Introduction
Breast cancer is the most common type of
non-cutaneous malignancy in females and is the second (only to lung
carcinoma) most common cause of cancer-related mortality (1). In the United States, females have an
estimated 12% lifetime risk of being diagnosed with breast cancer;
the risk of breast cancer-related mortality is estimated at 2.82%,
even after optimal treatment (2).
Conventional anticancer therapeutics have reached the limit of
their utility, necessitating a novel therapeutic strategy to
improve outcomes. PTEN (phosphatase and tens in homolog on
chromosome ten) is a tumor suppressor and encodes a
dual-specificity phosphatase (3).
Its primary substrate is the second messenger phosphatidylinositol
3,4,5 trisphosphate (PIP3) (4).
PTEN antagonizes the phosphatidylinositol 3-kinase (PI3K)/AKT
pathway and affects cellular processes including growth,
proliferation and survival (5).
PTEN is mutated in numerous types of cancer; the high
frequency of monoallelic mutations of PTEN has been
demonstrated in endometrial carcinoma, glioblastoma, and prostate,
breast, colon and lung tumors (6).
Complete loss of PTEN is generally associated with advanced
cancer and metastases in endometrial cancer and glioblastoma
(6).
In breast cancer, a recent study revealed that PTEN
loss is a common event in breast cancers caused by BRCA1
mutations (7). PTEN has also been
investigated for its prognostic power in several types of human
malignancy. Loss of PTEN expression is associated with the poor
survival of patients with basal-like breast cancer (8), and data from preclinical and clinical
studies implicate PTEN loss in constitutive PI3K/AKT/mTOR signaling
and de novo resistance to Herceptin 2-targeted therapy
(9). Delivery of the tumor
suppressor PTEN gene represents a powerful strategy for
breast cancer therapy, although virus-mediated gene therapy is
associated with safety problems and non-virus-mediated gene
therapies are inefficient (10).
Previous studies have shown that VP22 proteins from
HSV-1 have the capacity to cross cell membranes (11). In addition, VP22 proteins are
capable of transducing heterologous proteins, such as p53, p27, CD,
GFP and Hsp70 (12), across the
cell membrane, although the delivery mechanisms have not been fully
characterized. It was hypothesized that introducing VP22 proteins
as well as PTEN may improve cell penetration and increase antitumor
efficacy. In this study, VP22 was conjugated to the C terminus of
PTEN and the growth-inhibitory activity of the fused proteins was
observed in a breast carcinoma cell line.
Materials and methods
Cell lines and cell culture
The BT549 cell line was provided by Dr Bao Qian Jin
(North Sichuan Medical College, Sichuan, China). The cells were
grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL,
Carlsbad, CA, USA) supplemented with 10% bovine serum (Beyotime
Biotechnology, Shanghai, China).
Eukaryotic expression vector
construction
The vector pcDNA3-PTEN for the expression of
wild-type human PTEN was generated by PCR subcloning using a
full-length wild-type human PTEN cDNA as template. The
PTEN amplicon was digested with HindIII/XhoI
(Takara Biotechnology Co., Ltd., Dalian, China) and subcloned into
a eukaryotic expression vector pcDNA3 (Invitrogen LIfe
Technologies, Carlsbad, CA, USA). pcDNA3-VP22, which expressed
HSV-1 VP22, was constructed as described previously (13). pcDNA3-PTEN-VP22 was constructed for
the expression of N-terminal VP22-fused PTEN (PTEN-VP22) by
overlapping extension PCR. Briefly, human PTEN cDNA (Fisher
Scientific, Hanover Park, IL, USA) was amplified with forward
(5′-GTCGAATTCATGACAGCCATCATC-3′) and reverse primers
(5′-GAGAGGTCATGACTTTTGTAATTTGTGT-3′). VP22 was amplified
with forward (5′-TACAAAAGTCATGACCTCTCGCC-3′) and reverse primers
(5′-AATGAATTCTCACTCGACGGGC-3′). The reaction mixture (final volume,
50 μl) consisted of 25 μl Pfu PCR Master Mix (Tiangen
Biotech Co., Ltd., Beijing, China), 2 μl templates and 1
μl each primer (forward and reverse). The thermal cycling
conditions were: 94°C for 3 min, followed by 30 cycles of 94°C for
30 sec, 55°C for 30 sec and 72°C for 1 min, and a final step at
72°C for 5 min. The extension was then performed with the
PTEN and VP22 fragments as primers, with the
following conditions: 94°C for 3 min, followed by 10 cycles of 94°C
for 30 sec, 55°C for 30 sec and 72°C for 1 min, and a final step at
72°C for 5 min. After extension, upstream
(5′-GTCGAATTCATGACAGCCATCATC-3′) and downstream
(5′-AATGAATTCTCACTCGACGGGC-3′) primers were added, and 30 PCR
cycles were performed. The resulting chimeric PTEN-VP22 gene
was digested with EcoRI (Takara Biotechnology Co., Ltd. and
subcloned into pcDNA3.
