Introduction
The potential of gene therapy as a treatment of
cancer has been supported by preclinical and clinical experiments
(1,2). However, the detailed approach, such
as guidelines for the selection of suitable gene delivery vehicles
and therapeutic genes, remains to be further developed. As
mesoderm-derived stem cells, mesenchymal stem cells (MSCs) are
easily obtained from different tissues, including bone marrow,
adipose tissue and umbilical cord (3,4).
Following infusion into the body, MSCs were shown not to cause host
immunological rejection compared with oncolytic viruses and were
able to selectively migrate into tumor tissue (5–8). Due
to their low immunogenicity and specific tropism toward tumors,
MSCs have been used as potent cellular vehicles to treat tumors
following being engineered with different anti-cancer agents
(9,10).
Apoptin is a chicken anemia virus-derived
non-structural protein and composed of 121 amino acids, which
induces apoptosis in a broad range of transformed and cancerous
cells, but not in non-transformed and primary cells (11,12).
Although the mechanisms of cell death induced by apoptin remain to
be elucidated, previous studies have demonstrated that apoptin may
inhibit tumor growth in vivo (13,14).
In these studies, apoptin was delivered as a nucleotide by virus
carriers or directly injected into the body as a recombinant
protein. However, these administration routes may cause the
recipient to undergo a rejection reaction or may not reach
effective concentration due to a short half-life and the limitation
of the maximum tolerated dose (7,15).
Based on this background, it was the aim of the
present study to assess whether MSCs could be modified with apoptin
to inhibit tumor growth. In the present study, it was first
demonstrated that MSCs could be efficiently modified with apoptin
using a lentivirus system and delivery of apoptin could induce
apoptosis of lung cancer cells through activating caspase 3. In
vivo models further confirmed the anti-tumor effects of MSCs
modified with apoptin.
Materials and methods
Culture and preparation of human MSCs and
other cell lines
The present study was approved by the ethics
committee of the First Affiliated Hospital of Guangzhou Medical
University (Guangzhou, China). Human bone marrow-derived MSCs were
isolated, expanded and induced to differentiate as previously
described (16). In the current
study, the bone marrow samples were derived from two male
volunteers, who were 26 and 35 years old, respectively. The
individuals had been admitted to hospital due to a road traffic
accident. The bone marrow was collected between May 2012 and
January 2013. Informed consent was provided by all individuals. The
separated MSCs were sub-cultured at a concentration of
1×104 cells/cm2 in low-glucose Dulbecco’s
modified Eagle’s medium with 10% fetal bovine serum and were used
for experiments at passages 4–8. The human lung cancer cell lines
H460 and H1299 (American Type Tissue Collection, Rockville, MD,
USA), and normal fibroblast cells were cultured in RPMI 1640 media
(HyClone Laboratories, Inc., Logan, UT, USA) supplemented with 10%
fetal bovine serum and modified with humanized Renilla green
fluorescence protein (hrGFP; Invitrogen Life Technologies,
Carlsbad, CA, USA) as previously described (16). Subsequently, the cell lines were
termed H460 hrGFP, H1299 hrGFP and Fibroblast hrGFP,
respectively.
Construction of vectors
To prepare prokaryotic expression vector
pET28b-apoptin, an apoptin sequence derived from multiplex
polymerase chain reaction (PCR) was first amplified by PCR using
primer 1, 5′-CATGCCATGGTAAACGCTCTCCAAGAAG-3′ and primer 2,
5′-AAATATGCGGCCGCCAGTCTTATACACC-3′ (Invitrogen Life Technologies).
Subsequently, the PCR products were digested with NcoI and
NotI, gel-purified and then ligated into vector pET28b
prepared in the same manner. The ligation product was transformed
into DH5α and recombinant clones were selected. PCR, double enzyme
cutting and sequencing were used to further confirm the recombinant
vector pET28b-apoptin.
To prepare the lentiviral expression vector,
pLV/Final-puro-EF1α-apoptin, the apoptin sequence carrying the
secretory signal and the protein transduction domain (PTD) signal
was cloned into pDONRTM221 (Invitrogen Life
Technologies) using the BP recombination reaction to generate entry
clone pDown-apoptin. Subsequently, pUp-EF1α and pDown-apoptin were
recombined into pDest using the LR recombination reaction to
construct pLV/Final-puro-EF1α-apoptin according to the
manufacturer’s instructions (Invitrogen Life Technologies).
