Introduction
Glioma is a type of tumor, which grows from glial
cells, a supportive cell in the brain, and is the most
life-threatening type of brain tumor (1). Glioblastoma multiforme (GBM) is the
most advanced and aggressive subtype of glioma, and patients with
GBM have a median survival rate of ~14 months (2). As a complex medical condition, GBM is
considered to be result from the interaction between multiple
genetic and environmental factors (3). Currently, only a small number of low
penetrance variants attributing to cancer risk have been identified
using genome-wide association investigations (4), and the etiology remains to be
elucidated. Previous efforts to comprehensively characterize the
genomes of primary GBM have established that the disease is driven
by numerous and diverse genetic events in individual patients
(5,6).
It is generally accepted that somatic mutations are
important in the pathogenesis of cancer, and that cancer develops
via the accumulation of somatic mutations in certain
cancer-specific genes, including oncogenes and tumor suppressors,
depending on the type of tumor (7). Previous studies have demonstrated
that the frequency of somatic mutations in candidate cancer genes
is significantly higher than expected, and that the combination of
certain individual mutations may have a specific effect on the
properties of the tumor (8–11).
These mutations are considered to result from a combination of
environmental and genetic factors (12). Following the determination of the
human genome sequence, several investigations have been performed
to determine somatic mutations in various types of cancer. The
Sanger sequencing technique was used to directly sequence 13,023
genes, identifying 189 genes carrying excessive somatic mutations
in human breast and colorectal cancer (10). A mismatch repair detection method
was used to screen 22 cell lines and 93 matched tumor-control
sample pairs, for somatic mutations in 30 cancer-associated genes A
total of 152 somatic mutations were identified in breast and
colorectal cancer (13), including
genes, which are reported to be involved in the development of
cancer, including Kirsten rat sarcoma virus oncogene and v-raf
murine sarcoma viral oncogene homolog B (BRAF).
To further investigate the prevalence and
distribution of somatic mutations in glioma, the present study
aimed to examine 63 glioma tissues and their matched normal tissues
for somatic mutations in 20 genes, which have been reported to as
affirmatively or potentially associated with oncogenesis (14,15).
A total of six somatic mutations were identified, with three
repeated somatic mutations observed exclusively in myeloid cell
leukemia sequence 1 (Mcl-1). The effect of these somatic mutations
were subsequently investigated.
Materials and methods
Subjects and tumor samples
A total of 63 patients (42 male and 21 female) with
histologically confirmed GBM were recruited at the Department of
Neurosurgery, The Affiliated Hospital of Peking University
(Beijing, China), where the patients received surgical resection.
The median age of the patients was 55 years (range, 45–67 years)
and none had received any preoperative treatment. All the tumor
specimens were examined by two independent, experienced
pathologists prior to establishing the final diagnosis.
High-fidelity polymerase chain reaction (PCR) and direct sequencing
were performed to screen the coding region of 20 genes (Mcl-1,
Bcl-2, EGFR, KRAS, FGR, IKZF1, BTG1, TP53, PAX5, BRAF, BACH2, ARF,
Bcl6, VEGF, HER2, β-catenin, c-myc, Rb, EZH2, E2F) for potential
somatic mutations in 20 patients, and the gene of interest was
further screened in the remaining 43 patients. The size of the
tumor was determined by measuring the maximum tumor diameter
presented on radiographic images, including computed tomography
scans and magnetic resonance imaging. The tumor tissue samples were
rapidly frozen immediately following surgical resection in liquid
nitrogen and stored at -80°C for future investigations. Peripheral
blood samples (5 ml) were acquired from all patients. Informed
consent was obtained from the patient’s families, and the present
study was approved by the Review Board of the Hospital Ethics
Committee at Dalian Medical University (Dalian, China).
DNA extraction and nucleotide sequencing
analysis
Total genomic DNA was isolated from the GBM tumor
tissue samples and peripheral blood using a DNA extraction kit
(Invitrogen Life Technologies, Carlsbad, CA, USA). The isolated DNA
was dissolved in sterilized Milli-Q water (EMD Millipore,
Billerica, MA, USA) and quantified using a NanoDrop
spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA).
Agarose electrophoresis (Sigma-Aldrich, St. Louis, MO, USA) was
used to confirm the purity of the DNA. The chromosome segments
comprising the coding region of each gene were amplified by PCR
(primer set: forward 5′-GTATCGAGCTAGCGCTCCGCTATG-3′ and reverse
5′-GTACTCTTCAGCGAGCTAGATAT-3′; Sangong Biotech, Shanghai, China),
according to the following PCR cycling conditions: 95°C for 10 min;
35 cycles of 95°C for 30 sec, 58°C for 30 sec, 72°C for 1 min,
followed by 72°C for 10 min. The PCR products were purified using
an ExoSAP-IT purification kit (United States Biochemical Corp.,
Cleveland, OH, USA), prior to sequencing using an ABI sequencing
system by PerkinElmer Applied Biosystems (Foster City, CA, USA).
The sequences were subsequently aligned against the revised
Cambridge sequence in the MITOMAP database (www.mitomap.org) using the MegAlin program from the
DNAStar software package 12.1 (DNASTAR, Inc., Madison, WI, USA).
The sequence alterations identified in the tumor tissue samples,
but not in the matched peripheral blood samples, were recorded as
somatic mutations. The screened somatic mutations were confirmed at
least twice by additional independent PCR and resequencing.
