Introduction
Glioblastoma multiforme (GBM) is the most common and
aggressive form of brain cancer. GBM is a highly aggressive
neuroepithelial tumor, which grows almost exclusively in neural
tissue (1). The overall survival
rate of patients with GBM has increased in the last decade due to
the use of aggressive surgery combined with radiation, chemotherapy
or biological therapy. However, malignant glioma cell proliferation
and invasion remain the predominant factors affecting patient
mortality (2,3). At the time of GBM diagnosis, in the
majority of cases, the percentage of patients with a two-year
survival rate is <30% (4). This
severely malignant phenotype is at least partially due to acquired
excess cell proliferation. A recent development in cancer research
is the discovery of the tetraspanin proteins, which act as
molecular facilitators of adaptors to organize a network of
interactions among the cell surface molecules, known as the
‘tetraspanin web’. Members of the tetraspanin protein family are
involved in regulation of the cell cycle as well as cell growth and
proliferation. The mechanisms underlying the growth and
proliferation of GBM cells remain to be elucidated; therefore,
further study is required to improve therapeutic strategies.
CD9 is a cell surface glycoprotein of the
tetraspanin protein family, which has a characteristic structure of
four transmembrane domains and two extracellular loops.
Tetraspanins can interact with other functional proteins, including
growth factor receptors, intracellular signaling molecules and
integrins, to form multiprotein complexes at the
tetraspanin-enriched microdomain at the cell surface (5). Tetraspanins are involved in various
biological processes, including cell survival, growth and
migration. The tetraspanin CD9 was initially identified as a
metastasis suppressor of solid tumors and was shown to be expressed
in numerous types of cell, including platelets, pre-B cells and
Schwann cells. Clinical and pathological findings have demonstrated
that reduced CD9 gene expression is associated with poor prognoses
in breast (6,7), colon (8) and lung cancer (9). Recent studies have suggested that CD9
may have the potential to regulate motility, through other
transmembrane proteins (10,11).
Furthermore, it has previously been reported that CD9 is directly
associated with epidermal growth factor receptor (EGFR) and is able
to desta-bilize surface expression of EGFR and consequently
attenuate ligand-induced activation of the receptor (12).
A common characteristic of GBM histology is EGFR
amplification, which affects signal transduction processes
(13,14). EGFR signaling occurs through a
complex network of intermediates, including
phosphoinositide-3-kinase (PI3K), mitogen-activated protein kinase
(MAPK) and phospholipase C-γ (15). The dysregulation of signal
transduction processes affects various downstream biological
processes associated with cell proliferation, migration, adhesion
and invasion (16,17). Nakamura et al (18) previously reported that CD9 affects
EGFR ligand binding activities of the membrane-bound forms of
transforming growth factor-α and heparin-binding EGF-like growth
factor, which may contribute to human malignant glioma cell
growth.
The present study aimed to present a novel
mechanistic insight into the important role of tetraspanin CD9 in
regulating the proliferation of human glioblastoma cells through
epidermal growth factor receptor (EGFR) activity. The regulation of
the expression and phosphorylation of EGFR and EGFR-induced
signaling were examined in order to determined whether the function
of CD9 was linked the inhibition of proliferation in human
glioblastoma cells.
Materials and methods
Cell line and transfection
The LN229 human glioblastoma cells were obtained
from the American Type Culture Collection (Manassas, VA, USA). The
cells were cultured in Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal bovine serum (Gibco-BRL, Carlsbad, CA,
USA), 50 mg/ml streptomycin and 50 U/ml penicillin (Gibco-BRL), in
a humidified atmosphere containing 5% CO2 at 37°C. The
LN229 cells were stably transfected with a CD9/pcDNA3.1 plasmid
(LN/CD9) or a control pcDNA3.1 plasmid (LN/cont). The coding region
of CD9 cDNA was generated through reverse transcription
quantitative-polymerase chain reaction (RT-qPCR) from LN229 human
glioblastoma cells. The CD9 cDNA fragments were amplified by
RT-qPCR, were digested with BamHI and EcoRI, and the
purified cDNA fragments were ligated into the
BamHI-EcoRI-digested pcDNA3.1 (Invitrogen Life
Technologies, Carlsbad, CA, USA). The primers used in the PCR were
as follows: forward, 5′-CGGGATCCACCATGCCGGTCAAAGGAGGCA-3′ and
reverse, 5′-CGGAATTCCGCTAGACCATCTCGCGGTTCC-3′ (Takara Biotechnology
Co., Ltd., Dalian, China). The construct was verified by
sequencing. LN229 cell lines were transfected with a CD9/pcDNA 3.1
using the Lipofectamine 2000 reagent (Invitrogen Life Technologies)
followed by selection in Zeocin (Invitrogen Life Technologies). A
CD9 small hairpin (sh)RNA plasmid (LN/shCD9) was constructed in
order to generate CD9-depleted cells. The cells transfected with a
negative control (NC) shRNA plasmid were considered control cells
(shNC) and untransfected LN229 cells were considered the mock
control cells. A CD9 shRNA and a negative control plasmid were
constructed by GenePharma using pGPH1/Neo plasmid (GenePharma,
Shanghai, China) followed by selection in G418 (Gibco BRL). The 21
nt siRNA directed against CD9 was 5′-UUCUUGCUCGAAGAUGCUCTT-3′.
