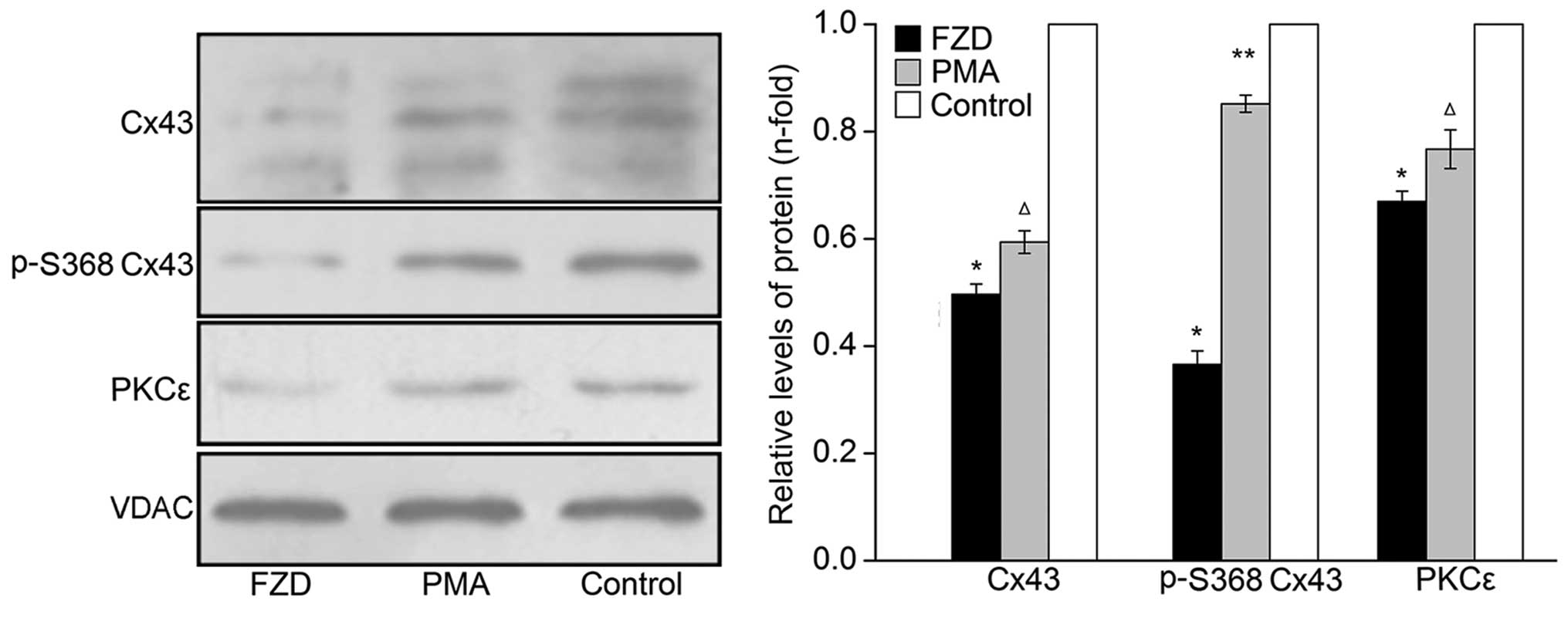
Fig. 3). As shown in Fig. 4, three typical Cx43-specific bands
were also detected in the myocardial mitochondrial protein extracts
and relatively low levels of overall Cx43 and p-S368 Cx43 were
observed in the FZD group compared with those in the control group
(P<0.05).
Changes of expression and activity of
PKCε in the myocardium and myocardial mitochondria
PKCε is an important cardioprotective mediator,
which interacts with Cx43 and is required for the phosphorylation
of Cx43 at the S368 PKC target site (8). In the present study, the expression
and activity of PKCε were examined using western blot analysis to
determine whether PKCε mediated the phosphorylation of Cx43 at S368
in the myocardium and mitochondria. MARCKS is a major substrate of
PKCε (9), therefore, p-MARCKS
levels were detected in order to assess PKCε activity. The results
showed that the immunoreactivity of PKCε in the myocardial protein
extracts was reduced by 25.1±2.7% in the FZD group compared with
that of the control group (P<0.05;Fig. 3), and the phosphorylation of MARCKS
was significantly decreased by 61.6±3.4% (P<0.05;Fig. 3). These changes indicated that FZD
induced a marked decrease of PKCε activity in the myocardium.