Cell lines and cell culture
The U251 GBM cell line was cultured in RPMI-1640
medium (Invitrogen Life Technologies) containing 10% fetal bovine
serum (Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada), 100
U/ml penicillin and 100 mg/ml streptomycin (Invitrogen Life
Technologies).
Apoptotic assay
Apoptosis was determined using an annexin
V/fluorescein isothiocyanate (FITC) apoptosis detection kit (Keygen
Biotech Co., Ltd., Nanjing, China), according to the manufacturer’s
instructions. Briefly, glioma cells (2×108 cells)
cultured in 10 cm dishes were trypsinized (Invitrogen Life
Technologies), washed with phosphate buffered saline (PBS) and
subsequently stained with FITC-conjugated anti-annexin V antibody
in the dark for 15 min at room temperature. The cells were then
analyzed by flow cytometry (FACSCanto II; BD Biosciences, San Jose,
CA, USA). All experiments were performed in triplicate.
Cell viability and proliferation
assay
Cell proliferation was measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphen-yltetrazolium bromide (MTT)
assay. The cells were seeded into 96-well plates at a density of
800 cells/well and were incubated at 75°C for 1, 2 or 3 days. MTT
(20 μl; 5 mg/ml; Sigma-Aldrich) was added to each well and
incubated for 4 h at room temperature. The supernatants were
subsequently removed and 150 μl dimethyl sulfoxide
(Sigma-Aldrich) was added. The absorbance value of each well was
measured at 490 nm (EnSight™ Multimode Plate Reader; PerkinElmer,
Inc., Boston, MA, USA). All experiments were performed in
triplicate.
Cloning of pcDNA4 and mutagenesis
The cDNA of full-length human Mcl-1 was subcloned
into the pcDNA4 vector (Invitrogen Life Technologies).
Site-directed mutagenesis was performed using a QuikChange
lightening kit (Stratagene, La Jolla, CA, USA), according to the
manufacturer’s instructions. The sequences of the primer sets used
for subcloning and mutagenesis are shown in Table I.
 | Table IPrimer sequences for the subcloning
and mutagenesis of Mcl-1. |
Table I
Primer sequences for the subcloning
and mutagenesis of Mcl-1.
| Primer | Sequence
(5′-3′) |
|---|
| Mcl-1 |
| Forward |
CGGGATCCCCCATGTACCCATACGATGTTCCAGATTACGCTTTTGGCCTCAAAAGAAACGCGGTA |
| Reverse |
CTATCTTATTAGATATGCCAAACCAGCTCCTAC |
| D155G |
| Forward |
CTGGTAATAACACCAGTACGGGCGGGTCACTACCCTCGACGCC |
| Reverse |
GGCGTCGAGGGTAGTGACCCGCCCGTACTGGTGTTATTACCAG |
| D155H |
| Forward |
CTGGTAATAACACCAGTACGCGCGGGTCACTACCCTCGACGCC |
| Reverse |
GGCGTCGAGGGTAGTGACCCGCGCGTACTGGTGTTATTACCAG |
| L174S |
| Forward |
CAGAGGAGGAGGAGGACGAGTCGTACCGGCAGTCGCTGGAGAT |
| Reverse |
ATCTCCAGCGACTGCCGGTACGACTCGTCCTCCTCCTCCTCTG |
Half-life determination
35S methionine (Guidechem, Hangzhou,
China) pulse-chase labeled glioma cells over-expressing wild-type
Mcl-1 (D155G, D155S or L174S) were plated into media deficient in
methionine for 1 h at 37°C. The cells were washed with PBS and
pulsed with labeling media containing 35S methionine, at
a final concentration of 100 mCi/ml−1, and the synthesis
of Mcl-1 was inhibited by exposure to ultraviolet light. The cells
were washed and harvested at 0, 0.5, 1 and 2 h. and treated with 1
mg/ml−1 recombinant TRAIL (R&D Systems, Minneapolis,
MN, USA) for 30 min.
Immunoprecipitation
The cells transfected with wild-type or mutant Mcl-1
were incubated with beads coupled to antibody. Protein A agarose
beads (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 0.5 ml
bed volume) were incubated in 1 mg/ml BSA in 1 ml
phosphate-buffered saline containing 100 μg polyclonal
anti-Mcl-1 antibody (Santa Cruz Biotechnology, Inc.) at 4°C
overnight. Following incubation, the beads were pelleted by
centrifugation and washed three times. The radiolabeled cells were
harvested and lysed in cold radioimmunoprecipitation (RIPA) lysis
buffer, containing 10 mM Tris-HCl (pH 7.4), 1% NP-40, 1 mM EDTA,
0.1% sodium dodecyl suplhate (SDS) and 150 mM NaCl, supplemented
with fresh proteinase inhibitor cocktail (Sigma-Aldrich), followed
by breaking through a 25 gauge syringe 20 times. Equivalent
quantities of the cell extract were adjusted to equal volumes using
RIPA buffer, precleared with protein A agarose beads and clarified
by centrifugation at 400 × g for 10 min at 4°C. The lysates were
subsequently incubated with anti-Mcl-1 antibody at 4°C overnight
and the immune complexes were precipitated using protein A agarose
beads for 6 h at 120°C. The immunoprecipitates were washed three
times with RIPA buffer and incubated at 95°C for 5 min in SDS
sample buffer. The protein samples were separated on an
SDS-polyacrylamide gel electrophoresis (PAGE) gel, followed by
autoradiography (Biospace Lab, Nesles la Vallée, France) to detect
the signals.