LN229 cells were transfected using Lipofectamine®2000
reagent (Invitrogen Life Technologies), according to the
manufacturer’s instructions.
Antibodies and reagents
Primary antibodies against the following antigens
were all purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA): Rabbit polyclonal CD9 immunoglobulin (Ig)G (1:500; sc9148),
goat polyclonal EGFR IgG (1:1,000; sc31157), goat polyclonal p-EGFR
(Tyr-1173) IgG (1:500; sc12351), mouse monoclonal p-EGFR (Tyr-1086)
IgG (1:500; sc81490), rabbit polyclonal p-Akt1/2/3 (Ser473) IgG
(1:1,000; sc33437), rabbit polyclonal p-Akt1/2/3 (Thr308) IgG
(1:1,000; sc135650), rabbit polyclonal Akt1/2/3 IgG (1:1,000;
sc8312), goat polyclonal p-Erk1/2 (Thr202/Tyr204) IgG (1:1,000;
sc16982), rabbit polyclonal Erk1/2 IgG (1:1,000; sc292838), goat
polyclonal β-actin IgG (1:1,000; sc1616). The PVDF membranes were
incubated with the primary antibodies overnight at 4°C. Secondary
antibodies (1:2,000), including goat anti-rabbit horseradish
peroxidase (HRP)-conjugated IgG, rabbit anti-goat HRP-conjugated
IgG and goat anti-mouse HRP-conjugated IgG-horseradish peroxidase
(Zhongshan Goldenbridge Biotechnology Co., Beijing, China), for 1h
at room temperature.
EGF was obtained from ProSpec-Tany TechnoGene Ltd.
(Rehovot, Israel). Akt inhibitor LY294002 and Erk inhibitor U0126
were purchased from Abcam (Hong Kong, China). Opti-MEM was obtained
from Invitrogen Life Technologies. Protease inhibitor cocktail was
obtained from Sigma-Aldrich (St. Louis, MO, USA).
RT-PCR
Total RNA was extracted from the cells using
TRIzol® (Invitrogen Life Technologies), and 1 μg
total RNA was reverse-transcribed using a cDNA Synthesis kit
(Invitrogen Life Technologies) with random hexamers. The PCR
cycling parameters were set as follows: 30 cycles of 40 sec at
94°C, 40 sec at 60°C and 60 sec at 72°C. The identity of the PCR
products was confirmed through sequencing by BGI Tech Solutions Co.
(Shenzhen, China) using T7 primer. The relative quantitative
analysis was normalized to the endogenous control GAPDH. The
following primer sequences were used: GAPDH forward,
TACTTATGCCGATGTCGTTGT and reverse, CCAGCCTCGTCCCGTAGA; CD9 forward,
TGCATCTGTATCCAGCGCCA and reverse, CTCAGGGATGTAAGCTGACT (Takara
Biotechnology Co., Inc.) (19).
Flow cytometry
Cell cycle distribution was analyzed using flow
cytometry with propidium iodide (PI) staining. Briefly,
1.5×105 cells transfected with CD9 and negative control
plasmids for four days were seeded in 6-cm dishes and cultured for
two days at 37°C. The cells were then treated with the desired
agents as described in the figure legends, in serum-free medium
overnight. The medium was refreshed and the cells were stimulated
with 50 ng/ml EGF for 10 min at room temperature. Cells
(1×104) were then harvested by adding 0.05%
trypsin/0.04% EDTA and washed twice with PBS. The cells were
subsequently fixed in ice cold 70% ethanol for 1 h and treated with
RNAase and PBS, containing 100 μg/ml DNase-free RNase and 40
μg/ml PI (Sigma-Aldrich), then incubated for 1 h at 37°C. A
total of 10,000 stained nuclei were analyzed using a FACSCalibur
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). To
identify the effects of the PI3K/Akt and MAPK/Erk pathways on the
regulation of cell proliferation, these two signaling pathway were
inhibited using 15 μM LY294002 or 20 μM U0126 for 6
h, respectively. The cells were then subjected to flow cytometry,
as described above.