Furthermore, decreased protein expression of PKCε was detected in
the myocardial mitochondrial protein extracts following treatment
with FZD compared with that of the untreated control (P<0.05;
Fig. 4).
Changes of protein expression in
FZD-treated cardiomyocytes
In order to investigate the possible mechanism
underlying the mitochondrial dysfunction in the FZD-induced DCM rat
model, neonatal rat cardiomyocytes were cultured and incubated with
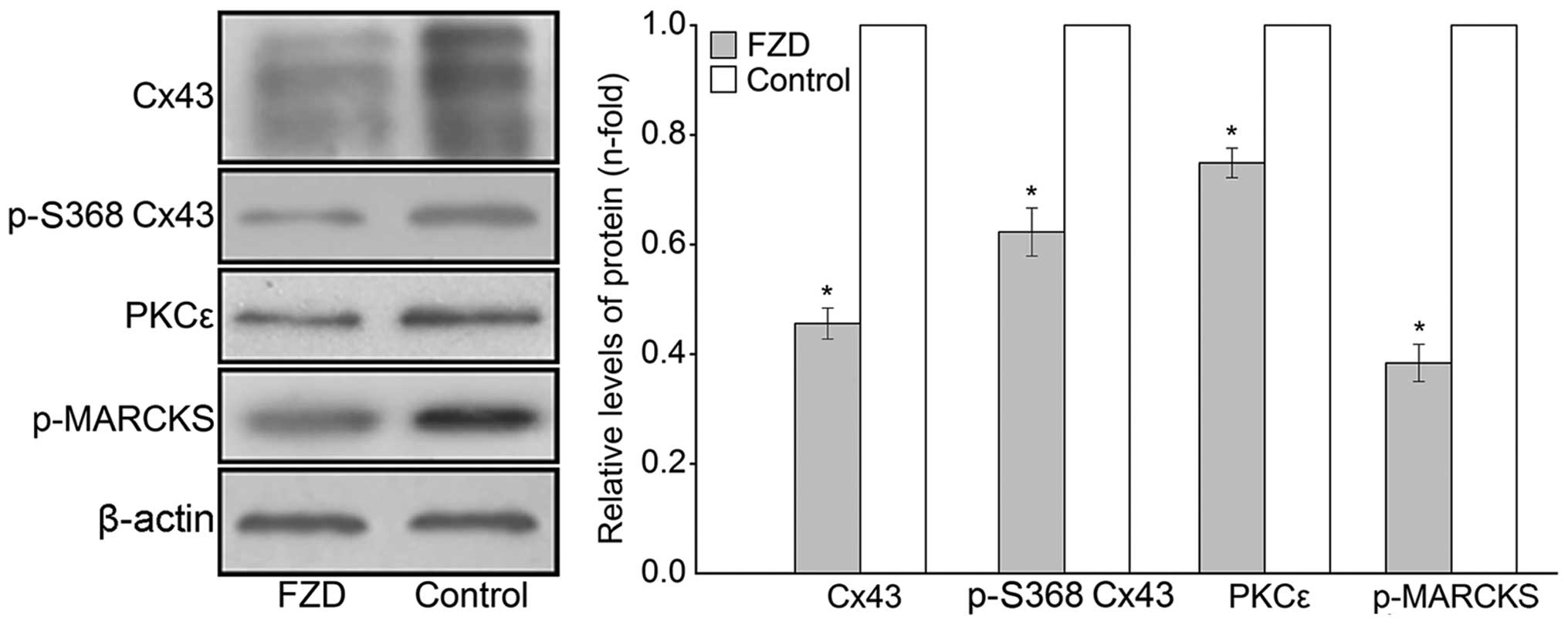
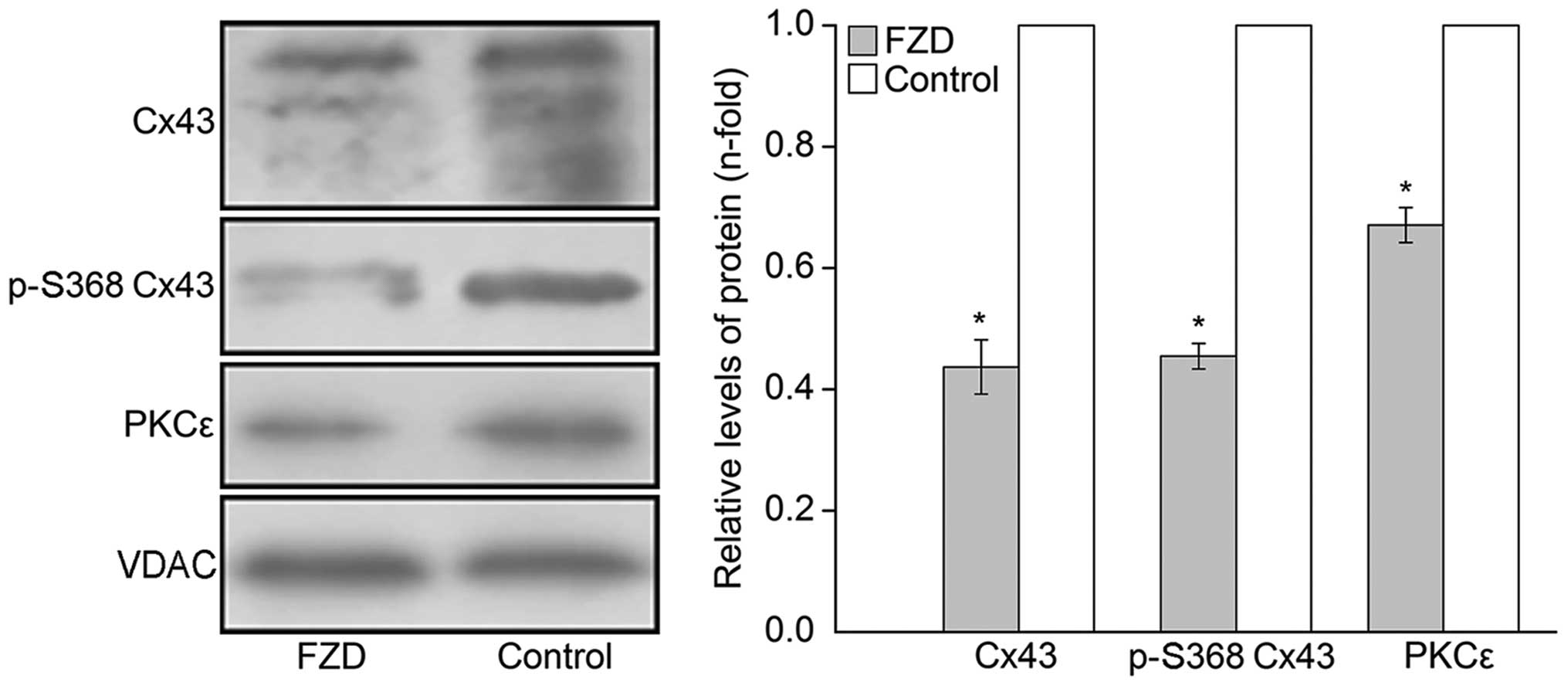
100 μmol/l FZD for 48 h. Western blot analyses revealed that
the protein levels of Cx43, p-S368 Cx43 and PKCε significantly
decreased in the the whole-cell and mitochondrial protein extracts
compared with those of the untreated control (P<0.05; Figs. 5 and 6). Furthermore, the phosphorylation of
MARCKS was downregulated by 43.6±2.3% in the cardiomyocytes
(P<0.05, Fig. 5).