Identification of the eukaryotic
expression vectors
Successful clones were identified by restriction
digestion with HindIII/XhoI (pcDNA3-PTEN) and
HindIII/XbaI (pcDNA3-PTEN-VP22) at 37°C for 4 h.
pcDNA3-PTEN and pcDNA3-PTEN-VP22 were sequenced by GenScript Co.,
Ltd. (Nanjing, China).
Cell transfection
BT549 cells were grown to 70% confluence and then
washed twice with phosphate-buffered saline prior to transfection
in serum-free DMEM containing TransIt-LT1 Transfection
Reagent (Mirus Bio LLC, Madison, WI, USA), as described by the
manufacturer. Cells were transfected with the same quantity of
pSV-β-Galactosidase (Promega Corporation, Madison, WI, USA) per
well to account for deviations generated by different transfection
efficiencies.
Western blot analysis
After transfection (72 h), the cells were harvested
in RIPA lysis buffer (Tiangen Biotech Co., Ltd.), homogenized, and
centrifuged at 15,000 × g for 10 min at 4°C; protein concentration
in the supernatants was measured by the Bradford assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and standardized. Proteins
were separated by 10% SDS-PAGE and immunoblotted with rabbit
anti-PTEN (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and
rabbit anti-phospho-AKT (Ser473) (Cell Signaling
Technology Inc., Beverly, MA, USA) polyclonal antibodies.
Horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz
Biotechnology Inc.) was used as the secondary antibody for the DAB
Detection system (Wuhan Boster Biological Technology, Ltd., Wuhan,
China). Antibodies for β-actin (Boster) or total AKT (Cell
Signaling Technology Inc.) were used as loading controls.
Immunofluorescence and quantitation
At 48 h post-transfection, cells were washed with
phosphate-buffered saline, fixed in cold methanol for 10 min at
room temperature (RT), and then permeabilized with 0.2% Triton
X-100 for 90 min at RT. After washing in PBS and blocking for 30
min in 5% non-fat milk at RT, cells were incubated with rabbit
anti-PTEN (1:200) antibody (Santa Cruz Biotechnology Inc.) at 4°C
overnight. After three washes, the secondary antibody fluorescein
isothiocyanate-conjugated sheep-anti-rabbit IgG (Santa Cruz
Biotechnology Inc.) was added for 1 h at RT. Cells were then
analyzed on a microplate reader (Fluoroskan Ascent FL; Thermo
Fisher Scientific, Waltham, MA, USA) and images were captured by
inverted fluorescence microscopy (TCS SP2; Leica Microsystems,
Wetzlar, Germany).
Cell proliferation assay
Cell proliferation was measured with the Cell
Counting kit-8 (CCK8) assay (Beyotime Biotechnology) according to
the manufacturer’s instructions. Briefly, transfected cells were
harvested at 10 h and plated in 96-well plates at a density of
3,000 cells/well for each treatment condition. At 24, 48, 72 and 84
h after transfection, 10 μl WST-8 dye (Beyotime
Biotechnology) was added to each well, then incubated at 37°C for 1
h, and absorbance (A) was measured at 450 nm using an iMark bio
microplate reader (Bio-Rad Laboratories, Inc.). Cell survival was
determined as Atreated/Acontrol.