Western blot analysis
Cells (1×105) were treated as described
above and seeded in 12-well plates (Falcon; Fisher Thermo
Scientific, Waltham, MA, USA). The cells were incubated overnight
in serum-free medium. The medium was replaced and the cells were
stimulated with 50 ng/ml EGF for 10 min at room temperature. The
cells were then harvested and lysed in 200 μl
radioimmunoprecipitation lysis buffer (50 mM Tris, pH 7.5, 150 mM
NaCl, 1% Triton X-100, 25 mM 0.5% sodium deoxycholate, 0.1% SDS, 5
mM pyrophosphate and 50 mM NaF; Sigma-Aldrich), supplemented with 1
mM Na3VO4, 1 mM dithiothreitol, 1% protease
inhibitor cocktail and 1% phosphatase inhibitor cocktail per well.
10 μl inhibitor cocktail (Sigma-Aldrich) was added to 1 ml
lysis buffer, all reagents described above were purchased from
Sangon Biotech Technogene Ltd. (Shanghai, China). The lysate was
determined using a bicinchoninic acid protein assay kit (Beyotime,
Jiangsu, China). Heat-denatured protein samples (20 μg per
lane) were separated using 12% SDS-PAGE (Invitrogen Life
Technologies) and the proteins were transferred to polyvinylidene
fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The
non-specific binding sites on the PVDF membranes were blocked with
5% nonfat dry milk in tris-buffered saline with 0.1% Tween-20. The
target protein bands in the PVDF membranes were revealed by
immunoblotting with primary antibodies, followed by an incubation
with the species-specific HRP-conjugated secondary antibodies.
Signal bands were detected using enhanced chemiluminescence
reagents (GE Healthcare Life Sciences, Little Chalfont, UK).
β-actin was used as a housekeeping antibody, to verify equal
amounts of protein were loaded in all of the lanes.
Statistical analysis
All of the experiments were repeated at least three
times and consistently yielded similar results. The data were
analyzed by GraphPad Prism version 5.0 (GraphPad Software Inc., La
Jolla, CA, USA). Statistical comparisons between groups were
performed using a one-way analysis of variance. P<0.01 was
considered to indicate a statistically significant difference
between values. Values are expressed as the mean ± standard error
of the mean.
Results
CD9 expression in LN229 cells
The CD9 gene was constructed into a pcDNA3.1 vector
and transfected into LN229 cells (LN/CD9). The protein expression
levels of CD9 were measured after 48 hours by western blotting. The
protein expression levels of CD9 were significantly enhanced in the
LN229 cells transfected with the pcDNA 3.1/CD9 vector, as compared
with the cells transfected with an empty pcDNA3.1 vector (Fig. 1A). The LN229 cells were also
trans-fected with CD9 shRNA plasmids in order to suppress CD9
expression (LN/shCD9). RT-PCR and western blot analyses indicated
that the expression levels of endogenous CD9 were stably decreased
by ~70% (Fig. 1B and C).
Overexpression of CD9 inhibits
EGF-stimulated LN229 cell proliferation EGF and affects the
phosphorylation of EGFR
To elucidate whether CD9 is able to inhibit cell
growth by affecting cell cycle progression, the cell cycle
distribution was assessed in the LN229 cells by flow cytometry. The
cell population in the LN/CD9 group displayed a significant
increase in the proportion of cells in G0/G1
phase, and a significant decrease in the number of cells in the S
phase, as compared with the control cells (Fig. 2A and B). These results suggested
that CD9 may inhibit the proliferation of LN229 cells via cell