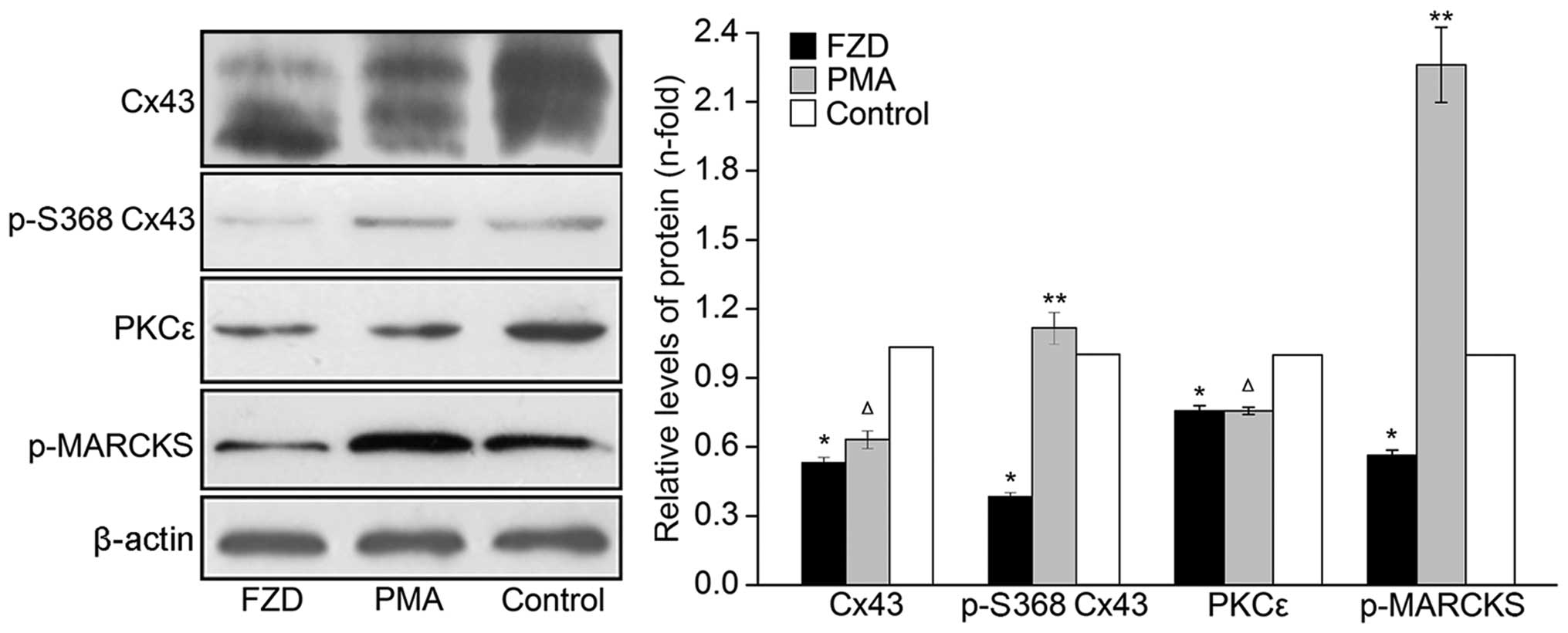 | Figure 5Western blot analysis of the protein
expression levels of Cx43, p-S368 Cx43, PKCε and p-MARCKS in the
rat cardiomyocytes of the FZD, PMA and control groups. Values are
presented as the mean ± standard deviation relative to β-actin.
*P<0.05 vs. control, **P<0.05 vs. FZD
and ΔP>0.05 vs. FZD. Cx43, connexin 43; p-S368 Cx43,
serine 368-phosphorylated Cx43; PKCε, protein kinase Cε; p-MARCKS,
phosphorylated myristoylated alanine-rich C kinase substrate; FZD,
furazolidone; PMA, phorbol-12-myristate-13-acetate. |
Effect of PKCε activation on the
phosphorylation of Cx43 at serine 368
As shown in Fig. 5,
the p-MARCKS immunoreactivity of the PMA group cells was enhanced
by 3.00±0.20-fold compared with that of the cells treated with FZD
only (P<0.05). In addition, the FZD-induced inhibition of Cx43
phosphorylation at S368 was eliminated by PMA in the cardiomyocytes
and mitochondria (P<0.05; Figs.
5 and 6). By contrast, no
significant differences were observed in the overall levels of Cx43
in either the cardiomyocyte or mitochondria between the PMA and FZD
groups (P>0.05; Figs. 5 and
6).
Role of mtCx43 in regulating
mitochondrial function
As shown in Fig. 7,
treatment with FZD resulted in a significant decrease in the level
of MMP and the activities of COX and SDH compared with those of the
control group (P<0.05). Notably, PMA treatment partially
reversed the FZD-induced decrease in MMP levels and COX and SDH
activities (P<0.05), and pretreatment with GA eradicated the
mitochondrial protective effects of PMA (P<0.05).