Apoptosis analysis
At 72 h post-transfection, cells were harvested,
washed with PBS, stained with Annexin V and propidium iodide, and
apoptosis was measured by flow cytometry (acquired 10,000
cells/cell; FACSVantage SE; BD Biosciences, Franklin Lakes, NJ,
USA).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical analysis was performed across multiple groups
using analysis of variance (ANOVA) and confirmed between individual
groups using Student-Newman-Keul’s method. P<0.05 was considered
to indicate a statistically significant difference.
Results
VP22 mediates PTEN intercellular
trafficking in BT549 cells
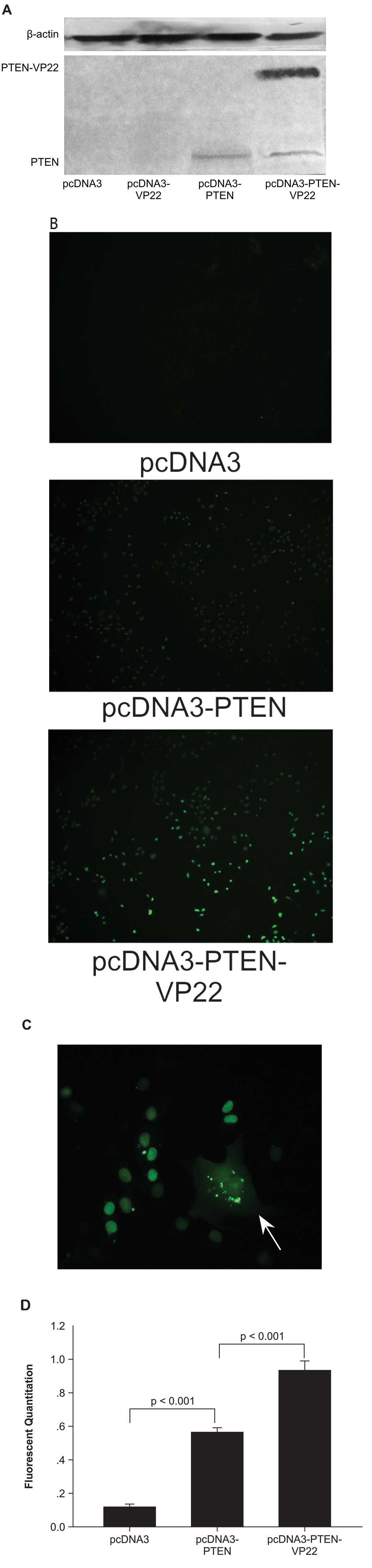
The trafficking ability of a fused PTEN-VP22
recombinant protein was measured. PTEN cDNA was fused to the
N-terminal of the VP22 cDNA to produce the pcDNA3-PTEN-VP22
fusion protein expression vector. Expression of the fusion protein
was monitored by western blotting. In transiently transfected BT549
cells, a PTEN-null breast carcinoma cell line (3), an anti-PTEN antibody clearly detected
the full-length PTEN-VP22 fusion protein at its expected size (~90
kDa), as well as PTEN (~60 kDa) (Fig.
1A). In pcDNA3-PTEN-VP22 transfected cells, western blotting
showed high expression of full-length PTEN-VP22 as well as a
truncated product with the molecular weight of PTEN, indicating
cleavage of the two proteins.
To investigate the trafficking property of the
fusion protein, BT549 cells were transfected with pcDNA3-PTEN-VP22
or pcDNA3-PTEN, and spreading was observed by fluorescence
microscopy (Fig. 1B). The results
showed that only a few cells per field were positive 48 h
post-transfection with PTEN (Fig.
1B). By contrast, when cells were transfected with
pcDNA3-PTEN-VP22, a larger number of positive cells (Fig. 1B) with a typical VP22 pattern
(i.e., primary transfected cells with cytoplasmic and nuclear
staining surrounded by recipient cells with nuclear staining) were
observed (Fig. 1C). This
phenomenon was confirmed by fluorescence quantitation after
immunofluorescence with the anti-PTEN antibody. The results showed
that the fluorescence of cells expressing PTEN-VP22 48 h after
transfection was 0.927±0.0196 versus 0.558±0.0105 in cells
expressing PTEN alone (P<0.001; Fig. 1D). Therefore, the fusion protein
PTEN-VP22 appears to have the same spreading abilities as VP22.