cycle regulation. To investigate the molecular mechanisms
underlying how CD9 affects EGF-stimulated cell growth, the
expression and phosphorylation status of EGFR was determined by
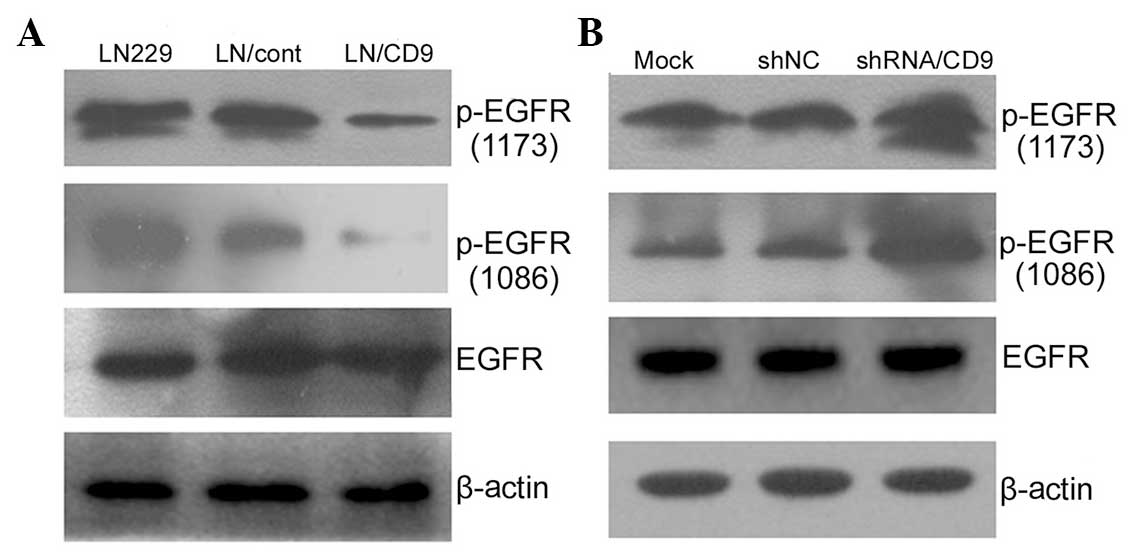
western blotting. Increased CD9 expression significantly reduced
EGF-stimulated phosphorylation of EGFR at Y1173 and Y1086 without
significantly affecting EGFR expression levels (Fig. 3A), while downregulation of CD9
elevated EGF-stimulated phosphorylation of EGFR at Y1173 and Y1086
(Fig. 3B).
 | Figure 2Effects of CD9 and inhibition of the
PI3K/Akt and MAPK/Erk signaling pathways using LY294002 and U0126,
respectively, on the proliferation of LN229 human glioblastoma
cells stimulated with EGF, using flow cytometry. Cells transfected
with CD9/pcDNA3.1 plasmid or an empty pcDNA3.1 plasmid were
serum-starved for 12 h in the presence of the desired regents and
then stimulated with 50 ng/ml EGF. (A and B) Flow cytometry was
performed to measure the proliferation efficiency of CD9 in LN229
cells transfected with a control pcDNA3.1 plasmid (LN/cont) or a
CD9/pcDNA3.1 plasmid (LN/CD9). (C–E) Treated cells were
serum-starved for 12 h in the presence of the desired regents and
the PI3K/Akt and MAPK/Erk signaling pathways were inhibited in the
cells using 15 μM LY294002 or 20 μM U0126 for 6 h,
respectively. The proliferation efficiency of the cells stimulated
with 50 ng/ml EGF was measured as described in the Materials and
Methods. (A) +EGF+pcDNA3.1 (control), (B) +EGF,+CD9/pcDNA3.1, (C)
+EGF (control), (D) +EGF+LY294002, (E) +EGF+U0126. MAPK,
mitogen-activated protein kinase; PI3K, phosphoinositide-3-kinase;
Erk, extracellular signal-regulated kinase; EGF, epidermal growth
factor. |
Effects of inhibiting the PI3K/Akt and
MAPK/Erk pathways
To investigate how CD9 modulated LN229 cell
proliferation and growth regulation via the PI3K/Akt and MAPK/Erk
pathways, the EGF-stimulated LN229 cells were transfected with an
empty pcDNA3.1 vector and were treated with LY294002 and U0126,
respectively. Inhibition of the PI3K/Akt and MAPK/Erk pathways,
using LY294002 and U0126, suppressed EGF-stimulated proliferation
(Fig. 2C–E). These results
suggested that the PI3K/Akt and MAPK/Erk pathways may be involved
in the modulation of LN229 cell growth regulation in
vitro.
CD9 attenuates EGFR signaling of PI3K/Akt
and MAPK/Erk pathways
Based on previous results, it was hypothesized that
PI3K/Akt and MAPK/Erk pathways may be associated with glioma cell
proliferation. To further investigate the mechanisms underlying the
suppressive effects of CD9 on EGF-stimulated growth in
vitro, these two signaling pathways, which are essential for
the modulation of tumor cell growth, were measured by western blot
analysis. Overexpression or suppression of CD9 directly affected
the activation of the PI3K/Akt and MAPK/Erk pathways. CD9
negatively affected EGFR-mediated activation of Akt and Erk
(Fig. 4). In the EGF-treated LN229
cells overexpressing CD9, the inhibitory effect of CD9 on
phosphorylation of Akt at Ser473 was enhanced, and the opposite
effect was observed on the phosphorylation of Akt at Ser473 and Erk
at Erk1 and 2 following knockdown of CD9 (Fig. 4A and B). However, alterations in
the cellular content of CD9 did not affect the level of EGF-induced
phosphorylation of Akt at Thr308. These results indicated that the
PI3K/Akt and MAPK/Erk signaling pathways may have an important role
in CD9-regulated cell proliferation.