Finally, PCR and sequencing were used to identify the correction of
the recombinant eukaryotic expression vector.
Preparation of anti-apoptin antibody
The recombinant pET28b-apoptin vector was
constructed in our laboratory and transformed into E.coli
BL21 (DE3; Fulengen Inc., Guangzhou, China). A positive clone was
induced to express target protein using isopropyl
β-D-1-thiogalactopyranoside (IPTG; 0.1 mM) and relatively low
temperature (26°C). Following sonication and centrifugation at
10,000 × g for 30 min, cell pellets were resolved with
phosphate-buffered saline (PBS) containing urea (8 M), applied to a
Ni2+-chelating column (GE Healthcare, Beijing, China),
then eluted using a stepwise gradient of PBS containing urea (8 M)
and different concentrations of imidazole (from 20 to 400 mM). The
eluates were collected and identified using SDS-PAGE analysis. The
fraction containing the recombinant protein were dialyzed with PBS
buffer, concentrated using a concentrator plus (Eppendorf, Hamburg,
Germany) and stored at −20°C for future use. A total of four
five-week-old male BALB/c mice were supplied by the Experimental
Animal Center of Guangdong Province (Foshan, China). The mice were
housed with ad libitum access to food and water at 22°C with
65% humidity and a 12 h light/dark cycle. After two days of
feeding, they were injected subcutaneously with purified apoptin
(0.03 mg/mouse) mixed with complete Freund’s adjuvant
(Sigma-Aldrich, St. Louis, MO, USA) in a 1:1 ratio. The mice were
subsequently injected three times with same quantity of protein
mixed with incomplete Freund’s adjuvant (Sigma-Aldrich) at two-week
intervals. At five days after the final injection, mouse blood was
harvested using the eyeball blood sampling method, and pooled. The
specificity of the antiserum was detected by western blotting, in
which the prepared apoptin was considered the antigen, and the
primary antibody the antiserum.
Lentivirus construction and transduction
of MSCs
Lentiviral particles carrying apoptin gene were
prepared by transient co-transduction of
pLV/Final-puro-EF1α-apoptin (Invitrogen Life Technologies) and a
lentiviral packaging mix (Invitrogen Life Technologies) into 293FT
cells (Invitrogen Life Technologies) using Lipofectamine 2000
(Invitrogen Life Technologies), according to manufacturer’s
instructions. At 48 h after transfection, viral particles were
harvested, filtered through a 0.45-μm polyethersulfone
membrane and concentrated by ultracentrifugation at 50,000 × g for
1 h at 4°C. Titers of concentrated lentivirus particles changed
from 4×107 to 9×107 U/ml. Human MSCs were
transduced with lentiviral particles at an infection multiplicity
of 50. Following two rounds of infection, puromycin (HyClone
Laboratories, Inc.) was added to the culture medium at a
concentration of 1–5 μg/ml and maintained for 2–3 days. The
obtained MSC lines were defined as MSCs APOPTIN. At the same time,
lentiviral particles carrying hrGFP were prepared with
pLV/Final-puro-EF1α-hrGFP (Invitrogen Life Technologies) and
transduced into MSCs in parallel to assess the modification
efficiency of apoptin.
Western blot analysis
MSCs APOPTIN or control cells were washed with cold
PBS and immediately lysed in Laemmli buffer (Nanjing Keygen
Biotech. Co. Ltd., Nanjing, China). Cell lysates were denatured at
100°C for 5 min and centrifuged at 10,000 × g for 5 min at 4°C.
Supernatants were recovered, separated on 12% SDS-PAGE and
transferred onto a 0.45-μm polyvinylidene difluoride
membrane (Millipore, Billerica, MA, USA). Following blocking the
membrane with Tris-buffered saline-Tween-20 containing 5% non-fat
milk (Mengniu Dairy, Inner Mongolia, China) for 1 h at room
temperature, the membrane was incubated with the appropriate
primary antibodies: Anti-apoptin polyclonal antiserum prepared in
the present study (1:500) and monoclonal anti-GAPDH antibody (cat.
no. ab8245; 1:10,000; Abcam, Cambridge, MA, USA), overnight at 4°C.