In addition, fluorescence microscopy of the BT549
human breast carcinoma cell line expressing exogenous PTEN or
PTEN-VP22 fusion protein revealed fluorescent PTEN or PTEN-VP22 in
the cytosolic and nuclear compartments, although PTEN and PTEN-VP22
were predominantly nuclear in localization (Fig. 1B and C).
VP22 enhances PTEN-mediated
antiproliferative activity in BT549 cells
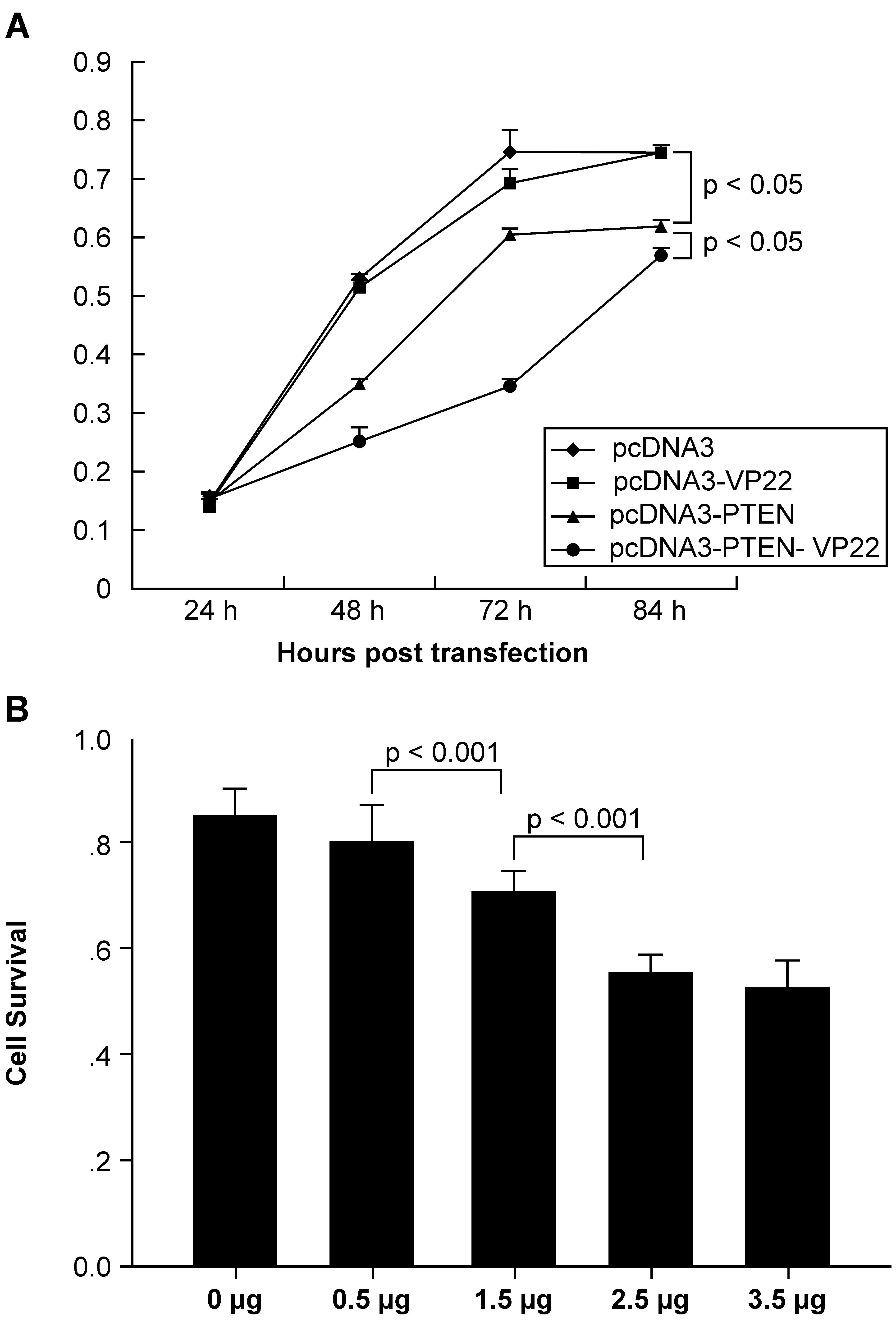
To determine whether fusion of PTEN to VP22 affected
its biological activity, the CCK-8 assay was used to measure the
effect on tumor cell proliferation. The results (Fig. 2) showed that addition of VP22 to
the C-terminus of PTEN enhanced antiproliferative activity in
general. Growth was similar between pcDNA3-VP22 and pcDNA3
transfected cells, indicating that VP22 expression is non-toxic
(Fig. 2A). Growth was inhibited
over time when BT549 cells were treated with 2.5 μg
pcDNA3-PTEN or pcDNA3-PTEN-VP22. The CCK-8 assay revealed that
pcDNA3-PTEN and pcDNA3-PTEN-VP22 did not inhibit BT549