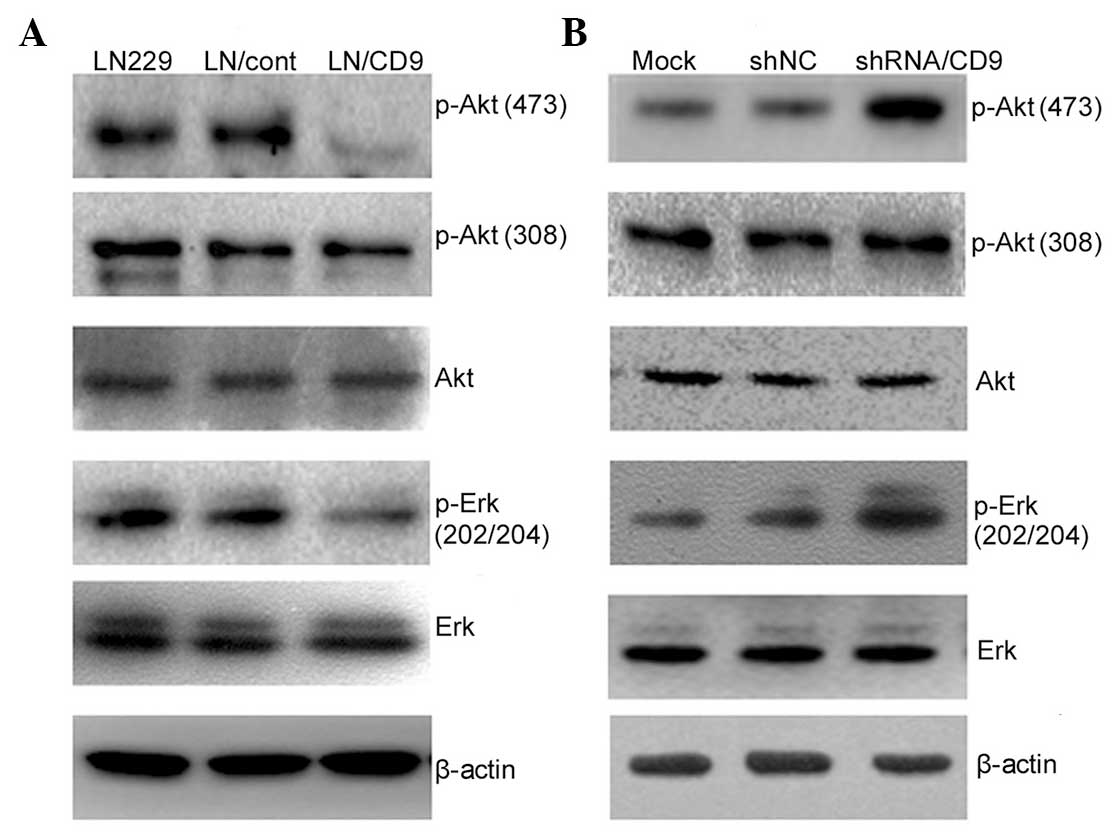 | Figure 4Effects of CD9 on the PI3K/Akt and
MAPK/Erk signaling pathways in LN229 human glioblastoma cells.
Treated cells were serum-starved for 12 h in the presence of the
desired regents and then stimulated with 50 ng/ml EGF for 10 min.
The cells were then harvested and subjected to western blot
analysis with antibodies targeting Akt 1/2/3, p-Akt1/2/3 (Thr308),
p-Akt 1/2/3 (Ser473), p-p44/42 MAPK (Thr202/Tyr204) and Erk1/2. (A)
Increased CD9 content inhibited EGF-stimulated phosphorylation of
Akt at Ser473 and Erk at Erk1 and 2. (B) Decreased CD9 content
upregulated EGF-stimulated phosphorylation of Akt at Ser473 and Erk
at Erk1 and 2. All experiments were performed at least three times.