Discussion
The results of the present study demonstrated that
the overall expression levels of mtCx43 and p-S368 mtCx43 were
downregulated in the myocardial mitochondria of the DCM rat model
as well as the mitochondria of the FZD-treated cardiomyocytes,
which was accompanied by suppression of the activity of PKCε and
mitochondrial function. In addition, the PKCε activator, PMA,
partially reversed FZD-induced mitochondrial dysfunction via a
p-S368 mtCx43-dependent mechanism. These data demonstrated a novel
mechanism of mitochondrial dysfunction in the DCM rat model, which
may involve the PKCε-mediated phosphorylation of Cx43.
Cx43 is the predominant protein forming gap
junctions in the myocardium, which are important in intercellular
electrical and metabolic coupling (10). A previous study demonstrated that
the myocardial expression of Cx43 was significantly downregulated
and the phosphorylation of Cx43 was also reduced in patients with
non-ischemic cardiomyopathy, and the conduction disorders, which
occur due to these abnormalities, may account for arrhythmias in
non-ischemic cardiomyopathy (11).
There is increasing evidence for the presence of
Cx43 in the inner mitochondrial membrane of cardiomyocytes, where
its level may be upregulated by ischemic preconditioning (2). It has been reported that inhibiting
heat shock protein 90 only eliminates the upregulation of mtCx43,
but also ablates the cardioprotection induced by diazoxide
(3). Direct single-channel
patch-clamp recordings have revealed that the suppression of
myocardial mtCx43 inhibited the function of mitochondrial adenosine
triphosphate (ATP)-sensitive K+ channels, which serve as
important effectors of cytoprotective signaling (12,13).
Accordingly, mtCx43 is important in cardioprotection by ischemic
preconditioning via the modulation of mitochondrial ATP-sensitive
K+ channels. However, whether the level of mtCx43 was
altered and correlated with mitochondrial dysfunction in the
pathogenesis of DCM remained to be elucidated. The results of the
present study revealed that mtCx43 was significantly downregulated
in the DCM rat myocardium, with a reduction in the overall levels
of Cx43. Furthermore, the expression of p-S368 mtCx43 was reduced
by ~36.8%. In addition, the level of MMP and the activities of COX
and SDH were significantly decreased. These findings indicated that
the suppression of mtCx43 and/or its phosphorylation may have been
associated with mitochondrial dysfunction in the DCM rats.
Cx43 is a phosphoprotein, with several
phosphorylation sites targeted by different kinases (14). The phosphorylation of Cx43 at
different sites and by different kinases may have different
effects. Phosphorylation of Cx43 at serine 279/282 results in
impaired cell communication (15),
while enhanced dephosphorylation of Cx43 contributes to slow
conduction in heart failure via increased activation of
p21-activated kinase 1 (16). In
addition, the phosphorylation status of Cx43 also affects its
suppressive effect on cell proliferation (17). PKCε is essential for the
phosphorylation of Cx43 at S368 and is important in ischemic
preconditioning-induced cardioprotection by suppressing chemical
coupling via Cx43 gap junction modulation (8). Similar to Cx43, PKCε is also located
in the mitochondria, which suggests a possible interaction between
Cx43 and PKCε in the mitochondria (18). Increasing evidence indicates that
the phosphorylation status of Cx43 is not static, but fluctuates in
response to cell stress (19–22).
In the present study, decreased protein expression of PKCε was
observed in the myocardial and mitochondrial protein extracts of
the DCM rats, and the PKCε activities also decreased by ~60%.
Furthermore, the PKCε activator, PMA, partially reversed the
dephosphorylation of Cx43 at S368, indicating that the
downregulation of PKCε activity was involved in the FZD-induced
dephosphorylation of Cx43.
The results of the present study suggested that the
PMA-induced activation of PKCε attenuated mitochondrial dysfunction
in the cardiomyocytes. Further investigations demonstrated that
inactivation of Cx43 eliminated PMA-induced mitochondrial
protection. As the cardiomyocytes were cultured sparsely without
cell to cell contact, the targeted inactivation of mtCx43 may have
led to the suppression of PMA-induced mitochondrial protection
(23,24). These results suggested that
activation of PKCε may have attenuated mitochondrial dysfunction in
a mtCx43-dependent manner.
