Introduction
Leukemia is a malignant blood disorder and serious
threat to human health (1). Tumor
cells can not be effectively attacked by the immune system in spite
of the presence of tumor-specific antigens, which is known as
immune evasion. Previous studies have shown that an important
mechanism of the immune evasion of tumor cells is the absent or low
expression of major histocompatibility complex (MHC) molecules and
co-stimulatory molecules (2–5).
However, it has rarely been studied whether or not leukemia cells
evade the immune system through this mechanism. The MHC is one of
the most important genetic systems for preventing pathogen invasion
and maintaining the immune system in higher developed animals
(6). As one of MHC class II gene
transcription factors, regulatory factor X activity is controlled
by class II transactivator (CIITA) (7). CIITA regulates MHC class II
expression as a transcriptional activator and as a general
transcription factor (8). It is
the ‘speed factor’ and ‘molecular switch’ of MHC class II, and
quantitatively controls MHC class II mRNA expression (9). Four types of CIITA promoter have been
identified in humans (10). In
certain instances, the silencing and knockdown of CIITA promoter IV
(CIITApIV) have been mostly responsible for failure of interferon
(IFN)-γ to induce MHC II gene transcription and the partial
silencing of MHCII molecules (11,12).
Epigenetic modifications in cells are closely
associated with the occurrence of leukemia. It has been reported
that epigenetic abnormalities occurred in human cancer cells and
may be the key to initiate tumorigenesis (13,14).
DNA methylation and histone modification are two main causes of
gene mutation (15). To date,
fifteen DNA methylation biomarkers for diagnosis and sub-typing of
pediatric acute lymphoblastic leukemia (ALL) have been found
(16), and DNA methylation was
shown to be an indicator of the ALL sub-type as well as clinical
outcome (17). The histone
deacetylase inhibitor (HDACi) belinostat (PXD101) inhibited cell
growth, induced apoptosis and increased the acetylation of histone
H3 and H4 in a dose-dependent manner in promyelocytic leukemia
HL-60 and NB4 cells; it is under development as an epigenetic drug
for anti-leukemia and differentiation therapy (18). In general, histone acetylation on
chromatin and DNA demethylation on cytosine-phosphate-guanine (CpG)
loci can loosen the chromatin structure, which aids in the binding
of transcription factors to gene control regions and promotes gene
expression. Conversely, histone deacetylation and methylation
inhibit gene expression (19).
Previous studies have shown that epigenetic regulation was able to
silence or reduce CIITA in cancer cells (20,21).
In a variety of MHC class II-negative tumor cells, elevated levels
of chromosomal histone deacetylation and CpG site methylation on
the CIITA promoter have been detected; however, treatment with
HDACi or DNA methyltransferase inhibitors (DNMTi) increased the
expression of MHC II, and the underlying mechanism may be the
transcriptional activation of the CIITA gene (21).
If treatment with HDACis or DNMTis has similar
effects on leukemia cells to those mentioned above, they may be
used as highly effective preventives or anti-leukemia agents. In
the present study, leukemia HL-60 cells were treated with the HDACi
5-aza-2′-deoxycytidine (5-Aza-CdR) and/or the DNMTi suberoylanilide
hydroxamic acid (SAHA) and then stimulated by IFN-γ to explore
their effect on CIITA methylation and the resulting MHC class II
expression. The results gave clues on the underlying mechanism of
the immune evasion of leukemia HL-60 cells.
Materials and methods
Cell lines and agents
The human HL-60 cell line was purchased from
Shanghai Institute of Biological Sciences, Chinese Academy of
Sciences (Shanghai, China). RPMI 1640 culture medium, fetal calf
serum, bovine serum albumin (BSA), dimethylsulfoxide (DMSO) and
trypsin were purchased from Gibco-BRL (Invitrogen Life
Technologies, Carlsbad, CA, USA). 5-Aza-CdR (Sigma Aldrich, St
Louis, MO, USA) and SAHA (Cayman Chemical Company, Ann Arbor, MI,
USA) were used for epigenetic modification. IFN-γ (PeproTech, Rocky
Hill, NJ, USA) was used for promoting the expression of MHC class
II molecules. TRIzol reagent was obtained from Invitrogen Life
Technologies. RevertAid™ First Strand cDNA kit and short DreamTaq™
Green polymerase chain reaction (PCR) Master Mix (2X) were products
of Fermentas (Thermo Fisher Scientific, Burlington, Canada).
PMD18-T cloning vector, Esherichia coli DH5α, proteinase K
and RNase A were purchased from Takara Biotechnology Co., Ltd.
(Dalian, China). Rabbit anti-human CIITA-1 antibody (cat. no.
A1709) was purchased from Wuhan Sino-US Sciences Co., Ltd (Wuhan,
China). β-actin rabbit polyclonal antibody (cat. no. sc-130657) was
purchased from Santa Cruz Biotechnologi, Inc. (Dallas, TX, USA). EZ
DNA methylation-Direct kit was purchased from Beijing Tianmo Sci
& Tech Development Co., Ltd (Beijing, China). DNA molecular
size standard was a product of New England Biolabs Inc (Ipswich,
MA, USA). A 100-bp DNA marker was purchased from Generay Biotech
(Shanghai, China). Protein standard was a product of Sangon Biotech
(Shanghai, China). Tween-20, nitrocellulose membrane, Ponceau S
solution and ethylene glycol tetraacetic acid (EGTA) were from
Amresco (Solon, OH, USA). Polyacrylamide gel, SDS,
isopropyl-β-d-thiogalactoside (IPTG), X-gal and Gel Extraction kit
were all purchsed from Shanghai Huashun Biotechnology Co., Ltd.
(Shanghai, China). Casein tryptone and yeast extract were from
Oxiod (Basingstoke, UK). Brilliant blue G 250 and ampicillin were
from Sigma Aldrich.
Cell culture
HL-60 cells were cultured in RPMI 1640 supplemented
with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml
streptomycin (Sigma-Aldrich) in a 5% CO2 atmosphere at
37°C. Cells were subcultured every two days.