The membrane was incubated with horseradish peroxidase-conjugated
secondary antibody (cat. no. sc-2005; 1:10,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 1 h at room temperature
and the bands were detected using the SignalFire™ Enhanced
Chemiluminescence reagent (Cell Signaling Technologies, Beverly,
MA, USA) in a dark room.
Co-culture experiments
A total of 1×105 MSCs APOPTIN or MSCs
were pre-plated in six-well plates overnight. Subsequently, three
types of cells, including fibroblast hrGFP (1×105), H460
hrGFP (4×105) and H1299 hrGFP (4×105), were
added to these wells. After 48 h of culture, the viability of total
cells was determined using a cell counting kit-8 (CCK-8; Dojindo
Laboratories, Kumamoto, Japan) and analyzed as a percentage against
control MSCs. The extent of apoptosis of cells with green
fluorescence was recorded via microscopy (BX51; Olympus
Corporation, Tokyo, Japan) following direct staining with DAPI
(Sigma-Aldrich). At the same time, the presence of activated
caspase-3 in H460 hrGFP was measured with an anti-active caspase-3
polyclonal antibody (Promega, Madison, WI, USA) at different
time-points during co-culture. The media derived from 24 h
incubation with MSCs APOPTIN and MSCs, were used to treat H460
cells for 48 h and the viability of the target cells was detected
using the CCK-8 kit.
Animal studies
Athymic nude mice (six weeks old) were purchased
from Guangdong Medical Laboratory Animal Center (Foshan, China) and
used in accordance with institutional guidelines under approved
protocols. A total of 1×106 H460 cells with or without
3×105 MSCs or MSCs APOPTIN were suspended in 100
μl PBS and subcutaneously injected into the flank area of
nude mice. At the 14th day after the first injection, the same
quantity of MSCs or MSCs APOPTIN were again injected into the same
position. Subsequently, mice were examined three times a week and
tumor sizes were calculated as previously described (10): volume = length ×
width2/2. On the 60th day, the tumor was excised after
mice were sacrificed by cervical dislocation and the tumor mass was
determined.
Statistical analysis
Values are expressed as the mean ± standard
deviation. The differences between experimental and control groups
were analyzed using a two-tailed Student’s t-test employing SPSS
version 12.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Identification of anti-apoptin polyclonal
antibody
To prepare the anti-apoptin polyclonal antibody, a
prokaryotic expression vector pET28b-apoptin was constructed by
double enzyme cutting and ligation reaction. As shown in Fig. 1A, the encoding fragment of apoptin
was correctly inserted into the multiple cloning sites of pET28b.
After pET28b-apoptin was transformed into host bacterial BL21
(DE3), the positive clone containing recombinant vector was induced
to express apoptin with IPTG and low temperature. Subsequently,
Ni+ ion affinity chromatography was used to purify the
target protein. As shown in Fig.
1B, apoptin with a purity >90% was collected. The eluate
containing apoptin was dialysed, concentrated and injected into
BALB/c mice to prepare anti-apoptin polyclonal antibody. The
western blotting results indicated that the polyclonal antibody
specifically bound apoptin derived from the prokaryotic expression
system (Fig. 1C).
 | Figure 1Preparation of anti-apoptin antibody
and apoptin-modified MSCs. (A) Recombinant vector pET28b-apoptin
was identified by PCR and double enzyme cutting. Lane 1, PCR
product; lane 2, double enzyme cutting product. (B) Apoptin protein
was induced and purified. Lane 1, total lysate of host BL21 (DE3)
bearing pET28b; lane 2, total lysate of host BL21 (DE3) bearing
pET28b-apoptin; lane 3, supernatant of BL21 (DE3) bearing
pET28b-apoptin; lane 4, sediment of BL21 (DE3) bearing
pET28b-apoptin; lane 5/9, protein marker; lane 6, sample used for
Ni+ affinity chromatography; lane 7, eluate with PBS
containing 100 mM imidazole; lane 8, eluate with PBS containing 200
mM imidazole. (C) The specificity of anti-apoptin antibody was
confirmed by western blot analysis. Lane 1, negative control; lane
2, total lysate of host bacteria containg pET28b-apoptin; lane 3,
purified apoptin. MSC, mesenchymal stem cell; PCR, polymerase chain
reaction; PBS, phosphate-buffered saline. |
Generation, identification and
characterization of apoptin-modified MSCs
Firstly, the correction of lentiviral expression
vector pLV/Final-puro-EF1α-apoptin was confirmed by PCR (Fig. 2A) and sequencing (data not shown).