proliferation 24 h post-transfection. By contrast, pcDNA3-PTEN and
pcDNA3-PTEN-VP22 exhibited significant antiproliferative activity
at 48, 72, and 84 h compared with pcDNA3-trans-fected cells
(pcDNA3-PTEN, P<0.001 at 48 and 84 h and P<0.01 at 72 h
versus pcDNA3 at the same time points; pcDNA3-PTEN-VP22, P<0.001
at 48, 72, and 84 h, versus pcDNA3 at the same time points).
Furthermore, the efficacy of pcDNA3-PTEN-VP22 inhibition of
proliferation was greater than that of pcDNA3-PTEN (P<0.001 at
48 and 72 h; P<0.05 at 84 h). These results indicated that the
conjugation of VP22 to the C-terminus of PTEN may enhance the basal
antiproliferative activity in the BT549 breast carcinoma cell
line.
To confirm these results, the antiproliferative
activity of various doses of PTEN-VP22 at 48 h post-transfection
were compared. As shown in Fig.
2B, cell growth was dose-dependent when BT549 cells were
treated with 0.5, 1.5, and 2.5 μg pcDNA3-PTEN-VP22
(P<0.001). Thus, addition of VP22 increased the
antiproliferative activity of PTEN in PTEN-deficient breast
carcinoma cells.
VP22 enhances PTEN-mediated apoptotic
induction in BT549 cells
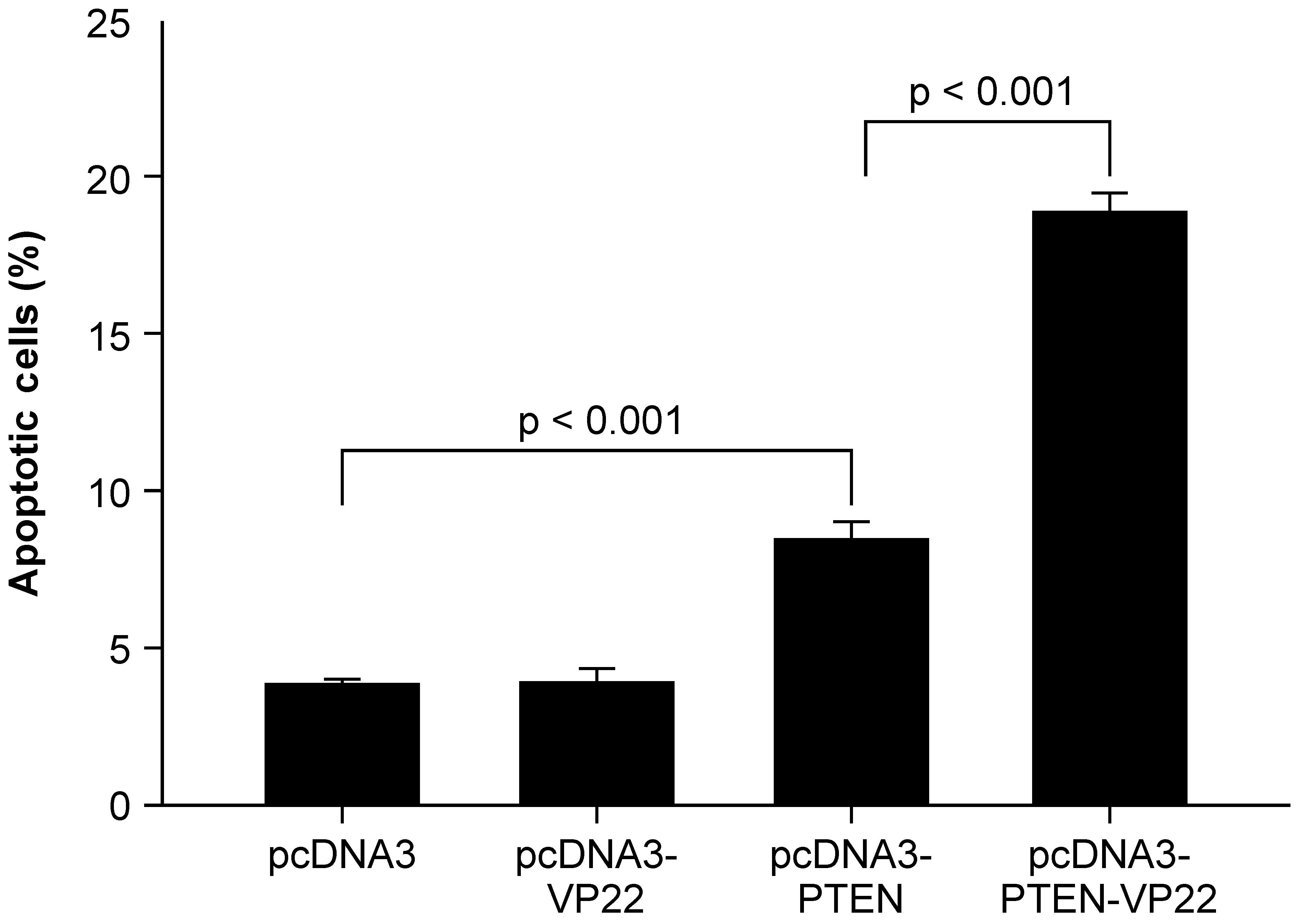
Previous studies have shown that transduction of the
wild-type PTEN gene into cancer cells induces apoptosis (14–16).
To investigate whether intercellular spread of PTEN-VP22 enhances
its apoptotic capacity, apoptotic induction by PTEN-VP22 and PTEN
was compared in BT549 cells (Fig.
3). It was observed that 5 μg pcDNA3-VP22 did not induce
apoptosis compared with 5 μg pcDNA3 (the negative control);
however, apoptotic rates in cells transfected with 5 μg
pcDNA3-PTEN differed significantly from those for the negative
control (P<0.001), revealing that transfection of pcDNA3-PTEN
induced apoptosis. In addition, a significant increase in apoptosis
was detected in cells transfected with 5 μg pcDNA3-PTEN-VP22
versus 5 μg pcDNA3-PTEN (P<0.001), indicating that
VP22-mediated spreading of PTEN-VP22 is associated with an enhanced
rate of PTEN-mediated apoptosis in BT549 cells.