Experimental groups
According to the various final concentrations of
drugs in the media, cells were divided into five groups: A) control
group treated with phosphate-buffered saline (PBS; Sigma-Aldrich)
only for 120 h; B) 1,000 U/ml IFN-γ for 48 h; C-E) 5-Aza-CdR (0.1
μM, 1 μM or 10 μM, respectively, for 48 h)
followed by SAHA (0.5 μmol/l) for 24 h, then stimulation
with IFN-γ (1,000 U/ml) for 48 h. Experimental conditions in each
group were repeated five times. Following treatment, growth
conditions and morphological changes of cells were observed.
Reverse-transcription quantitative
PCR
Following treatment, cells from all groups were
washed in PBS. Total RNA was isolated using TRIzol and identified
using an ultraviolet (UV) spectrophotometer (UV2550; Shimadzu,
Kyoto, Japan). cDNA was synthesized using the RevertAid™ First
Strand cDNA Synthesis kit. Consulting the sequences in GeneBank,
primers for MHC II, MHC, CD40, CD80 and were designed using Primer
5.0 software and synthesized by Invitrogen (Shanghai, China).
Primer sequences were as follows: MHC II (HLA-DRA) forward,
5′-GAAATGGAAAACCTGTCACCAC-3′; MHC II reverse,
5′-AAACTCCCAGTGCTTGAGAAGA-3′; MHC I (HLA-A) forward,
5′-GTATTTCTTCACATCCGTGTCC-3′; MHC I reverse,
5′-TTCACATTCCGTGTCTCCTG-3′; CD40 forward, 5′-ACCTCGCTATGGTTCGTC-3′;
CD40 reverse, 5′-AAGGCATTCCGTTTCAGT-3′; CD80 forward,
5′-ACCATCCAAGTGTCCATACCTC-3′; CD80 reverse,
5′-CAGCACCATTTTCTTCTCCTTT-3′; β-actin forward,
5′-AAGTACTCCGTGTGGATCGG-3′; β-actin reverse,
5′-ATGCATTCACCTCCCCTGTG-3′. Genes above were quantified using
DreamTaq™ Green PCR Master Mix. ABI 7500 Fast Real-Time PCR
platform (Applied Biosystems, Thermo Fisher Scientific, Waltham,
MA, USA) conditions were as follows: 94°C for 5 min; 94°C for 40
sec, 55°C for 40 sec and 72°C for 40 sec, for 34 cycles for MHC I
and 36 cycles for all others, followed by 72°C for 7 min. The
amplified products were separated on a 3% agarose gel and
visualized after 5 μg/ml ethidium bromide (Sigma-Aldrich)
staining for 10 min.
Western blot analysis
HL-60 cells from all five groups were lysed in
ice-cold Laemmli lysis buffer (cat. no. 38733; Sigma-Aldrich). The
protein concentrations were measured using the coomassie brilliant
blue method (Brilliant Blue G-250; Sigma-Aldrich) (22). Protein samples were separated using
SDS-PAGE and then transferred to nitrocellulose membranes at a
voltage of 100 V for 100 min. Following staining with Ponceau S
solution, samples were blocked with 5% skimmed milk in PBS with
Tween 20 at room temperature for 2 h. The membranes were incubated
with rabbit anti-human CIITA (1:1,000) and β-actin (1:1,000)
primary antibodies at 4°C overnight, and subsequently with a
horseradish peroxidase-conjugated polyclonal goat anti-rabbit
secondary antibody (1:3,000; cat. no. A24537; Invitrogen Life
Technologies) at room temperature for 3 h. Blots were visualized
using an enhanced chemiluminescence reagent (Invitrogen) and a
LAS-3000mini luminoimage analyzer (Fujifilm, Tokyo, Japan).
DNA bisulfite treatment
Genomic DNA of HL-60 cells was isolated with
proteinase K (0.5%)/SDS (20 mg/ml) and identified using a UV
spectrophotometer (Shimadzu UV2550) as previously described
(23). The DNA was then treated
with sodium bisulfite using the EZ DNA methylation-Direct kit
according to the manufacturer’s instructions. Briefly, 500 ng DNA
was denatured for 10 min at 9°C and incubated for 2.5 h at 64°C in
130 μl CT Conversion Reagent. Subsequently, 600 μl
M-Binding Buffer was added and the mixture was centrifuged (12,000
× g) for 2 min prior to the addition of 200 μl M-Wash
Buffer. Following centrifugation, the samples were incubated with
200 μl M-Desulphonation Buffer for 15-20 min at room
temperature and washed with M-Wash Buffer twice. Finally, 10
μl M-Elution Buffer was added and the DNA precipitate was
eluted by centrifugation (12,000 × g, 4 min).
Bisulfite-sequencing PCR (BSP)
analysis
Using CpG Island Searcher (http://cpgislands.usc.edu/), a DNA sequence of ~1,000
bp was analyzed, which was located on the transcription start site
of CIITapIV. A CpG island comprising >200 bp (observed
CpGs/expected CpGs>0.6, GC>50%) was selected (Fig. 1). Primers (Sangon Biotech Co.,
Ltd.) were as follows: Forward, 5′-TTGGGATGTTATTTTTGATAAAGTA-3′ and
reverse, 5′-ACAAAAAAAACTTTAATCACCTACC-3′. Using DreamTaq™ Green PCR
Master Mix, PCR (ABI 7500 Fast Real-Time PCR platform) was
performed in a volume of 20 μl containing 2 μl buffer
(10X), 0.5 μl deoxynucleotide triphosphates (10 mM), 0.5
μl Taq enzyme, 0.5 μl Primer F (10 mM), 0.5 μl
of Primer R (10 mM), 14 μl ddH2O and 2 μl
DNA template. Reaction conditions were as follows: 94°C for 3 min;
94°C for 30 sec, 53°C 30 sec, 72°C for 40 sec for 35 cycles,
followed by 72°C for 5 min. The amplified products were separated
on a 3% agarose gel and visualized after ethidium bromide
(Sigma-Aldrich) staining.