MAPK, mitogen-activated protein kinase; PI3K,
phosphoinositide-3-kinase; Erk, extracellular signal-regulated
protein kinase; EGF, epidermal growth factor; p, phosphorylated;
shRNA, small hairpin RNA; NC, negative control. |
Discussion
The present study demonstrated that CD9 decreased
the phosphorylation of EGFR at specific sites. CD9 attenuated EGFR
signaling of PI3K/Akt and MAPK/Erk, which are associated with cell
growth and proliferation. Conversely, shRNA-mediated knockdown of
CD9 expression enhanced the activation of the EGFR signal
transduction pathway, including PI3K/Akt and MAPK/Erk, which
enhanced cell proliferation. This activation was blocked by
treatment with PI3K and MAPK inhibitors.
The tetraspanin CD9 was initially characterized as a
cell surface antigen on lymphohemopoietic cells, platelets,
eosinophil and pre-B cells (20,21).
CD9 is also normally expressed in mature oligodendrocytes and
Schwann cells, and is implicated in neurite outgrowth and
myelination (22,23). Recently, numerous clinical studies
have demonstrated that CD9 expression is inversely correlated with
patient survival and metastatic potential in various types of human
cancer (6–9,24).
Conversely, Kawashima et al (25) demonstrated that among the
neuroepithelial tumors; high-grade astrocytic tumors, including
glioblastomas, had higher immunoreactivity of CD9, as compared with
low-grade cerebral astrocytomas, thus suggesting that CD9
expression in astrocytic tumors may be correlated with their
malignancy (25). Tetraspanin
members, such as CD82, can associate with EGFR and attenuate EGFR
signaling by accelerating EGFR internalization, and redistribute
EGFR into distinct cellular compartments following endocytosis.
This process significantly impacts the early events associated with
receptor activation, including ligand binding, dimerization and
re-localization of activated receptors into clathrin-coated pits
(26,27). For numerous reasons, the present
study hypothesized that CD9 may affect glioma tumor biology. To
fully understand the roles of CD9 on glioma tumor biology, CD9
expression was modulated in the LN229 human glioblastoma cell line,
and it was determined whether CD9 was able to drive cell
proliferation. Numerous studies have suggested that CD9 provides a
bridge among cell surface proteins, including growth factor
receptors, to generate functional complexes involved in cell
proliferation, division migration and apoptosis (28,29).
To explore the precise roles and mechanistic action of CD9 on the
proliferation of glioblastomas, the present study focused on the
association between CD9 and EGFR, and the EGFR signals mediated by
CD9 (30). CD9 was shown to
decrease the phosphorylation of particular tyrosine residues and
reduce the EGFR signals, which usually mediate the proliferation
and survival of glioblastoma.
The EGFR is the prototypical member of the ErbB/EGFR
family, which is a primary contributor to glioblastoma initiation
and progression. To clarify the role of the association between
EGFR and CD9, CD9 content was altered in LN229 cells by
transfection with CD9 and shRNA-CD9. The results demonstrated that
CD9 inhibited EGF-stimulated phosphorylation at Y1173 and Y1086.
EGFR phosphorylation leads to the recruitment of complex effector
proteins through recognition and binding of Src homology 2 and
phosphotyrosine binding domains on the effector proteins, to
phosphotyrosine motifs on the receptor (31–33).
Consequently, various downstream signaling cascades, including the
PI3K and MAPK are activated.
The results of the present study demonstrated that
inhibitors of PI3K or MAPK blocked the proliferation of LN229
cells, which indicates that the PI3K/Akt and MAPK/Erk pathways are
important for cell growth and survival (34,35).
The effects of CD9 were then determined on the activity of these
two signaling pathways stimulated by EGF. The results suggested
that CD9 negatively regulated EGF-stimulated activation of PI3K/Akt
phosphorylation at Ser473 and MAPK phosphorylation, but did not
affect PI3K/Akt phosphorylation at Thr308. Flow cytometric analyses
confirmed that CD9 had negligible effects on EGF-stimulated cell
proliferation. Phosphorylation of PI3K/Akt by
phos-phoinositide-dependent protein kinase 1 at Thr308 and at
Ser473 by the mammalian target of rapamycin complex 2 is required
for full kinase activity (36).
The results of the present study suggested that Akt Ser473
phosphorylation contributes to CD9-mediated cell proliferation in
response to EGF. In addition, CD9 affects the MAPK/Erk signaling
pathway. Activation of the MAPK/Erk pathway is triggered by growth
factor receptor-bound protein 2 binding directly to the receptor at
Y1068, and indirectly through Src homology domain-containing
adaptor protein C binding at Y1173 and Y1148 (37). Initial experiments demonstrated
that CD9 suppressed EGF-stimulated EGFR phosphorylation at Y1173
and Y1086. Therefore, it may be hypothesized that CD9 can regulate
the phosphorylation of Y1173 and Y1086 residues on EGFR, which is
responsible for the EGF-induced activation of PI3K/Akt and
MAPK/Erk. The changes to the levels of phosphorylation at the Y1173
and Y1086 residues were consistent with those of the cellular
content of CD9.