VP22 enhances PTEN-mediated decreases in
the level of phosphorylated AKT
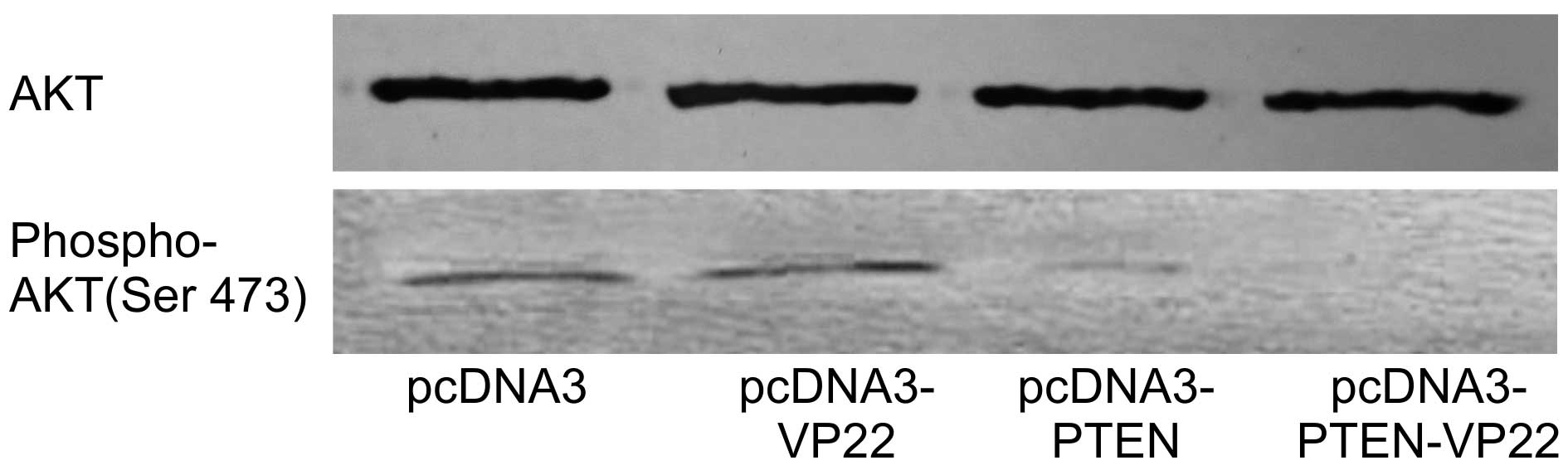
AKT is activated following phosphorylation by PIPs
and AKT phosphorylation is inversely associated with PTEN
expression. The correlation between AKT phosphorylation state and
PTEN-VP22 expression was investigated in BT549 cells. As expected,
levels of phospho-AKT were the same in pcDNA3 and pcDNA3-VP22
transfected cells and the high level of phospho-AKT was abrogated
in pcDNA3-PTEN transfected cells, whereas the higher level
of phospho-AKT was abrogated in pcDNA3-PTEN-VP22 transfected cells
(Fig. 4). These results suggest
VP22-mediated spreading of PTEN-VP22 is associated with decreased
expression of phosphorylated AKT.
Discussion
A present limitation of gene therapy is the ability
to deliver sufficient quantities of active proteins to target
cells. While secreted proteins can overcome this limitation to a
certain extent, it is particularly a problem for nonsecreted
proteins, such as PTEN as these proteins are only active in the
cells that they are initially delivered to. It has been suggested
that VP22 fusion proteins may increase distribution through
inter-cellular transport. Thus increased numbers of cells may reach
therapeutic steady state, leading to an overall increase in drug
efficacy in the target cell population.
Only N-terminal fusion was investigated as the
C-terminal extremity of VP22 is essential for cell-to-cell
transport (11,17). Expression vectors for wild-type
PTEN and PTEN-VP22 were constructed and their activities were
compared in the PTEN-null BT549 breast carcinoma cell line.
Wild-type PTEN protein and high levels of PTEN-VP22 fusion protein
expression in vitro were observed (Fig. 1A); both proteins were
transcriptionally active (Fig.
1B). The present study shows that VP22 transduces PTEN across
the cell membrane, resulting in a wider distribution in the BT549
cells (Fig. 1B and C). In
addition, VP22 does not change the characteristics of PTEN
localization in cells, and PTEN-VP22 was predominantly nuclear in
localization, which is similar to PTEN (Fig. 1B). Furthermore, it was demonstrated
that this fusion protein is functional and that the
PTEN-VP22 gene transfer induces a stronger antiproliferative
effect than PTEN alone in a time- and dose-dependent manner
in vitro (Fig. 2A and B).
VP22 did not display toxicity in these experiments (Fig. 2A), suggesting that the increased
activity is solely due to the transport properties of VP22.
PTEN acts as a phosphatidylinositol phosphatase with
a possible role in the phosphatidylinositol 3-kinase (PI3K)/AKT
pathway (5). Introduction of PTEN
into PTEN-deficient cells inhibits the activation of AKT, which is
a serine-threonine kinase downstream in the PI3K pathway, is
involved in proliferative and anti-apoptotic pathways and exhibits
tumor suppressive properties (18). In order to demonstrate how
PTEN-VP22 gene transfer could induce this antiproliferative
effect, it was evaluated whether higher protein transport levels
were correlated with increased apoptotic activity and decreased
levels of phosphorylated AKT in BT549 cells. It was observed that
PTEN-VP22 transfection enhanced apoptosis relative to
PTEN, while VP22 did not alter apoptotic activity
(Fig. 3). These results suggest
that PTEN-VP22 can induce apoptosis in BT549 cells and
VP22-mediated spreading of PTEN correlates with enhanced apoptosis.