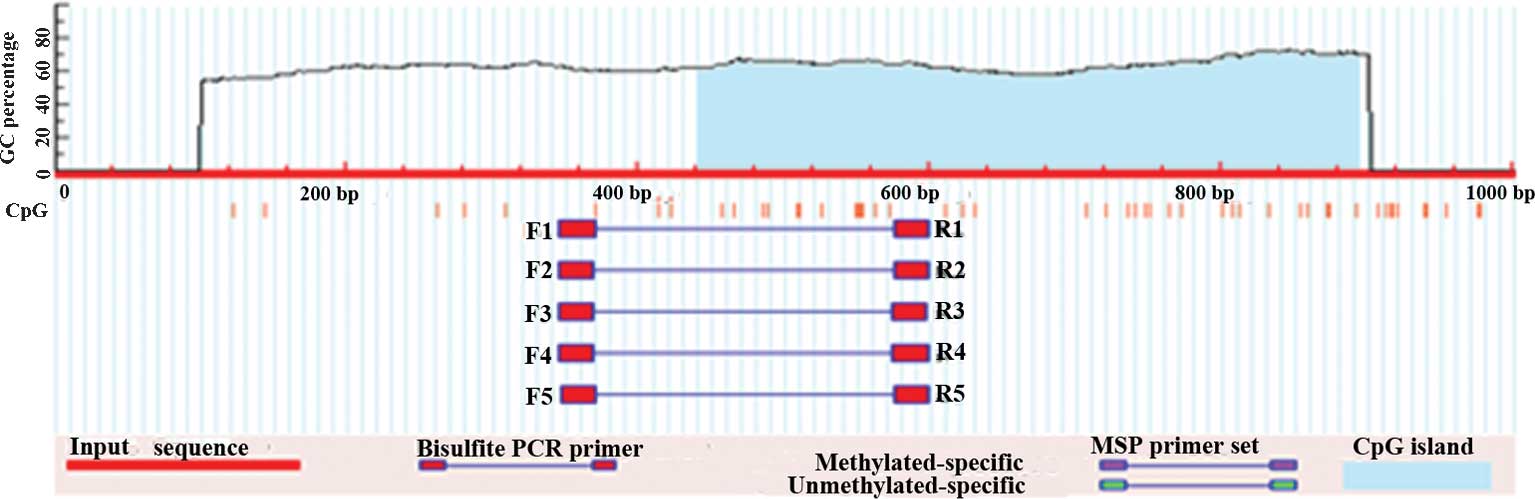 | Figure 1CpG sites located on the
transcription start site in proximity to CIITApIV. Criteria used:
Island size, >200; GC percentage, >50.0%; observed/expected,
>0.6. CpG island 1: Start, 441; end, 895; size, 455 bp;
observed/expected, 0.65; GC percentage, 55%. CIITApIV, class II
transactivator promoter IV; CpG, cytosine-phosphate-guanine; PCR,
polymerase chain reaction; MSP, monosulfite PCR. |
Cloning and sequence analysis
To sequence the bisulfite-PCR products, amplified
fragments were spliced into pMD18-T vector using pMD18-T Vector
Cloning kit from Takara Company. The cloning was performed in a
volume of 10 μl containing 1 μl pMD18-T vector, 1
μl PCR product, 3 μl dH2O and 5 μl
Solution I at 16°C for 60-120 min. Following mixing with 100
μl DH5a competent cells, the mixture above was put on ice
for 30 min, followed by heating at 42°C for 60 sec. Finally, the
mixture was cultured with agitation in 890 μl super optimal
broth medium at 37°C for 8 h, after which individual bacterial
colonies were formed in LB-agar medium containing LB-agar medium
containing 20mg/ml X-gal, 24 mg/ml IPTG and 100 mg/ml ampicillin
(24). The next day, the growth of
bacterial colonies was observed, and positive clones as blue or
white plaques were screened. The selected clones were inoculated
with agitation at 37°C overnight in 5 ml Luria-Bertani medium
containing ampicillin (1:1,000). Universal primers of recombinant
plasmids were as follows: Forward, 5′-GAGCGGATAACAATTTCACACAGG-3′
and reverse, 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′. Recombinant plasmid
was detected by PCR. Five positive clones were selected randomly to
be sequenced from each recombinant colony. The DNA was sequenced
using an ABI 3100 automated sequencer (Applied Biosystems) and gene
sequence alignment was performed using DNASTAR-Lasergene v6
software (DNASTAR, Inc., Madison, WI, USA).
Statistical analysis
All values are expressed as the mean ± standard
error of the mean and analyzed using SPSS 10.0 software (SPSS,
Inc., Chicago, IL, USA). The t-test was used for comparison
of two groups, while single factor analysis of variance was used
for comparison of multiple groups. P<0.05 was considered to
represent a significant difference.
Results
Effect of epigenetic modification on
expression of MHC molecules, CD40 and CD80
Using RT-PCR, the expression of MHC molecules as
well as CD40 and CD80 was examined in HL-60 cells. mRNA levels of
MHC class I, MHC class II, CD40 and CD80 were calculated as the
integrated optical density (IOD) ratio to β-actin. The results
showed that mRNA levels of MHCI, CD40 and CD80 were all
significantly increased in the three epigenetic modification groups
compared with those in the IFN-γ and control groups (P<0.05)
(Table I).
 | Table ImRNA levels of MHC class I, II, CD40
and CD80. |
Table I
mRNA levels of MHC class I, II, CD40
and CD80.
| IOD ratio | Group A | Group B | Group C | Group D | Group E |
|---|
| MHC-I/β-actin | 0.036±0.011 | 0.046±0.012 | 0.092±0.008 | 0.146±0.010 | 0.218±0.022 |
| MHC-II/β-actin | 0 | 0 | 0.146±0.011 | 0.314±0.011 | 0.368±0.019 |
| CD40/β-actin | 0.058±0.016 | 0.060±0.014 | 0.170±0.019 | 0.202±0.017 | 0.258±0.021 |
| CD80/β-actin | 0.052±0.008 | 0.058±0.009 | 0.112±0.015 | 0.160±0.016 | 0.178±0.013 |
The expression of MHC class II gene was not
detectable in the control and IFN-γ groups (Fig. 2 and Table I). However, following treatment
with 5-Aza-CdR + SAHA + IFN-γ, HL-60 cells re-expressed MHC class
II (0.146±0.011 in group C, 0.314±0.011 in group D and 0.368±0.019
in group E), and the expression of MHC class II was increased by
5-Aza-CdR in a concentration-dependent manner (P<0.05) (Table I).