In cardiomyocytes, Cx43 is phosphorylated at
different sites by various kinases, including mitogen-associated
protein kinase (25), protein
kinase A (26), protein kinase B
(27), casein kinase 1 (28) and the src tyrosine kinase (29). Further studies are required to
elucidate whether the phosphorylation states of Cx43, at sites
other than S368, alter or are involved in the pathogenesis of DCM.
As a regulator of the mitochondrial physiology, cardiac mtCx43 may
target mitochondrial adenosine triphosphate-sensitive K+
channels (30). Furthermore,
reactive oxygen species formation is reported to be impaired in the
cardiomyocytes of Cx43−/− mice (4) and cytochrome c and
Ca2+ are induced to release from isolated mitochondria
by the Cx43 gap junction inhibitor (31). The present study demonstrated a
significant downregulation of p-S368 mtCx43 in the DCM rat
myocardium due to the suppression of PKCε activity; however, the
precise molecular mechanisms by which mtCx43 affects mitochondrial
function require further elucidation.
In conclusion, the present study revealed that the
phosphorylation of mtCx43 at S368 was suppressed in the myocardium
of DCM rats and was required for the mitochondrial protective
effects of the PKCε activator in FZD-treated cardiomyocytes. These
findings suggested a possible mechanism involved in mitochondrial
dysfunction in the pathogenesis of DCM and revealed a novel
mitochondrial protective role of the PKCε activator, PMA, which may
offer therapeutic potential for the treatment of DCM.
Acknowledgments
The present study was supported by The National
Science Foundation of China (grant no. 81170209) awarded to Jin
Wei.
References
|
1
|
Jeyaraman MM, Srisakuldee W, Nickel BE and
Kardami E: Connexin43 phosphorylation and cytoprotection in the
heart. Biochim Biophys Acta. 2009–2013:1818s2012.
|
|
2
|
Boengler K, Dodoni G, Rodriguez-Sinovas A,
et al: Connexin 43 in cardiomyocyte mitochondria and its increase
by ischemic preconditioning. Cardiovasc Res. 67:234–244. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rodriguez-Sinovas A, Boengler K,
Cabestrero A, et al: Translocation of connexin 43 to the inner
mitochondrial membrane of cardiomyocytes through the heat shock
protein 90-dependent TOM pathway and its importance for
cardioprotection. Circ Res. 99:93–101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heinzel FR, Luo Y, Li X, et al: Impairment
of diazoxide-induced formation of reactive oxygen species and loss
of cardioprotection in connexin 43 deficient mice. Circ Res.
97:583–586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu G, Haider HK, Porollo A and Ashraf M:
Mitochondria-specific transgenic overexpression of connexin-43
simulates preconditioning-induced cytoprotection of stem cells.
Cardiovasc Res. 88:277–286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jefferies JL and Towbin JA: Dilated
cardiomyopathy. Lancet. 375:752–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang M, Wei J, Shan H, et al:
Calreticulin-STAT3 Signaling Pathway Modulates Mitochondrial
Function in a Rat Model of Furazolidone-Induced Dilated
Cardiomyopathy. PLoS One. 8:e667792013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Naitoh K, Yano T, Miura T, et al: Roles of
Cx43-associated protein kinases in suppression of gap
junction-mediated chemical coupling by ischemic preconditioning. Am
J Physiol Heart Circ Physiol. 296:H396–H403. 2009. View Article : Google Scholar
|
|
9
|
Heidkamp MC, Iyengar R, Szotek EL, et al:
Protein kinase Cε-dependent MARCKS phosphorylation in neonatal and
adult rat ventricular myocytes. J Mol Cell Cardiol. 42:422–431.
2007. View Article : Google Scholar :
|
|
10
|
van Veen TA, van Rijen HV and Opthof T:
Cardiac gap junction channels: modulation of expression and channel
properties. Cardiovasc Res. 51:217–229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Glukhov AV, Fedorov VV, Kalish PW, et al:
Conduction remodeling in human end-stage nonischemic left
ventricular cardiomyopathy. Circulation. 125:1835–1847. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rottlaender D, Boengler K, Wolny M, et al:
Connexin 43 acts as a cytoprotective mediator of signal
transduction by stimulating mitochondrial KATP channels in mouse
cardiomyocytes. J Clin Invest. 120:1441–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahmad Waza A, Andrabi K and Ul Hussain M:
Adenosine-triphos phate-sensitive K+ channel (Kir6. 1):
A novel phosphospecific interaction partner of connexin 43 (Cx43).