In addition, western blotting also revealed that apoptin protein
was expressed in MSCs APOPIN and not in control MSCs (Fig. 2B). Lentiviral particles containing
apoptin or hrGFP gene were prepared in 293FT cells. Subsequently,
MSCs were transfected with the lentiviral particles. Following
puromycin selection, >90% of hrGFP-modified MSCs expressed hrGFP
(Fig. 2C). Due to the same
experimental conditions, it was hypothesized that the modification
efficiency in MSCs APOPTIN was similar. Furthermore, it was
demonstrated that MSCs APOPTIN may be induced into adipocytes or
osteoblasts in vitro under the appropriate conditions
(Fig. 2D).
 | Figure 2Construction of apoptin-modified MSCs.
(A) Lentiviral vector pLV/Final-puro-EF1α-apoptin was identified by
PCR. Lane 1/3, negative control; lane 2, amplification product of
fragment of apoptin; lane 4, amplification product of fragment of
EF1α promoter. (B) Expression of apoptin was confirmed by western
blot analysis. Lane 1, negative control; lane 2, MSCs APOPTIN 1;
lane 3, MSCs APOPTIN 2. (C) Morphology of MSCs hrGFP and MSCs
APOPTIN. (D) Adipogenesis and osteogenesis differentiation of MSCs
APOPTIN. Magnification, ×200. MSCs APOPTIN, mesenchymal stem cells
expressing apoptin; PCR, polymerase chain reaction; hrGFP,
humanized Renilla green fluorescence protein. |
MSCs APOPTIN induce lung tumor cell
apoptosis by activating caspase-3 within target cells
As shown in Fig.
3A–D, MSCs APOPTIN induced apoptosis in the lung cancer cell
lines H460 and H1299, as represented by detachment of adherent
GFP-positive cells and the appearance of cellular debris in
DAPI-GFP double positive cells, but not in fibroblast cells. The
percentage of viable cells detected using the CCK-8 kit also
indicated that MSCs APOPTIN inhibited the proliferation of
transformed cells (P<0.05). In addition, control MSCs had no
detectable effect on these cells, whereas media derived from MSCs
APOPTIN inhibited H460 cell proliferation (Fig. 3E and F).
To further examine the mechanism of action of
apoptin, the activation of caspase-3 within target cells was
measured. As shown in Fig. 4, the
percentage of H460 cells positive for activated caspase-3 in the
MSCs APOPTIN group was markedly higher than that in the control
MSCs group (16.5±2.9% at 24 h and 27.3±2.0% at 48 h vs. 3.4±1.1% at
24 h and 2.2±0.6% at 48 h). It was observed that the percentage of
target cells positive for activated caspase-3 evidently increased
with prolongation of co-culture time in the MSCs APOPTIN group,
whereas there was no marked change in the control group. From these
results, it was concluded that apoptin derived from MSCs APOPTIN
effectively induced tumor cell apoptosis via activation of
caspase-3 within target cells.