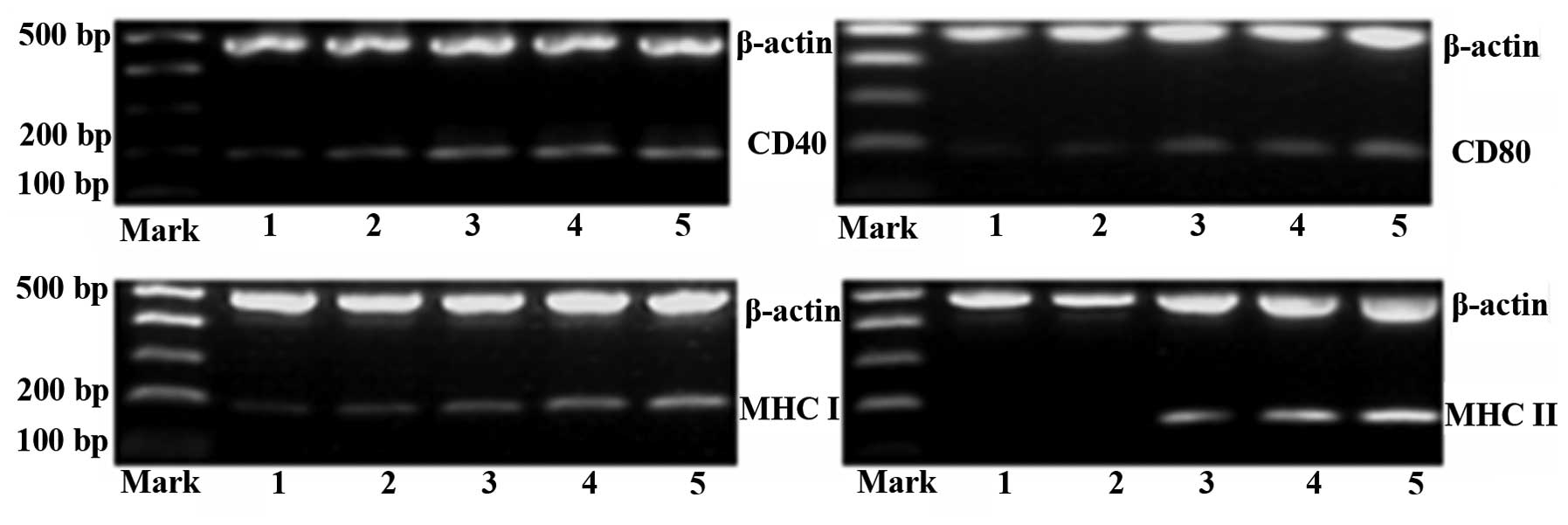 | Figure 2Electrophoretic analysis of the
expression of MHC Class I and II as well as CD40 and CD80 in HL-60
cells. Lanes 1-5 represent groups A-E, respectively. Groups: A,
phosphate-buffered saline; B, IFN-γ; C, 5-Aza-CdR (0.1 μM) +
SAHA (0.5 μM) + IFN-γ; D, 5-Aza-CdR (1 μM) + SAHA
(0.5 μM) + IFN-γ; E, 5-Aza-CdR (10 μM) + SAHA (0.5
μM) + IFN-γ. MHC, major histocompatibility complex; IFN,
interferon; SAHA, suberoylanilide hydroxamic acid; 5-Aza,
5-aza-2′-deoxycytidine. |
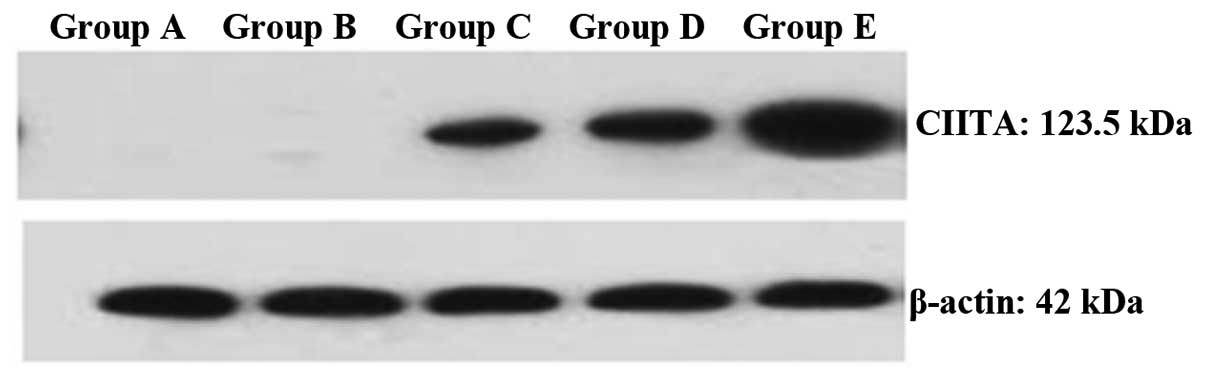
Effect of epigenetic modification on the
expression of CIITA protein
CIITA protein was not detectable in the control and
IFN-γ groups (Fig. 3). However,
the expression of CIITA protein increased dramatically following
epigenetic modification, and the expression was significantly
higher in group E compared with that in the other two epigenetic
modification groups. This 5-Aza-CdR concentration-dependent
increase in CIITA expression was in parallel to that of MHC class
II, which implies that there may be a link between MHC class II and
CIITA.