Exp Cell Res. 318:2559–2566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen VC, Gouw JW, Naus CC and Foster LJ:
Connexin multi-site phosphorylation: mass spectrometry-based
proteomics fills the gap. Biochim Biophys Acta. 23–34:pp.
1828s2013
|
|
15
|
Chen SC, Kennedy BK and Lampe PD:
Phosphorylation of connexin43 on S279/282 may contribute to
laminopathy-associated conduction defects. Exp Cell Res.
319:888–896. 2013. View Article : Google Scholar :
|
|
16
|
Ai X, Jiang A, Ke Y, Solaro RJ and Pogwizd
SM: Enhanced activation of p21-activated kinase 1 in heart failure
contributes to dephosphorylation of connexin 43. Cardiovasc Res.
92:106–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dang X, Jeyaraman M and Kardami E:
Regulation of connexin-43-mediated growth inhibition by a
phosphorylatable amino-acid is independent of gap junction-forming
ability. Mol Cell Biochem. 289:201–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Budas GR, Churchill EN, Disatnik M-H, Sun
L and Mochly-Rosen D: Mitochondrial import of PKCε is mediated by
HSP90: a role in cardioprotection from ischaemia and reperfusion
injury. Cardiovasc Res. 88:83–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun L-Q, Gao J-L, Cui C-M, et al:
Astrocytic p-connexin 43 regulates neuronal autophagy in the
hippocampus following traumatic brain injury in rats. Mol Med Rep.
9:77–82. 2014.
|
|
20
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zador Z, Weiczner R and Mihaly A:
Long-lasting dephosphorylation of connexin 43 in acute seizures is
regulated by NMDA receptors in the rat cerebral cortex. Mol Med
Rep. 1:721–727. 2008.PubMed/NCBI
|
|
22
|
Popolo A, Morello S, Sorrentino R and
Pinto A: Antiadrenergic effect of adenosine involves connexin 43
turn-over in H9c2 cells. Eur J Pharmacol. 715:56–61. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li K, Chi Y, Gao K, et al: Connexin43
hemichannel-mediated regulation of connexin43. PLoS One.
8:e580572013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trudeau K, Muto T and Roy S:
Downregulation of mitochondrial connexin 43 by high glucose
triggers mitochondrial shape change and cytochrome C release in
retinal endothelial cells. Invest Ophthalmol Vis Sci. 53:6675–6681.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnstone SR, Kroncke BM, Straub AC, et
al: MAPK phosphorylation of connexin 43 promotes binding of cyclin
E and smooth muscle cell proliferation. Circ Res. 111:201–211.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shah MM, Martinez A-M and Fletcher WH: The
connexin43 gap junction protein is phosphorylated by protein kinase
A and protein kinase C: in vivo and in vitro studies. Mol Cell
Biochem. 238:57–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park DJ, Wallick CJ, Martyn KD, Lau AF,
Jin C and Warn-Cramer BJ: Akt phosphorylates Connexin43 on Ser373,
a “mode-1” binding site for 14-3-3. Cell Commun Adhes. 14:211–226.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cooper CD and Lampe PD: Casein kinase 1
regulates connexin-43 gap junction assembly. J Biol Chem.
277:44962–44968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin R, Martyn KD, Guyette CV, Lau AF and
Warn-Cramer BJ: v-Src tyrosine phosphorylation of connexin43:
regulation of gap junction communication and effects on cell
transformation. Cell Commun Adhes. 13:199–216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rottlaender D, Boengler K, Wolny M, et al:
Glycogen synthase kinase 3β transfers cytoprotective signaling
through connexin 43 onto mitochondrial ATP-sensitive K+ channels.
Proc Natl Acad Sci USA. 109:E242–E251. 2012. View Article : Google Scholar
|
|
31
|
Goubaeva F, Mikami M, Giardina S, Ding B,
Abe J and Yang J: Cardiac mitochondrial connexin 43 regulates
apoptosis. Biochem Biophys Res Commun. 352:97–103. 2007. View Article : Google Scholar :
|