Western blot analysis
U251 cells were transfected with wild-type or mutant
Mcl-1 and were lysed in lysis buffer (Beyotime, Shanghai, China) on
ice 72 h after transfection. The cell lysates were loaded onto 10%
SDS-PAGE gels (Invitrogen Life Technologies) and the separated
proteins were transferred onto a polyvinylidene fluoride membrane
(EMD Millipore), which was subsequently blocked with Tris-buffered
saline, containing 5% non-fat dry milk at room temperature for 1 h.
The membrane was subsequently incubated with the mouse monoclonal
anti-haemagglutinin tag antibody (cat. no. 12ca5; Roche
Diagnostics, Mannheim, Germany; 1:1,000) and rabbit polyclonal
anti-Mcl-1 antibody (cat. no. sc-819; Santa Cruz Biotechnology,
Inc.; 1:2,000) at 4°C overnight, followed by incubation with
horseradish peroxidase-anti-rabbit secondary antibody (Cell
Signaling Technology, Inc., Danvers, MA, USA; cat. no. 7074S) and
goat-anti-mouse (cat. no. sc-2031; 1:10,000; Santa Cruz
Biotechnology, Inc.) at room temperature for 1.5 h. Chemical
fluorescence was detected using an enhanced chemilluminescence kit
(Amersham Biosciences, Piscataway, NJ, USA), according to the
manufacturer’s instructions. The target bands were
densitometrically analyzed and normalized against actin.
Statistical analysis
Statistical analysis was performed using SPSS 19.0
software (SPSS, Inc., Chicago, IL, USA). The association between
somatic mutations and clinicopathologic parameters of GBM was
examined using Fisher’s exact test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Identification of genetic
determinants
To identify the genetic determinants of GBM, the
present study initially performed Sanger sequencing of 20
cancer-associated genes in the tumor tissue samples and the
corresponding blood genomic DNA from 20 patients with
histologically confirmed GBM. Comparison between the tumor and
corresponding blood DNA revealed five somatic mutations in the
coding regions of four of the 20 candidate genes, with two somatic
mutations located in the proline, glutamic acid, serine,
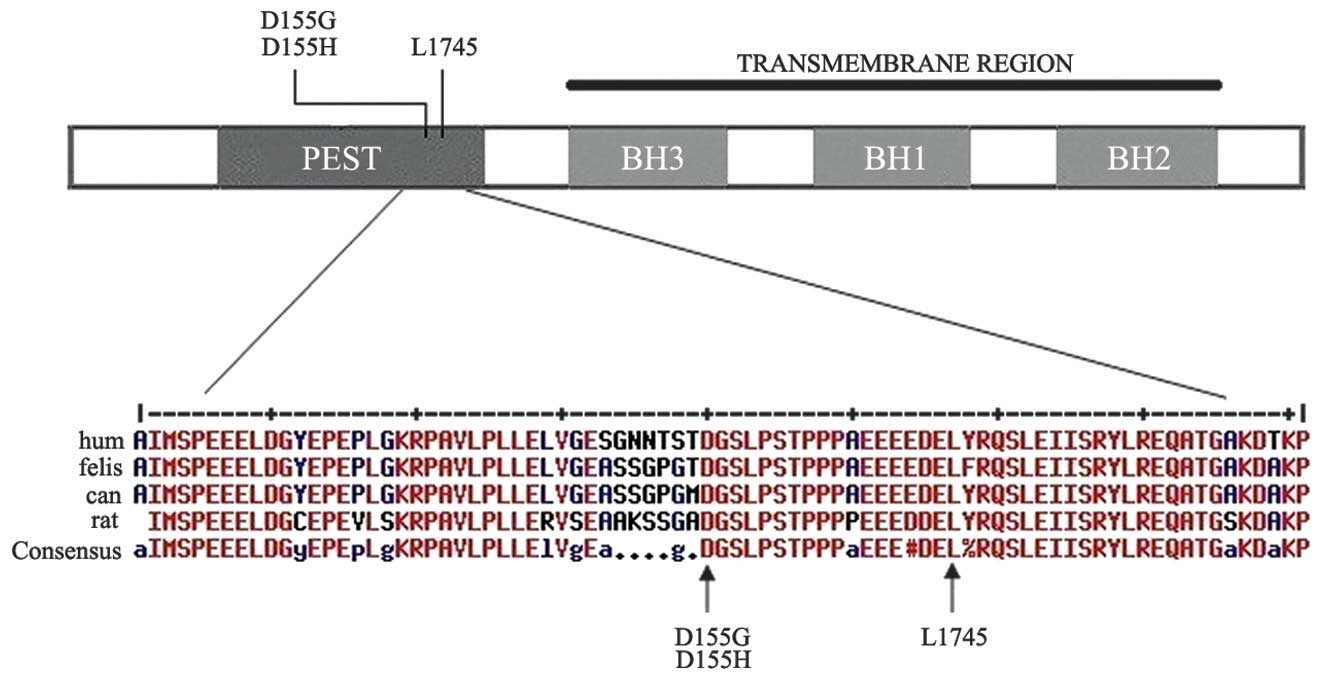
threonine-rich (PEST) region of Mcl-1 (Fig. 1). The five somatic mutations were
non-synonymous missense point mutations, as shown in Table II. In addition, four of the
mutations were heterozygous (three Mcl-1 mutations and one ASAP3
mutation) and the other was homozygous. Notably, within this small
set of genetic alterations, multiple somatic mutations were
revealed exclusively in Mcl-1, a member of the B-cell lymphoma
(Bcl-2) family. Missense variants in Mcl-1 were present in two of
the 20 cases (c.A447G and c.T521C), leading to p.D155G and p.L174S
substitutions, respectively. The present study then focussed on
Mcl-1 and expanded the investigation into the remaining 43 GBM
tissue samples. This revealed another somatic mutation (c.G446C,
p.D155H), which was in an identical amino acid position as one of
the originally identified somatic mutations in Mcl-1.