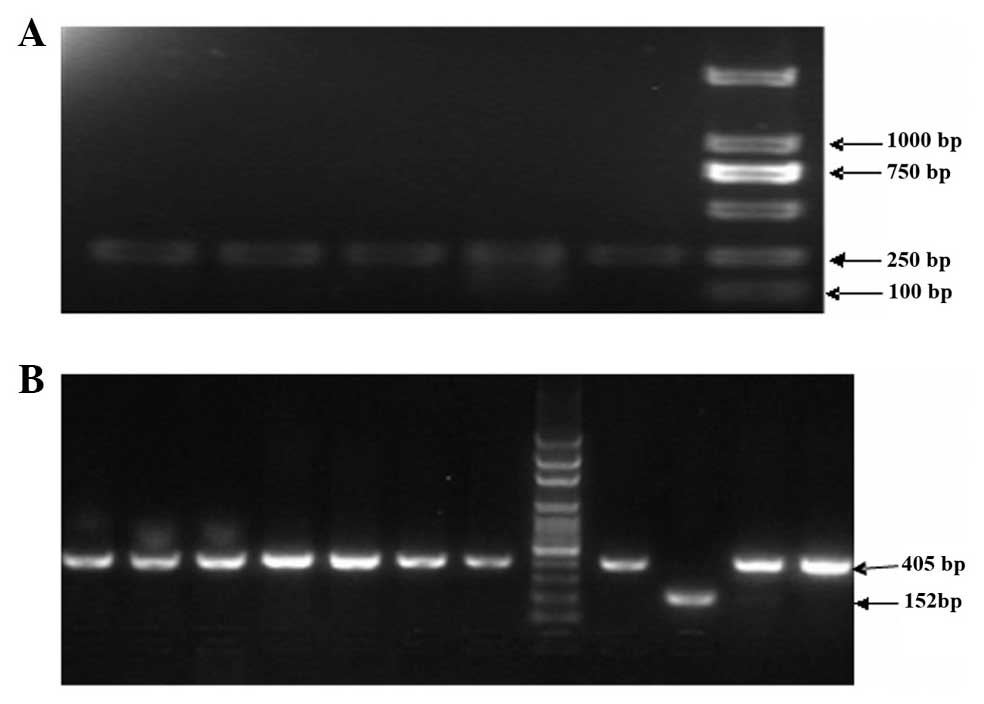
PCR identification of CIITApIV gene and
recombinant PMD18-T vector
Following treatment with bisulfite, total DNA of
leukemia cells was analyzed by BSP, and amplified BSP products were
electrophoresed on 1.5% agarose gel. The length of BSP product was
253 bp, which was the expected length of the amplified fragment
(Fig. 4A). Using colony PCR, it
the correctness of the recombinant plasmids was further confirmed.
The results showed that the amplified 152-bp fragment was the
PMD18-T vector, while the 405-bp fragment was the recombinant
plasmid, which had been inserted into the target gene (Fig. 4B).
Effect of epigenetic modification on CpG
island methylation of CIITA-pIV
DNA sequencing results of CIITApIV showed that there
were 16 CpG island sites, which were able to be methylated. Under
the premise of at least five clones being sequenced for each group,
it was found that methylation of the 162-, 164-, 179-, 202- and
206-bp sites occurred more frequently and the 162- and 164-bp sites
of all five clones were methylated in groups A and B. The
methylation rates of groups A and B (35/80 for group A and 33/80
for group B) were significantly higher than those in the the three
epigenetics modification groups (P<0.05) (Table II and Fig. 5). The methylation rate decreased
with increasing of 5-Aza-CdR concentration, and when the
concentration of 5-Aza-CdR increased to 10 μM, CIITApIV was
completely demethylated (13/80 for group C, 6/80 for group D and 0
for group E) (Fig. 5 and Table II).
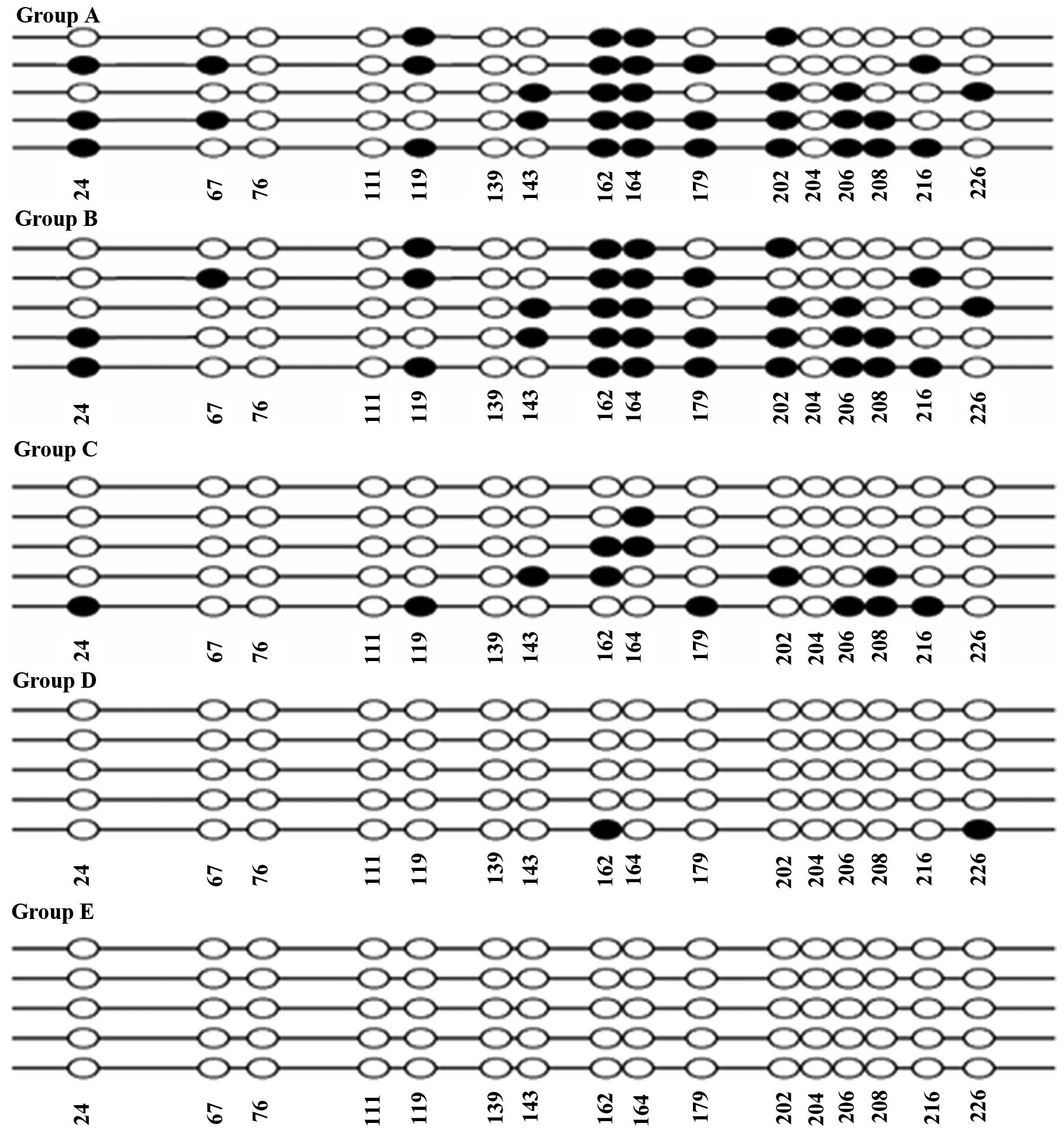 | Figure 5Bisulfite-sequencing of CIITApIV in
bacterial colonies of different groups. Sites in black represent
DNA hypermethylation, while white represents normal non-methylated
sites. Groups: A, phosphate-buffered saline; B, IFN-γ; C, 5-Aza-CdR
(0.1 μM) + SAHA (0.5 μM) + IFN-γ; D, 5-Aza-CdR (1
μM) + SAHA (0.5 μM) + IFN-γ; E, 5-Aza-CdR (10
μM) + SAHA (0.5 μM) + IFN-γ. IFN, interferon; SAHA,
suberoylanilide hydroxamic acid; 5-Aza-CdR, 5-aza-2′-deoxycytidine;
CIITApIV, class II transactivator promoter IV. |
| Table IIEffect of epigenetics modification on
CpG island methylation of CIITApIV. |
Table II
Effect of epigenetics modification on
CpG island methylation of CIITApIV.
| Group | CpG methylation
rate |
|---|
| A | 35/80a |
| B | 33/80a |
| C | 13/80 |
| D | 6/80 |
| E | 0/80 |
Discussion
Antigen-specific T cells are a major force to induce
anti-tumor immune response, and its activation depends on a dual
signal (25). Following antigen
presentation by MHC molecules, tumor antigens are recognized by the
T-cell receptor (TCR) and hence the first signal for T-cell
activation is transmitted (26).
The transmission of the second signal depends on the mutual
recognition between tumor cells and T-cell co-stimulatory molecules
(27). If the number of first
signals is not sufficient or if the second signal is absent, T
cells are disabled (28). Antigen
presentation by MHC class I molecules can activate CD8+ T cells,
which is the main anti-tumor immune effector in cells (29). However, thorough activation of