MSCs APOPTIN reduces subcutaneous lung
tumor growth in vivo
To further confirm the in vitro finding of
MSCs APOPTIN in vivo, a xenotransplantation model of lung
carcinoma was established and the kinetics of tumor growth were
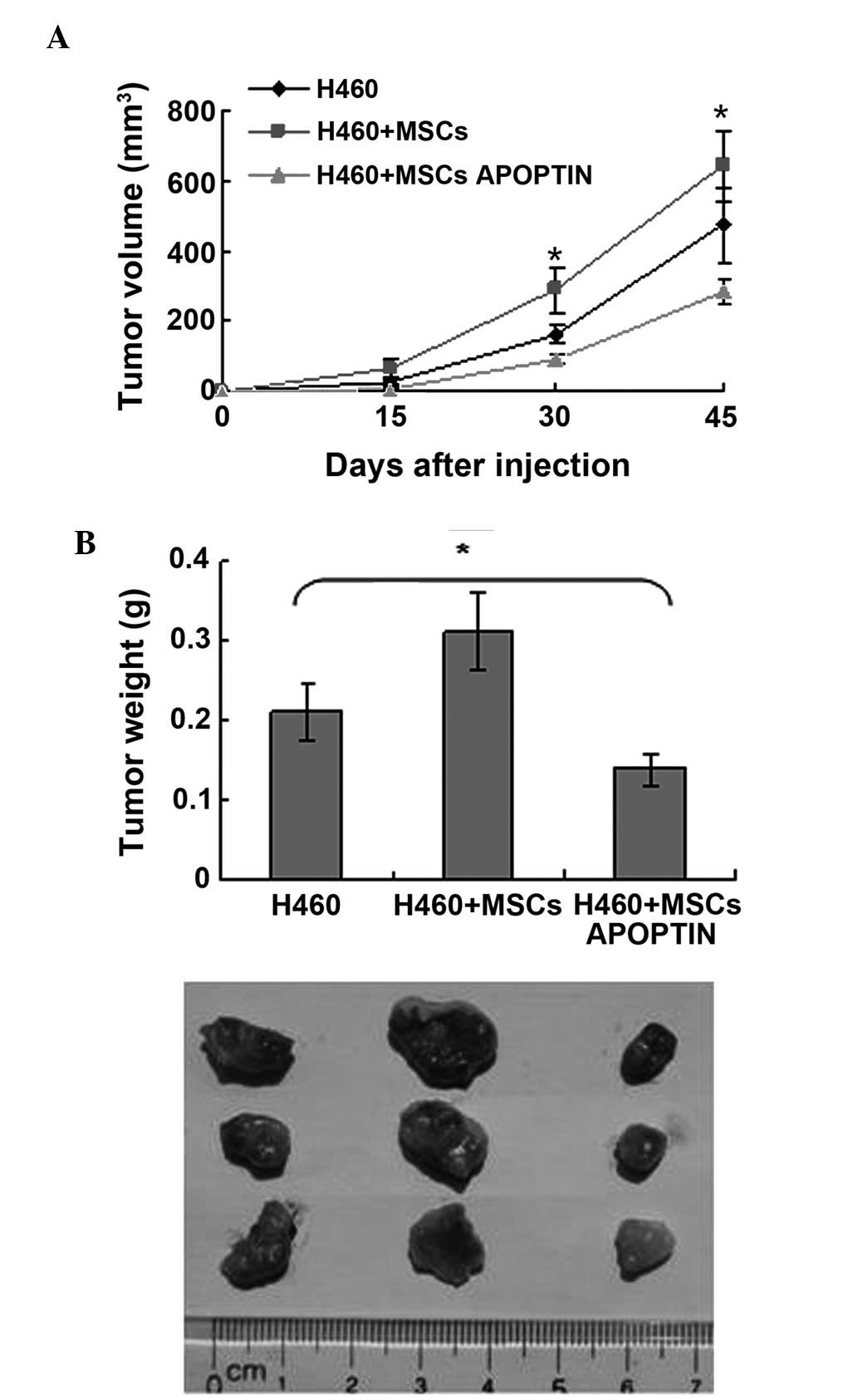
assessed. As shown in Fig. 5,
tumor masses began to appear at ~23 days following inoculation in
the single H460 group. MSCs APOPTIN delayed the appearance and
inhibited the growth of the tumor mass. The average weight of tumor
mass in the H460+MSCs APOPTIN group, the H460 group, and the
H460+MSCs group was 0.14±0.02, 0.21±0.04 and 0.31±0.05 g,
respectively. By contrast, MSCs accelerated the growth of the tumor
mass in the early phase of subcutaneous tumor growth (up to 50
days). However, the difference in tumor size and weight between the
tumor cell group containing MSCs and tumor cell group alone was
eliminated at approximately 60 days.
Discussion
MSCs have the advantage of being an optimal cell
delivery vehicle for gene therapy (17). As a small molecular protein,
apoptin has been used to suppress tumor cell growth (18). However, the limitations of the
protein drug, such as the short half life, indicated a requirement
for a novel method of its delivery (15). Therefore, it was assessed whether
apoptin-modified MSCs may inhibit tumor growth.
Initially, a specific anti-apoptin antibody was
prepared by immunizing mice with purified apoptin protein derived
from a prokaryotic expression system.