| Table IIDescription of the somatic mutations
identified. |
Table II
Description of the somatic mutations
identified.
| Gene | Nucleotide | Type | Amino acid
change |
|---|
| Mcl-1 |
GTACGGA>GCGGGTC | Missense | D>D/G
(157aa) |
| Mcl-1 |
GTACGG>CACGGGTC | Missense | D>D/H
(157aa) |
| Mcl-1 |
ACGAGTT>CGTACCG | Missense | L>S (174aa) |
| ZMYM3 |
GGGTCG>CTCCTG | Missense | R>R/P
(1363aa) |
| SORCS1 |
GCCGAG>CGCCCT | Missense | G>G/R
(460aa) |
| ASAP3 |
TCAATG>CAGGTC | Missense | E>E/Q
(504aa) |
Sequence comparisons
As shown in Fig. 1,
sequence comparisons demonstrated that the affected amino acids in
Mcl-1 are highly conserved among species. Projection of the somatic
mutations onto the amino acid sequence of Mcl-1 revealed that all
three alterations were located in the PEST region of the enzyme.
These three missense point mutations may exhibit functional
consequences by affecting the degradation of the protein, since
they are all accumulated at highly evolutionarily conserved amino
acid residues of the respectively affected subunits (Fig. 1). The PEST region is a well known
degradation regulatory element in certain proteins (16). The recurrence of mutations
affecting these highly conserved regions involved in the regulation
of degradation is suggestive of a gain-of-function effect.
Therefore, these mutations are hypothesized to compromise the
degradation of the protein, resulting in the intracellular
accumulation of Mcl-1.
Mcl-1 post-translational
modifications
Mcl-1 is reported to undergo various
post-translational modifications and the present study hypothesized
that the amino acid alternations identified may affect the
phosphorylation of the residual amino acids, thereby preventing the
degradation of the protein. However, no differences in the
phosphorylation of Mcl-1 were identified between the wild-type and
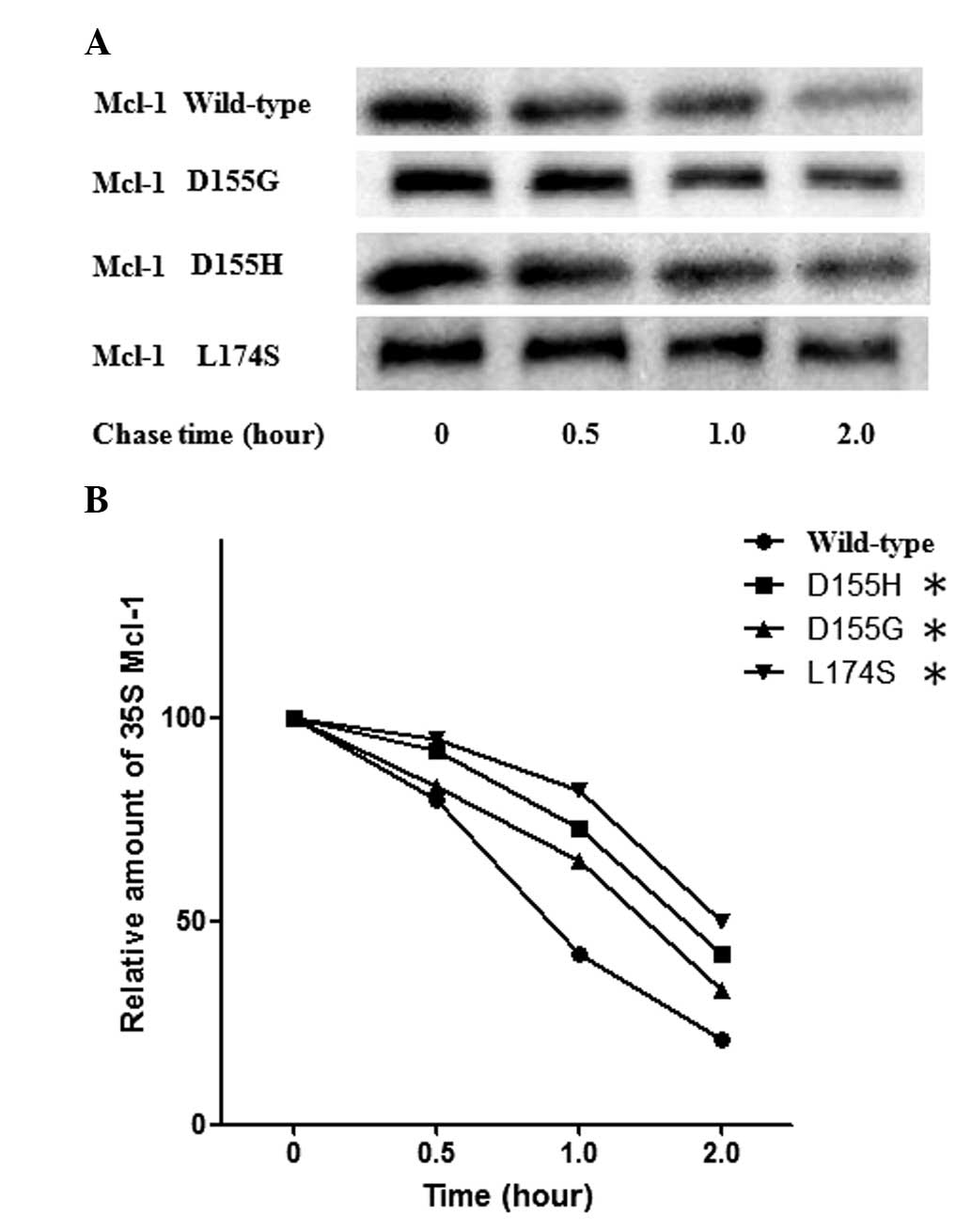
the mutants (data not shown). The half-life of the Mcl-1 protein
was previously reported to be between 30 and 90 min (16–18).