cytotoxic T-lymphocytes (CTL), the participation of CD4+ T cells is
also required, whose receptor (CTL) recognizes the presenting
antigens via MHC class II (30).
Thus, once tumor antigens are not presented effectively by MHC
molecules, antigen-specific T cells cannot be activated, and
consequently, tumor cells evade being attacked by the immune
system.
In a previous study, following treatment with the
HDAC-1-specific inhibitor MS-275, the expression of CIITA and MHC
class II in diffuse large B-cell lymphoma (DLBCL) cells was
upregulated (21). Furthermore,
the addition of HDACi trichostatin A (TSA) enhanced the expression
of MHC class I and II, as well as the co-stimulatory molecule CD40
on the human neuroblastoma tumor cell line SK-N-MC (31). The MHC surface expression on tumor
cells not only enhanced the anti-tumor immune response but also
reduced tumorigenicity (32,33).
There is currently no research regarding whether leukemia cells
evade immune responses through reduced expression of MHC and
co-stimulatory molecules. Therefore, the present study determined
the expression of MHC molecules on the leukemia cell line HL-60 and
found that the expression of MHC class II was very low at
undetectable levels, even following stimulation with IFN-γ. This
indicated that tumor antigen-specific CD4+ T cells cannot be
effectively activated following contact with leukemia cells.
Furthermore, activation of CTL and antibody production were
affected (30). However, when
mouse tumor-infiltrated CD11b myeloid cells were treated with DNMTi
5-Aza-CdR, cells were able to differentiate into mature
antigen-presenting cells (34).
Here, when HL-60 cells were pre-treated with 5-Aza-CdR + SAHA
followed by IFN-γ stimulation, the expression of MHC class I, CD40+
and CD80+ significantly increased and expression of MHC class II
genes was restored. This showed that the effect of 5-Aza-CdR and
SAHA on HL-60 cells is non-specific. By elevating the expression of
MHC class I, II and co-stimulatory molecules in leukemia cells,
they may be transformed into antigen-presenting cells in
vivo, which may be employed as an efficient anti-leukemia
therapy. In this way, the proliferation and activation of CD4+ T
cells and CD8+ T cells may be enhanced, which then activates the
anti-tumor immune response. This may provide novel ways for the
immunotherapy of leukemia.
At the same time, the demonstrated feasibility of
restoring the expression of MHC class II by the epigenetic
modification of 5-Aza-CdR + SAHA + IFN-γ on HL-60 cells suggested
that there may be a direct association between absence of MHC class
II and epigenetic abnormalities. A previous studies has shown that
DNA hypermethylation and histone deacetylation in tumor cells may
inhibit not only the expression of MHC II, but also certain
co-stimulatory molecules and tumor-associated antigens (35). As a molecular switch of MHC II,
CIITA may quantitatively control the expression of MHC II in a
variety of cells (36,37). Therefore, the present study
assessed the effect of changes in the methylation status of CIITA
on the expression of class II MHC molecules in HL-60 cells.