In conclusion, upregulated CD9 expression in LN229
cells inhibited cell proliferation, decreased EGF-induced
phosphorylation of PI3K/Akt at Ser473 and Erk1/2, and reduced
EGF-induced phosphorylation at Y1173 and Y1086 residues on EGFR.
Conversely, knockdown of CD9 expression resulted in attenuation of
EGF-induced activation of PI3K/Akt and MAPK/Erk, and enhanced the
EGF-induced phosphorylation of EGFR at Y1173 and Y1086. These
results suggested that the mechanism by which CD9 affects EGFR
signaling appears rather complex and requires further study.
References
|
1
|
Thorsen F and Tysnes BB: Brain tumor cell
invasion, anatomical and biological considerations. Anticancer Res.
17:4121–4126. 1997.
|
|
2
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parsons DW, Jones S, Zhang X, et al: An
integrated genomic analysis of human glioblastoma multiforme.
Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, et al
European Organisation for Research and Treatment of Cancer Brain
Tumor and Radiotherapy Groups; National Cancer Institute of Canada
Clinical Trials Group: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Regina Todeschini A and Hakomori SI:
Functional role of glycosphingolipids and gangliosides in control
of cell adhesion, motility, and growth, through glycosynaptic
microdomains. Biochim Biophys Acta. 1780:421–433. 2008. View Article : Google Scholar
|
|
6
|
Miyake M, Nakano K, Itoi SI, Koh T and
Taki T: Motility-related protein-1 (MRP-1/CD9) reduction as a
factor of poor prognosis in breast cancer. Cancer Res.
56:1244–1249. 1996.PubMed/NCBI
|
|
7
|
Huang CI, Kohno N, Ogawa E, Adachi M, Taki
T and Miyake M: Correlation of reduction in MRP-1/CD9 and KAI1/CD82
expression with recurrences in breast cancer patients. Am J Pathol.
153:973–983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mori M, Mimori K, Shiraishi T, et al:
Motility related protein 1 (MRP1/CD9) expression in colon cancer.
Clin Cancer Res. 4:1507–1510. 1998.PubMed/NCBI
|
|
9
|
Higashiyama M, Doi O, Kodama K, et al:
Immunohistochemically detected expression of motility-related
protein-1 (MRP-1/CD9) in lung adenocarcinoma and its relation to
prognosis. Int J Cancer. 74:205–211. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Halova I, Dráberová L, Bambousková M, et
al: Cross-talk between tetraspanin CD9 and transmembrane adaptor
protein non-T cell activation linker (NTAL) in mast cell activation
and chemotaxis. J Biol Chem. 288:9801–9814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang CL, Liu D, Masuya D, et al:
MRP-1/CD9 gene transduction downregulates Wnt signal pathways.
Oncogene. 23:7475–7483. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murayama Y, Shinomura Y, Oritani K, et al:
The tetraspanin CD9 modulates epidermal growth factor receptor
signaling in cancer cells. J Cell Physiol. 216:135–143. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Bäcklund LM, Nilsson BR, et al:
Clinical significance of EGFR amplification and the aberrant
EGFRvIII transcript in conventionally treated astrocytic gliomas. J
Mol Med (Berl). 83:917–926. 2005. View Article : Google Scholar
|
|
14
|
Sugawa N, Yamamoto K, Ueda S, et al:
Function of aberrant EGFR in malignant gliomas. Brain Tumor Pathol.
15:53–57. 1998. View Article : Google Scholar
|
|
15
|
Wang HX, Li Q, Sharma C, Knoblich K and
Hemler ME: Tetraspanin protein contributions to cancer. Biochem Soc
Trans. 39:547–552. 20w11. View Article : Google Scholar
|
|
16
|
Huang X, Li Y, Zhang J, Xu Y, Tian Y and
Ma K: Ganglioside GM3 inhibits hepatoma cell motility via
down-regulating activity of EGFR and PI3K/AKT signaling pathway. J
Cell Biochem. 114:1616–1624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Huang X, Zhong W, Zhang J and Ma K:
Ganglioside GM3 promotes HGF-stimulated motility of murine hepatoma
cell through enhanced phosphorylation of cMet at specific tyrosine
sites and PI3K/Akt-mediated migration signaling. Mol Cell Biochem.