The present study subsequently determined whether the mutations
identified affected the half-life of Mcl-1. The wild-type Mcl-1 or
the mutant forms were expressed in the GBM cell line and the
half-life of Mcl-1 was determined using 35S methionine
pulse-chase labeling. Consistent with the previous findings, the
wild-type Mcl-1 had a half-life of ~1 h in the U251 cells (Fig. 2). By contrast, the three mutants
were more stable compared with the wild-type and exhibited a
half-life of ~2 h (Fig. 2).
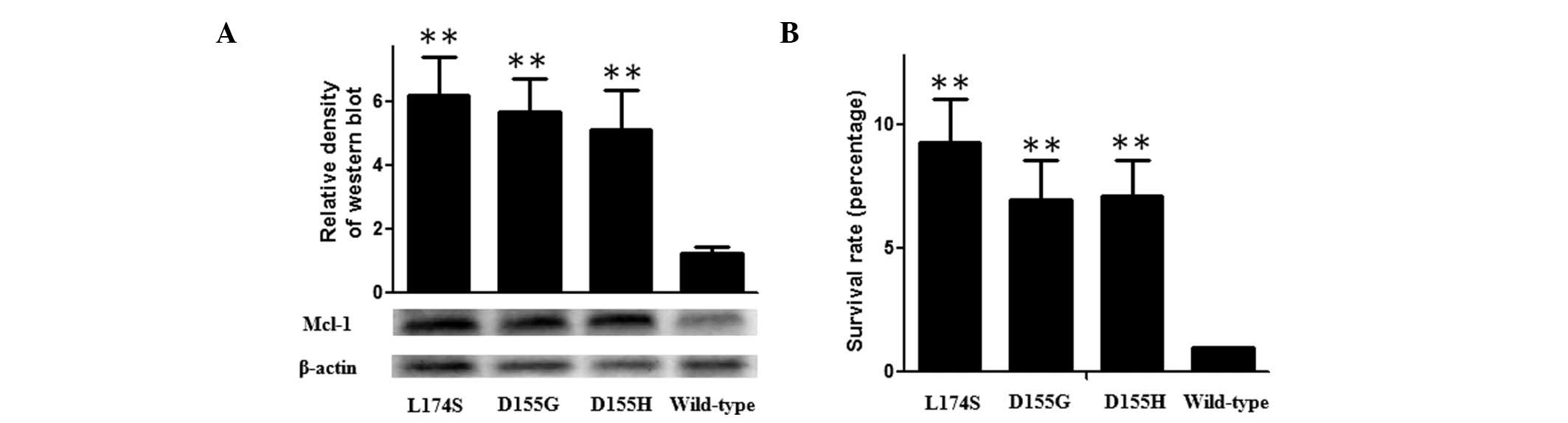
Cell proliferation
Since Mcl-1 functions as an anti-apoptotic protein
and the somatic mutations resulted in the intracellular
accumulation of the enzyme, the present study expressed the mutants
and wild-type Mcl-1 transiently in U251 cells and examined the cell
proliferation rate using an MTT assay. As shown in Fig. 3, the ectopic expression levels of
the three mutants were significantly higher compared with the
wild-type, and the survival rate of the U251 cells transfected with
wild-type Mcl-1 was significantly higher compared with the mutants
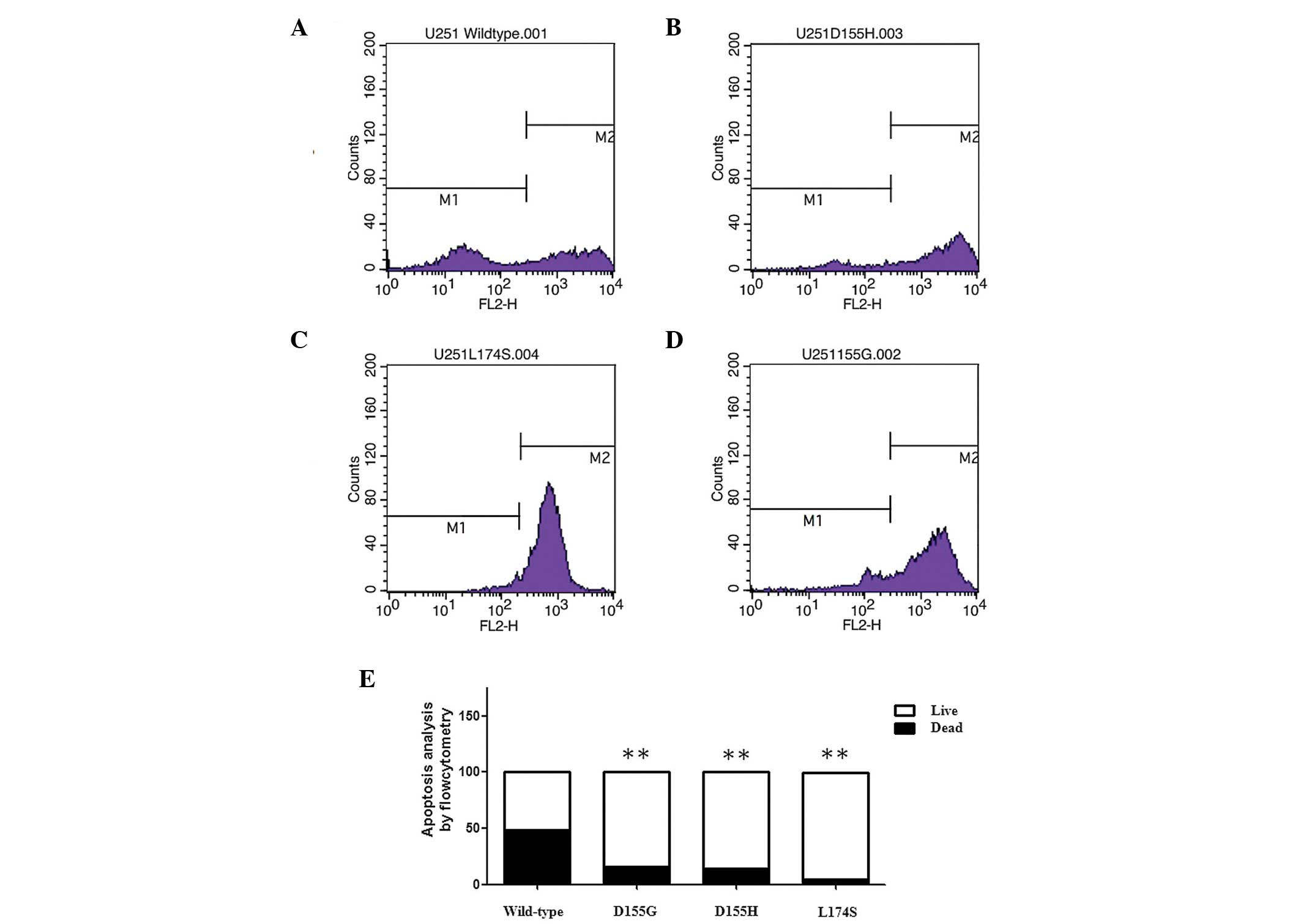
treated with TRAIL. To investigate the underlying mechanism, the
present study examined whether mutant Mcl-1 had a more marked
effect on TRAIL-induced apoptosis in glioma cells using flow
cytometry. The Mcl-1 mutants suppressed TRAIL-induced apoptosis to
a greater extent compared with the wild-type in the glioma cell
line (Fig. 4)
Discussion
The identification of cancer specific somatic
mutations may improve current understanding of the molecular
mechanisms underlying tumorigenesis and the progression of
malignancy in human cells. This has substantially contributed to
the development of highly targeted treatments using monoclonal
antibodies or specific inhibitors, which are characterized by a
marked improvement in clinical efficacy and an evident reduction in
adverse effects compared with conventional chemotherapeutic agents
(19,20). Positive results from several
clinical trials have led to the approval of similar therapeutic
agents for melanoma (vemurafenib/BRAF), non-small cell lung cancer
(crizotinib/anaplastic lymphoma kinase) and myelo-dysplasia
(ruxolitininb/Janus kinase 2; JNK2) (18), and the improved capability to