It is important to maximally improve the
modification efficiency of MSCs in order to realize the expected
goal. Compared with other gene modification systems, including
adenoviral or retroviral vectors, the human immunodeficiency
virus-based lentivirus system may stably transduct cells at a
different mitotic stage, which is very applicable to MSCs, which
are often quiescent (19,20). Several studies have reported that
lentiviral vectors may effectively deliver target genes into MSCs
(19,21). Correspondingly, the present results
also indicated that the apoptin gene may be stably integrated into
MSCs via a lentiviral vector and expressed by host cells. In
addition, the differentiation capacity of MSCs was not affected by
apoptin or the lentiviral vector.
A further problem which requires development is to
guarantee that apoptin enters target cells following its expression
by MSCs. Although the detailed mechanism of apoptin-induced
apoptosis remains to be elucidated, it is certain that
apoptin-induced apoptosis depends on the entering of apoptin into
target cells (22). As apoptin
itself has no associated signal sequences, one secreting signal and
one PTD were introduced into the N-terminus of apoptin during
construction of the lentiviral expression vector according to
previous studies (23,24). The two signal sequences may assist
apoptin to be secreted from host cells and then transferred into
target cells. As demonstrated by the present results, apoptin may
be successfully secreted into the extracellular space and enter
target cells.
According to associated studies, >70 human tumor
cell lines are susceptible to the pro-apoptotic effect of apoptin
(11,12). From these studies, it was found
that apoptin exerts its apoptosis-inducing function mainly through
direct expression in tumor cells or in the form of a fusion
protein. It is likely that these modes may narrow its clinical
application. The present results indicated that apoptin-modified
MSCs may induce apoptosis in lung cancer cell lines, while sparing
normal cells. It is evident that the combination of MSCs and
apoptin may broaden their application. Previous studies have
indicated that cell apoptosis induced by apoptin is associated with
the activation of typical caspases (25,26).
This theory was further confirmed by the present results, which
demonstrated that apoptin-modified MSCs markedly activated
caspase-3 within target cells.
Of note, apoptin-modified MSCs may decrease the
growth of tumors in a xenograft mouse model, although native MSCs
promoted their growth in comparison with tumor cells alone, which
is in agreement with the results of a previous study (27). The possible explanation may be that
the pro-apoptotic capacity of apoptin overcomes the
tumor-supportive capacity of MSCs, which eventually results in the
inhibition of tumor growth by apoptin-modified MSCs.
In conclusion, the present study indicated that MSCs
may be effectively modified by insertion of the apoptin gene via a
lentiviral transduction system. The apoptin-engineered MSCs may
inhibit tumor cell growth in vitro and in vivo. Thus,
apoptin-modified MSCs may be a novel option in cancer therapy.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81101730) and the
PhD Start-up Fund of Guangzhou Medical University (grant no.
2012C80).
References
|
1
|
Cotrim AP and Baum BJ: Gene therapy: some
history, applications, problems and prospects. Toxicol Pathol.
36:97–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Verma IM and Somia N: Gene therapy –
promise, problems and prospects. Nature. 389:239–242. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee RH, Kim B, Choi I, et al:
Characterization and expression analysis of mesenchymal stem cells
from human bone marrow and adipose tissue. Cell Physiol Biochem.
14:311–324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erices A, Conget P and Minguell JJ:
Mesenchymal progenitor cells in human umbilical cord blood. Br J
Haematol. 109:235–242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Le Blanc K, Rasmusson I, Sundberg B, et
al: Treatment of severe acute graft-versus-host disease with third
party haploidentical mesenchymal stem cells. Lancet. 363:1439–1441.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kidd S, Spaeth E, Dembinski JL, et al:
Direct evidence of mesenchymal stem cell tropism for tumor and
wounding microenvironments using in vivo bioluminescent imaging.
Stem Cells. 27:2614–2623. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Raper SE, Chirmule N, Lee FS, et al: Fatal
systemic inflammatory response syndrome in a ornithine
transcarbamylase deficient patient following adenoviral gene
transfer. Mol Genet Metab. 80:148–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Msaouel P, Iankov ID, Dispenzieri A and
Galanis E: Attenuated oncolytic measles virus strains as cancer
therapeutics. Curr Pharm Biotechnol. 13:1732–1741. 2012. View Article : Google Scholar :
|
|
9
|
Kucerova L, Altanerova V, Matuskova M,
Tyciakova S and Altaner C: Adipose tissue-derived human mesenchymal
stem cells mediated prodrug cancer gene therapy. Cancer Res.