In a variety of tumor cells, stimulation with IFN-γ
increases the expression of MHC II through the activation of
CIITApIV (12), while
hypermethylation and deacetylation were shown to block the
inductive effects of IFN-γ on CIITA in promyelocytic cells and
breast cancer cells (38,39), which thereby enabled tumor cells to
evade immune surveillance. Therefore, the inhibition of inductive
effects of IFN-γ is closely associated with hypermethylation of
CIITApIV. Previous studies showed that epigenetic modifications
contribute to transcriptional silencing of CIITA in human tumor
cells (38,40). However, this effect can be reversed
by inhibitors of epigen-etic modifications. The HDACi TSA restored
the expression of CIITA in rhabdomyosarcoma RD cells, and
co-treatement of a DNMTi and TSA restored CIITA expression in
SJRH30 cells (20). Simultaneous
treatment of IFN-γ and TSA activated CIITA transcription in mouse
trophoblasts (41). Previous
studies also found that the hypermethylated tumor-suppressor genes
MlH1, TIMP3, p15 and p16 were not able to be activated by TSA alone
in tumour cells; however, when a low dose of 5-Aza-CdR for slight
demethylation was added, TSA treatment resulted in the restoration
of the expression of all genes stated above (42). Furthermore, the combination of DNA
methylation inhibitor 5-Aza-CdR with histone deacetylase inhibitor
TSA or FR901228 produced a greater inhibition of growth and DNA
synthesis and a greater loss of clonogenicity than either agent
alone in myeloid leukemic cells (43). In conclusion, DNA demethylation and
HDAC inhibition have a synergistic effect on the restoration of the
expression of antioncogenes, which had been de-activated by
methylation or acetylation. Cells were treated with 5-Aza-CdR, SAHA
and IFN-γ cooperatively for the reversal of epigenetic modification
in the present study, leading to efficient restoration of the
expression of molecules required for immune recognition.
The present study showed that the CIITA protein on
leukemia HL-60 cells was not detected following stimulation with
IFN-γ. Furthermore, hypermethylation of CpG islands in the CIITApIV
gene promoter was not significantly changed following IFN-γ
treatment. However, following deacetylation and demethylation with
5-Aza-CdR + SAHA prior to stimulation with IFN-γ significantly
decreased CpG island methylation, and CIITA and expression was
restored in parallel with that of MHC class II. This indicated that
the loss in expression of MHC II was caused by the absence of
CIITA, which was epigenetically regulated by CpG island
hypermethylation of the CIITApIV promoter in leukemia HL-60 cells.
The results of the present study indicated that treatment with
5-Aza-CdR + SAHA + IFN-γ may be an efficient strategy to restore
the immune recognition of leukemia cells as a treatment
strategy.
Acknowledgments
The authors of the present study would like to thank
The Fifth People’s Hospital of Shanghai Affiliated to Fudan
University and Shanghai Daopei Hospital for providing
laboratory-related equipment.
References
|
1
|
McKenna SJ: Leukemia. Oral Surg Oral Med
Oral Pathol Oral Radiol Endod. 89:137–139. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klippel ZK, Chou J, Towlerton AM, et al:
Immune escape from NY-ESO-1-specific T-cell therapy via loss of
heterozygosity in the MHC. Gene Ther. 21:337–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siddle HV, Kreiss A, Tovar C, et al:
Reversible epigenetic down-regulation of MHC molecules by devil
facial tumour disease illustrates immune escape by a contagious
cancer. Proc Natl Acad Sci USA. 110:5103–5108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wolkersdörfer T, Fussel M, Kiesslich T, et
al: MHC class II genotype- and MHC class I and II phenotype-related
parameters in sporadic colorectal cancer. Oncol Rep. 26:1165–1171.
2011.PubMed/NCBI
|
|
5
|
Xu WC, Li ZB, Chen YR, et al: Expression
and distribution of S-100, CD83 and costimulatory molecules (CD80
and CD86) in tissues of thyroid papillary carcinoma. Cancer Invest.
29:286–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fernando MM, Stevens CR, Walsh EC, et al:
Defining the role of the MHC in autoimmunity: A review and pooled
analysis. PLoS Genet. 4:e10000242008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi NM, Majumder P and Boss JM:
Regulation of major histocompatibility complex class II genes. Curr
Opin Immunol. 23:81–87. 2011. View Article : Google Scholar :
|
|
8
|
Devaiah BN and Singer DS: CIITA and its
dual roles in MHC gene transcription. Front Immunol. 4:4762013.
View Article : Google Scholar
|
|
9
|
Otten LA, Steimle V, Bontron S and Mach B:
Quantitative control of MHC class II expression by the
transactivator CIITA. Eur J Immunol. 28:473–478. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Green MR, Yoon H and Boss JM: Epigenetic
regulation during B cell differentiation controls CIITA promoter
accessibility. J Immunol. 177:3865–3873. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen H, Gilbert CA, Hudson JA, Bolick SC,
Wright KL and Piskurich JF: Positive regulatory domain I-binding
factor 1 mediates repression of the MHC class II transactivator
(CIITA) type IV promoter. Mol Immunol. 44:1461–1470. 2007.
View Article : Google Scholar :
|
|
12
|
Pisapia L, Pozzo GD, Barba P, Citro A,
Harris PE and Maffei A: Contrasting effects of IFNalpha on MHC
class II expression in professional vs. nonprofessional APCs: Role
of CIITA type IV promoter. Results Immunol. 2:174–183. 2012.