identify novel targets is likely to assist in expanding the list of
targeted therapies. GBM is one of the most life-threatening
malignancies and is the most common type of primary brain tumor in
adults (21). The standard
treatments, including surgery or chemoradiotherapy, are rarely
curative, and the majority of tumors recur within a few months. A
previous comprehensive genomic study demonstrated that the genetic
landscape of GBM is heterogeneous, with 80% of patients affected in
one of the three main signaling pathways: p53,
phosphatidylinositol-4,5-bisphosphate 3-kinase α and retinoblastoma
(6). The present study identified
a number of protein-altering somatic mutations by directly
sequencing 20 cancer-associated genes (Table II). Notably, within this small set
of genetic alterations, multiple somatic mutations were identified
exclusively in Mcl-1. Missense variants in Mcl-1 were present in
two of the 20 cases (D155G and L174S). Investigating Mcl-1 in the
remaining 43 GBM cases, revealed another somatic mutation (D155H),
which was close to the former two variants.
Mcl-1, initially identified in differentiating
myeloid cells, is a member of the anti-apoptotic Bcl-2 family with
unique structural features (22).
Although the C-terminal sequence of Mcl-1 shares similarities with
other Bcl-2 family members, the N-terminus lacks the characteristic
BH4 domain. Instead, it contains two highly conserved proline,
glutamic acid, serine and threonine-rich PEST sequences (23), with two caspase cleavage sites,
Asp127 and Asp157, and several phosphorylation sites, which are
involved in regulating the degradation and stability of Mcl-1
(24–27). Thr163 is the major phosphorylation
site in Mcl-1, and extracellular signal-regulated kinase-mediated
phosphorylation at Thr163 and Thr92 increases its binding to Pin-1,
thereby increasing its stability and its antiapoptotic function
(28). By contrast,
phosphorylation of Ser121 and Thr163 suppresses the anti-apoptotic
function of Mcl-1 (29,30) and joint phosphorylation of Thr163
and Ser159, mediated by JNK and glycogen synthase kinase3β,
promotes the degradation of Mcl-1 and interferes with its
interaction with the pro-apoptotic protein, Bim (31).