67:6304–6313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grisendi G, Bussolari R, Cafarelli L, et
al: Adipose-derived mesenchymal stem cells as stable source of
tumor necrosis factor-related apoptosis-inducing ligand delivery
for cancer therapy. Cancer Res. 70:3718–3729. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maddika S, Mendoza FJ, Hauff K, Zamzow CR,
Paranjothy T and Los M: Cancer- selective therapy of the future:
apoptin and its mechanism of action. Cancer Biol Ther. 5:10–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Backendorf C, Visser AE, de Boer AG, et
al: Apoptin: therapeutic potential of an early sensor of
carcinogenic transformation. Annu Rev Pharmacol Toxicol.
48:143–169. 2008. View Article : Google Scholar
|
|
13
|
Li X, Liu Y, Wen Z, et al: Potent
anti-tumor effects of a dual specific oncolytic adenovirus
expressing apoptin in vitro and in vitro. Mol Cancer. 9:102010.
View Article : Google Scholar
|
|
14
|
Schoop RA, Baatenburg de Jong RJ and
Noteborn MH: Apoptin induces apoptosis in an oral cancer mouse
model. Cancer Biol Ther. 7:1368–1373. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Torchilin VP and Lukyanov AN: Peptide and
protein drug delivery to and into tumors: challenges and solutions.
Drug Discov Today. 8:259–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du J, Zhou L, Chen X, et al: IFN-γ-primed
human bone marrow mesenchymal stem cells induce tumor cell
apoptosis in vitro via tumor necrosis factor-related
apoptosis-inducing ligand. Int J Biochem Cell Biol. 44:1305–1314.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aboody KS, Najbauer J and Danks MK: Stem
and progenitor cell-mediated tumor selective gene therapy. Gene
Ther. 15:739–752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun J, Yan Y, Wang XT, et al: PTD4-apoptin
protein therapy inhibits tumor growth in vivo. Int J Cancer.
124:2973–2981. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kyriakou CA, Yong KL, Benjamin R, et al:
Human mesenchymal stem cells (hMSCs) expressing truncated soluble
vascular endothelial growth factor receptor (tsFLk-1) following
lentiviral-mediated gene transfer inhibit growth of burkitt’s
lymphoma in a murine model. J Gene Med. 8:253–264. 2006. View Article : Google Scholar
|
|
20
|
Chang LJ and Gay EE: The molecular
genetics of lentiviral vectors - current and future perspectives.
Curr Gene Ther. 1:237–251. 2001. View Article : Google Scholar
|
|
21
|
Loebinger MR, Eddaoudi A, Davies D and
Janes SM: Mesenchymal stem cell delivery of TRAIL can eliminate
metastatic cancer. Cancer Res. 69:4134–4142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Danen-Van Oorschot AA, Zhang YH, Leliveld
SR, et al: Importance of nuclear localization of apoptin for
tumor-specific induction of apoptosis. J Biol Chem.
278:27729–27736. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Daugherty BL, Zavodny SM, Lenny AB, et al:
The uses of computer-aided signal peptide selection and polymerase
chain reaction in gene construction and expression of secreted
proteins. DNA Cell Biol. 9:453–459. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Flinterman M, Farzaneh F, Habib N, Malik
F, Gäken J and Tavassoli M: Delivery of therapeutic proteins as
secretable TAT fusion products. Mol Ther. 17:334–342. 2009.
View Article : Google Scholar
|
|
25
|
Danen-van Oorschot AA, van Der Eb AJ and
Noteborn MH: The chicken anemia virus-derived protein apoptin
requires activation of caspases for induction of apoptosis in human
tumor cells. J Virol. 74:7072–7078. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maddika S, Booy EP, Johar D, Gibson SB,
Ghavami S and Los M: Cancer-specific toxicity of apoptin is
independent of death receptors but involves the loss of
mitochondrial membrane potential and the release of mitochondrial
cell-death mediators by a Nur77-dependent pathway. J Cell Sci.
118:4485–4493. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu W, Xu W, Jiang R, et al: Mesenchymal
stem cells derived from bone marrow favor tumor cell growth in
vivo. Exp Mol Pathol. 80:267–274. 2006. View Article : Google Scholar
|