View Article : Google Scholar
|
|
13
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sandoval J and Esteller M: Cancer
epigenomics: beyond genomics. Curr Opin Genet Dev. 22:50–55. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burke MJ and Bhatla T: Epigenetic
modifications in pediatric acute lymphoblastic leukemia. Front
Pediatr. 2:422014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chatterton Z, Burke D, Emslie KR, et al:
Validation of DNA methylation biomarkers for diagnosis of acute
lymphoblastic leukemia. Clin Chem. 60:995–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nordlund J, Backlin CL, Wahlberg P, et al:
Genome-wide signatures of differential DNA methylation in pediatric
acute lymphoblastic leukemia. Genome Biol. 14:r1052013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Savickiene J, Treigyte G, Valiuliene G,
Stirblyte I and Navakauskiene R: Epigenetic and molecular
mechanisms underlying the antileukemic activity of the histone
deacetylase inhibitor belinostat in human acute promyelocytic
leukemia cells. Anticancer Drugs. 25:938–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Londhe P, Zhu B, Abraham J, Keller C and
Davie J: CIITA is silenced by epigenetic mechanisms that prevent
the recruitment of transactivating factors in rhabdomyosarcoma
cells. Int J Cancer. 131:E437–E448. 2012. View Article : Google Scholar :
|
|
21
|
Cycon KA, Mulvaney K, Rimsza LM, Persky D
and Murphy SP: Histone deacetylase inhibitors activate CIITA and
MHC class II antigen expression in diffuse large B-cell lymphoma.
Immunology. 140:259–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Candiano G, Bruschi M, Musante L, et al:
Blue silver: A very sensitive colloidal Coomassie G-250 staining
for proteome analysis. Electrophoresis. 25:1327–1333. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dodd KW, Burns TC, Wiesner SM, et al:
Transgenic mice expressing luciferase under a 4.5 kb tyrosine
hydroxylase promoter. Cureus. 3:e342011.
|
|
24
|
Baev MV, Baev D, Radek AJ and Campbell JW:
Growth of Escherichia coli MG1655 on LB medium: Monitoring
utilization of sugars, alcohols, and organic acids with
transcriptional micro-arrays. Appl Microbilol Biot. 71:310–316.
2006. View Article : Google Scholar
|
|
25
|
Cohn M: How does the immune response get
started? Cell Immunol. 254:91–93. 2009. View Article : Google Scholar :
|
|
26
|
Weiss A, Imboden J, Hardy K, et al: The
role of the T3/antigen receptor complex in T-cell activation. Annu
Rev Immunol. 4:593–619. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Santos DO, Miranda A, Suffys P, et al:
Current understanding of the dendritic cells and their
co-stimulatory molecules as a key in generating efficient T cell
responses in lepromatous leprosy. Curr Immunol Rev. 3:77–85. 2007.
View Article : Google Scholar
|
|
28
|
Shafer-Weaver K, Anderson M, Malyguine A
and Hurwitz AA: T cell tolerance to tumors and cancer
immunotherapy. Adv Exp Med Biol. 601. pp. 357–368. 2007
|
|
29
|
Ueki T, Murata S, Kitamura N, Mekata E and
Tani T: Pre-treatment with cyclophosphamide or OX40 (CD134)
costimulation targeting regulatory T cell function enhances the
anti-tumor immune effect of adoptively transferred CD8+ T cells
from wild-type mice. Mol Med Rep. 2:615–620. 2009.PubMed/NCBI
|
|
30
|
Hung K, Hayashi R, Lafond-Walker A, et al:
The central role of CD4(+) T cells in the antitumor immune
response. J Exp Med. 188:2357–2368. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Magner WJ, Kazim AL, Stewart C, et al:
Activation of MHC class I, II and CD40 gene expression by histone
deacetylase inhibitors. J Immunol. 165:7017–7024. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garrido C, Paco L, Romero I, et al: MHC
class I molecules act as tumor suppressor genes regulating the cell
cycle gene expression, invasion and intrinsic tumorigenicity of
melanoma cells. Carcinogenesis. 33:687–693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Seliger B: The link between MHC class I
abnormalities of tumors, oncogenes, tumor suppressor genes and
transcription factors. J Immunotoxicol. 11:308–310. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Daurkin I, Eruslanov E, Vieweg J and
Kusmartsev S: Generation of antigen-presenting cells from
tumor-infiltrated CD11b myeloid cells with DNA demethylating agent
5-aza-2′-deoxycytidine. Cancer Immunol Immunother. 59:697–706.
2010. View Article : Google Scholar
|
|
35
|
Sigalotti L, Coral S, Fratta E, et al:
Epigenetic modulation of solid tumors as a novel approach for
cancer immunotherapy. Semin Oncol. 32:473–478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reith W, Muhlethaler-Mottet A, Masternak
K, Villard J and Mach B: The molecular basis of MHC class II
deficiency and transcriptional control of MHC class II gene
expression. Microbes Infect. 1:839–846. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ulbricht T, Alzrigat M, Horch A, et al:
PMl promotes MHC class II gene expression by stabilizing the class
II transactivator. J Cell Biol. 199:49–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Truax AD, Thakkar M and Greer SF:
Dysregulated recruitment of the histone methyltransferase EZH2 to
the class II transactivator (CIITA) promoter IV in breast cancer
cells. PLoS One. 7:e360132012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
De Lerma Barbaro A, De Ambrosis A, Banelli
B, et al: Methylation of CIITA promoter IV causes loss of HLA-II
inducibility by IFN-gamma in promyelocytic cells. Int Immunol.
20:1457–1466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Radosevich M, Song Z, Gorga JC, Ksander B
and Ono SJ: Epigenetic silencing of the CIITA gene and
posttranscriptional regulation of class II MHC genes in ocular
melanoma cells. Invest Ophthalmol Vis Sci. 45:3185–3195. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Holtz R, Choi JC, Petroff MG, Piskurich JF
and Murphy SP: Class II transactivator (CIITA) promoter methylation
does not correlate with silencing of CIITA transcription in
trophoblasts. Biol Reprod. 69:915–924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cameron EE, Bachman KE, Myohanen S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shaker S, Bernstein M, Momparler LF and
Momparler RL: Preclinical evaluation of antineoplastic activity of
inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone
deacetylation (trichostatin A, depsipeptide) in combination against
myeloid leukemic cells. Leuk Res. 27. pp. 437–444. 2003, View Article : Google Scholar
|