The present study demonstrated that the affected
amino acids are highly conserved among species (Fig. 1). Projection of the somatic
mutations onto the amino acid sequence of Mcl-1 revealed that the
three alterations were located in the PEST region of the enzyme.
The recurrence of mutations affecting these highly conserved
regions, which are involved in the regulation of degradation, is
suggestive of a gain-of-function effect. To investigate the effect
of these somatic mutations on the function of Mcl-1, the coding
sequences of wild-type Mcl-1 and the mutants corresponding to the
amino acid alternations were cloned into pcDNA4. These constructs
were subsequently expressed in the GBM cell line and the half-life
of Mcl-1 was determined by 35S methionine pulse-chase
labeling. Wild-type Mcl-1 exhibited a half-life of ~1 h in the U251
cells (Fig. 2). By contrast, the
mutants were more stable, with a half-life of ~2 h (Fig. 2).
Mcl-1 is readily induced by various survival
regulators, including epidermal growth factor, vascular endothelial
growth factor, granulocyte-macrophage colony-stimulating factor,
mitogen activated protein kinase and JAK/signal transducer and
activator of transcription signaling cascades, and is also rapidly
degraded by certain apoptosis-inducing signals (32,33).
The rapid induction and degradation of Mcl-1 suggested that Mcl-1
functions as sensor and reactor of environmental stimuli to
maintain a balance between cell survival and death, suggesting that
Mcl-1 is an essential regulator of proliferation or differentiation
of human cells (34,35). Inhibition or elimination of Mcl-1
in response to cytotoxic signals is considered critical in cell
death in a number of normal and malignant cells (34,36).
In addition, increased expression of Mcl-1 may not only promote
short-term survival in a wide range of cells, but may also cause
long-term immortalization and the malignant transformation of
certain human cells (37–39). Previous studies have suggested that
overexpression of Mcl-1 may be involved in the mechanism underlying
chemoresistance in a number of human malignancies, including breast
cancer, leukemia, melanoma, pancreatic cancer and hepatocellular
carcinoma (33,40). The present study examined the
effect of the identified mutants on the growth of glioma cells and
TRAIL-induced apoptosis in U271 cells, and found that the survival
rate of the cells transfected with wild-type Mcl-1 was
significantly lower compared with the Mcl-1 mutants. Additionally,
the Mcl-1 mutants suppressed TRAIL-induced apoptosis to a greater
extent compared with the wild-type in the glioma cell lines
(Figs. 3 and 4).
In conclusion, despite the small sample size, the
present study identified a panel of somatic mutations in 20
cancer-associated genes, and found that several mutations may be
pathogenic with potential detrimental impacts on gliomagenesis,
within which multiple somatic mutations were identified in Mcl-1.
The preliminary functional investigations suggested that the
mutations increased the stability of Mcl-1 and contributed to the
pathogenesis of GBM. Future studies using a larger number of
patients from different ethnic backgrounds are required to confirm
the findings of the present study.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, et al: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hemminki K and Li X: Familial risks in
nervous system tumors. Cancer Epidemiol Biomarkers Prev.
12:1137–1142. 2003.PubMed/NCBI
|
|
4
|
Scheurer ME, Etzel CJ, Liu M, El-Zein R,
Airewele GE, Malmer B, Aldape KD, Weinberg JS, Yung WK and Bondy
Ml: Aggregation of cancer in first-degree relatives of patients
with glioma. Cancer Epidemiol Biomarkers Prev. 16:2491–2495. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, et al: An integrated genomic analysis of human
glioblastoma multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sjöblom T, Jones S, Wood LD, Parsons DW,
Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al:
The consensus coding sequences of human breast and colorectal
cancers. Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lichtenstein P, Holm NV, Verkasalo PK,
Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A and Hemminki
K: Environmental and heritable factors in the causation of
cancer-analyses of cohorts of twins from Sweden, Denmark and
Finland. N Engl J Med. 343:78–85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bentivegna S, Zheng J, Namsaraev E,
Carlton VE, Pavlicek A, Moorhead M, Siddiqui F, Wang Z, Lee L,
Ireland JS, et al: Rapid identification of somatic mutations in
colorectal and breast cancer tissues using mismatch repair
detection (MRD). Hum Mutat. 29:441–450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Di Lonardo A, Nasi S and Pulciani S:
Cancer: we should not forget the past. J Cancer. 6:29–39. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Erstad DJ and Jr JC: Mutational analysis
of merkel cell carcinoma. Cancers (Basel). 6:2116–2136. 2014.