382:83–92. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakamura K, Mitamura T, Takahashi T,
Kobayashi T and Mekada E: Importance of the major extracellular
domain of CD9 and the epidermal growth factor (EGF)-like domain of
heparin-binding EGF-like growth factor for up-regulation of binding
and activity. J Biol Chem. 275:18284–18290. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Funakoshi T, Tachibana I, Hoshida Y, et
al: Expression of tetraspanins in human lung cancer cells: frequent
downregulation of CD9 and its contribution to cell motility in
small cell lung cancer. Oncogene. 22:674–687. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boucheix C and Benoit P: CD9 antigen: will
platelet physiology help to explain the function of a surface
molecule during hemopoietic differentiation? Nouv Rev Fr Hematol.
30:201–202. 1988.PubMed/NCBI
|
|
21
|
Masellis-Smith A and Shaw AR:
CD9-regulated adhesion. Anti-CD9 monoclonal antibody induce pre-B
cell adhesion to bone marrow fibroblasts through de novo
recognition of fibronectin. J Immunol. 152:2768–2777.
1994.PubMed/NCBI
|
|
22
|
Schmidt C, Künemund V, Wintergerst ES,
Schmitz B and Schachner M: CD9 of mouse brain is implicated in
neurite outgrowth and cell migration in vitro and is associated
with the alpha 6/beta 1 integrin and the neural adhesion molecule
L1. J Neurosci Res. 43:12–31. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chernousov MA, Stahl RC and Carey DJ:
Tetraspanins are involved in Schwann cell-axon interaction. J
Neurosci Res. 91:1419–1428. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murayama Y, Miyagawa J, Shinomura Y, et
al: Significance of the association between heparin-binding
epidermal growth factor-like growth factor and CD9 in human gastric
cancer. Int J Cancer. 98:505–513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawashima M, Doh-ura K, Mekada E, Fukui M
and Iwaki T: CD9 expression in solid non-neuroepithelial tumors and
infiltrative astrocytic tumors. J Histochem Cytochem. 50:1195–1203.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takahashi M, Sugiura T, Abe M, Ishii K and
Shirasuna K: Regulation of c-Met signaling by the tetraspanin
KAI-1/CD82 affects cancer cell migration. Int J Cancer.
121:1919–1929. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Odintsova E, Voortman J, Gilbert E and
Berditchevski F: Tetraspanin CD82 regulates compartmentalisation
and ligand-induced dimerization of EGFR. J Cell Sci. 116:4557–4566.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gustafson-Wagner E and Stipp CS: The
CD9/CD81 tetraspanin complex and tetraspanin CD151 regulate
alpha3beta1 integrin-dependent tumor cell behaviors by overlapping
but distinct mechanisms. PloS One. 8:e618342013. View Article : Google Scholar
|
|
29
|
Nakagawa T, Higashiyama S, Mitamura T,
Mekada E and Taniguchi N: Amino-terminal processing of cell surface
heparin-binding epidermal growth factor-like growth factor
up-regulates its juxtacrine but not its paracrine growth factor
activity. J Biol Chem. 271:30858–30863. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Murayama Y, Miyagawa J, Oritani K, et al:
CD9-mediated activation of the p46 Shc isoform leads to apoptosis
in cancer cells. J Cell Sci. 117(Pt 15): 3379–3388. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jones RB, Gordus A, Krall JA and MacBeath
G: A quantitative protein interaction network for the ErbB
receptors using protein microarrays. Nature. 439:168–174. 2006.
View Article : Google Scholar
|
|
32
|
Lindemann C, Hackmann O, Delic S, Schmidt
N, Reifenberger G and Riemenschneider MJ: SOCS3 promoter
methylation is mutually exclusive to EGFR amplification in gliomas
and promotes glioma cell invasion through STAT3 and FAK activation.
Acta Neuropathol. 122:241–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jin W, Xu X, Yang T and Hua Z: p53
mutation, EGFR gene amplification and loss of heterozygosity on
chromosome 10, 17 p in human gliomas. Chin Med J (Engl).
113:662–666. 2000.
|
|
34
|
Cecconi S, Mauro A, Cellini V and
Patacchiola F: The role of Akt signalling in the mammalian ovary.
Int J Dev Biol. 56:809–817. 2012. View Article : Google Scholar
|
|
35
|
Golding SE, Morgan RN, Adams BR, Hawkins
AJ, Povirk LF and Valerie K: Pro-survival AKT and ERK signaling
from EGFR and mutant EGFRvIII enhances DNA double-strand break
repair in human glioma cells. Cancer Biol Ther. 8:730–738. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schulze WX, Deng L and Mann M:
Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol
Syst Biol. 1:2005.00082005. View Article : Google Scholar
|