PTEN decreased the expression of phospho-AKT; however, the
phosphorylation status of AKT was lower in the presence of
PTEN-VP22 versus PTEN. The phospho-AKT states of
BT549 cells were not altered by VP22 alone (Fig. 4). Thus, this demonstrated that
PTEN-VP22 can effectively block the (PI3K)/AKT pathway and
VP22-mediated spreading of PTEN-VP22 is correlated with the
enhanced rate of PTEN-mediated decreases in phospho-AKT status and
induction of apoptosis in BT549 cells.
In conclusion, VP22-mediated transport of the PTEN
tumor suppressor protein could enhance the biological functions of
PTEN, providing a strategy for enhancing the efficacy of gene
therapy of breast cancer.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81102288).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Horner MJ, Ries L, Krapcho M, et al: SEER
cancer statistics review, 1975–2006. National Cancer Institute;
Bethesda, MD: 2009
|
|
3
|
Baker SJ: PTEN enters the nuclear age.
Cell. 128:25–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stambolic V, Suzuki A, De La Pompa JL, et
al: Negative regulation of PKB/Akt-dependent cell survival by the
tumor suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ali IU, Schriml LM and Dean M: Mutational
spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid
phosphatase activity. J Natl Cancer Inst. 91:1922–1932. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saal LH, Gruvberger-Saal SK, Persson C, et
al: Recurrent gross mutations of the PTEN tumor suppressor gene in
breast cancers with deficient DSB repair. Nat Genet. 40:102–107.
2008. View Article : Google Scholar
|
|
8
|
Toft DJ and Cryns VL: Minireview:
Basal-like breast cancer: from molecular profiles to targeted
therapies. Mol Endocrinol. 25:199–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharial MM, Crown J and Hennessy B:
Overcoming resistance and restoring sensitivity to HER2-targeted
therapies in breast cancer. Ann Oncol. 23:3007–3016. 2012.
View Article : Google Scholar
|
|
10
|
Verma IM and Somia N: Gene
therapy-promises, problems and prospects. Nature. 389:239–242.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elliott G and O’Hare P: Intercellular
trafficking and protein delivery by a herpes virus structural
protein. Cell. 88:223–233. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nishikawa M, Otsuki T, Ota A, et al:
Induction of tumor-specific immune response by gene transfer of
Hsp70-cell-penetrating peptide fusion protein to tumors in mice.
Mol Ther. 18:421–428. 2010. View Article : Google Scholar :
|
|
13
|
Yu X, Liu L, Wu L, et al: Herpes simplex
virus type 1 tegument protein VP22 is capable of modulating the
transcription of viral TK and gC genes via interaction with viral
ICP0. Biochimie. 92:1024–1030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Persad S, Attwell S, Gray V, et al:
Inhibition of integrin-linked kinase (ILK) suppresses activation of
protein kinase B/Akt and induces cell cycle arrest and apoptosis of
PTEN-mutant prostate cancer cells. Proc Natl Acad Sci USA.
97:3207–3212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steelman LS, Bertrand FE and McCubrey JA:
The complexity of PTEN: mutation, marker and potential target for
therapeutic intervention. Expert Opin Ther Targets. 8:537–550.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Z, Liu GX, Liu YL, et al: Effect of
adenovirus-mediated PTEN gene on ulcerative colitis-associated
colorectal cancer. Int J Colorectal Dis. 28:1107–1115. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aints A, Güven H, Gahrton G, Smith CIE and
Dilber MS: Mapping of herpes simplex virus-1 VP22 functional
domains for inter- and subcellular protein targeting. Gene Ther.
8:1051–1056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davies MA, Koul D, Dhesi H, et al:
Regulation of Akt/PKB activity, cellular growth and apoptosis in
prostate carcinoma cells by MMAC/PTEN. Cancer Res. 59:2551–2556.
1999.PubMed/NCBI
|