View Article : Google Scholar
|
|
16
|
Schubert KM and Duronio V: Distinct roles
for extracellular-signal-regulated protein kinase (ERK)
mitogen-activated protein kinases and phosphatidylinositol 3-kinase
in the regulation of Mcl-1 synthesis. Biochem J. 356:473–480. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chao JR, Wang JM, Lee SF, Peng HW, Lin YH,
Chou CH, et al: Mcl-1 is an immediate-early gene activated by the
granulocyte-macrophage colony-stimulating factor (GMCSF) signaling
pathway and is one component of the GM-CSF viability response. Mol
Cell Biol. 18:4883–4898. 1998.PubMed/NCBI
|
|
18
|
Nijhawan D, Fang M, Traer E, Zhong Q, Gao
W, Du F and Wang X: Elimination of Mcl-1 is required for the
initiation of apoptosis following ultraviolet irradiation. Genes
Dev. 17:1475–1486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Varela I, Tarpey P, Raine K, Huang D, Ong
CK, et al: Exome sequencing identifies frequent mutation of the
SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 469:539–542.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jones S, Wang TL, Shih IM, Mao TL,
Nakayama K, et al: Frequent mutations of chromatin remodeling gene
ARID1A in ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wen PY and Kesari S: Malignant Gliomas in
Adults. N Engl J Medicine. 359:492–507. 2008. View Article : Google Scholar
|
|
22
|
Kozopas KM, Yang T, Buchan HL, Zhou P and
Craig RW: MCL1, a gene expressed in programmed myeloid cell
differentiation, has sequence similarity to Bcl-2. Proc Natl Acad
Sci USA. 90:3516–3520. 1993. View Article : Google Scholar
|
|
23
|
Day CL, Chen L, Richardson SJ, Harrison
PJ, Huang DCS and Hinds MG: Solution structure of prosurvival Mcl-1
and characterization of its binding by proapoptotic BH3-only
ligands. J Biol Chem. 280:4738–4744. 2005. View Article : Google Scholar
|
|
24
|
Clohessy JG, Zhuang J and Brady HJM:
Characterisation of Mcl-1 cleavage during apoptosis of
haematopoietic cells. Br J Haemato. 125:655–665. 2004. View Article : Google Scholar
|
|
25
|
Domina AM, Smith JH and Craig RW: Myeloid
cell leukemia 1 is phosphorylated through two distinct pathways,
one associated with extracellular signal regulated kinase
activation and the other with G2/M accumulation or protein
phosphatase 1/2A inhibition. J Biol Chem. 275:21688–21694. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Domina AM, Vrana JA, Gregory MA, Hann SR
and Craig RW: MCL1 is phosphorylated in the PEST region and
stabilized upon ERK activation in viable cells and at additional
sites with cytotoxic okadaic acid or taxol. Oncogene. 23:5301–5315.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhong Q, Gao W, Du F and Wang X:
Mule/ARFBP1, a BH3-only E3 ubiquitin ligase, catalyzes the
polyubiquitination of Mcl-1 and regulates apoptosis. Cell.
121:1085–1095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao
Y, Chang CJ, Yang Y, Lai CC, Lee DF, Yen CJ, Chen YJR, Hsu JM, Kuo
HP, Lin CY, Tsai FJ, Li LY, Tsai CH and Hung MC: Down-regulation of
myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by
sorafenib facilitates chemosensitization in breast cancer. Cancer
Res. 68:6109–6117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Inoshita S, Takeda K, Hatai T, Terada Y,
Sano M, Hata J, Umezawa A and Ichijo H: Phosphorylation and
inactivation of myeloid cell leukemia 1 by JNK in response to
oxidative stress. J Biol Chem. 277:43730–43734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morel C, Carlson SM, White FM and Davis
RJ: Mcl-1 integrates the opposing actions of signaling pathways
that mediate survival and apoptosis. Mol Cell Biol. 29:3845–3852.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maurer U, Charvet C, Wagman AS, Dejardin E
and Green DR: Glycogen synthase kinase-3 regulates mitochondrial
outer membrane permeabilization and apoptosis by destabilization of
Mcl-1. Mol Cell. 21:749–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Michels J, Johnson PW and Packham G:
Mcl-1. Int J Biochem Cell Biol. 37:267–71. 2005. View Article : Google Scholar
|
|
33
|
Craig RW: Mcl-1 provides a window on the
role of the Bcl-2 family in cell proliferation, differentiation and
tumorigenesis. Leukemia. 16:444–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rinkenberger JL, Horning S, Klocke B, Roth
K and Korsmeyer SJ: Mcl-1 deficiency results in periimplantation
embryonic lethality. Genes Dev. 14:23–27. 2000.PubMed/NCBI
|
|
35
|
Opferman JT, Letai A, Beard C, Sorcinelli
MD, Ong CC and Korsmeyer SJ: Development and maintenance of B and T
lymphocytes requires antiapoptotic Mcl-1. Nature. 426:671–676.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Willis SN, Chen L, Dewson G, Wei A, Naik
E, Fletcher JI, et al: Proapoptotic Bak is sequestered by Mcl-1 and
Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes
Dev. 19:1294–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McDonnell TJ and Korsmeyer SJ: Progression
from lymphoid hyperplasia to high-grade malignant lymphoma in mice
transgenic for the t(14; 18). Nature. 349:254–256. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou P, Levy NB, Xie H, et al: Mcl-1
transgenic mice exhibit a high incidence of B-cell lymphoma
manifested as a spectrum of histologic subtypes. Blood.
97:3902–3909. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Townsend KJ, Zhou P, Qian L, et al:
Regulation of Mcl-1 through a serum response factor/Elk-1-mediated
mechanism links expression of a viability-promoting member of the
Bcl-2 family to the induction of hematopoietic cell
differentiation. J Biol Chem. 274:1801–1813. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thallinger C, Wolschek MF, Maierhofer H,
et al: Mcl-1 is a novel therapeutic target for human sarcoma:
synergistic inhibition of human sarcoma xenotransplants by a
combination of mcl-1 antisense oligonucleotides with low-dose
cyclophosphamide. Clin Cancer Res. 10:4185–4191. 2004. View Article : Google Scholar : PubMed/NCBI
|