Introduction
Clear cell renal cell carcinoma (ccRCC) is the most
common type of cancer of the renal parenchyma. Although current
ccRCC treatment involves surgery combined with chemotherapy and
radiotherapy, the median survival rate of patients with ccRCC
remains low and a significant proportion of patients with ccRCC are
at a high risk of relapse (1–3).
Therefore, the development of effective therapeutic targets for
ccRCC is urgently required.
MicroRNAs (miRNAs) are non-coding single-stranded
RNAs between 19 and 25 nucleotides in length. They negatively
regulate the expression of target genes by binding to target mRNAs
at a post-transcriptional level. miRNAs are involved in various
biological processes, including tumorigenesis, but may also
function as tumor suppressors or promoters (4). Similarly to protein-coding genes, the
expression of miRNAs may also be mediated by epigenetic processes,
including DNA methylation, which may also be involved in the
development and progression of human malignancies (5,6).
miRNA (miR)-492, has been observed to be associated
with multiple types of cancer, including retinoblastoma,
hepatoblastoma, non-small cell lung cancer, pancreatic cancer and
oropharyngeal carcinoma (7–14).
Recently, it was reported that the expression levels of miR-492
were reduced in rectal cancer tissues compared with those of the
normal rectal mucosa, suggesting that miR-492 may have an
inhibitory role in the regulation of the development and
progression of rectal cancer (10,14).
In addition, numerous downregulated miRNAs in cancer have been
demonstrated to be associated with epigenetic mechanisms, for
example hypermethylation of their promoters (15,16).
However, the detailed mechanism of miR-492 function, as well as the
epigenetic regulatory processes in ccRCC have remained to be
elucidated.
The present study aimed to investigate the
epigenetic regulation of the expression of miR-492 in ccRCC. The
effect of miR-492 on cell proliferation, apoptosis, invasion and
adhesion in ccRCC cells was also examined.
Materials and methods
Reagents and materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal
bovine serum (FBS), TRIzol reagent, the TaqMan MicroRNA assay kit,
the bicinchoninic acid (BCA) protein assay kit and
Lipofectamine® 2000 were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). The demethylation drug
5-Aza-2′-deoxycytidine (Aza) and the histone deacetylase inhibitor
4-phenylbutyric acid (PBA) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). The miRNeasy mini kit was purchased from Qiagen
(Valencia, CA, USA). Mouse monoclonal anti-E-cadherin (1:500; cat.
no. YM0208), mouse monoclonal Vimentin (1:500; cat. no. YM0645),
rabbit polyclonal Caspase 3 (1:500; cat. no. YT0656), mouse
monoclonal BH3 interacting-domain death agonist (Bid; 1:500; cat.
no. YM0062), rabbit polyclonal phosphorylated B-cell lymphoma-2
(Bcl-2; 1:500; cat. no. YP0031) and mouse monoclonal β-actin
(1:500; cat. no. YM3028) antibodies were purchased from ImmunoWay
(Cambridge, UK). Goat anti-mouse (1:1,000; cat. no. SA001) and goat
anti-rabbit (1:1,000; cat. no. SA009) IgG(H+L)-HRP secondary
antibodies were purchased from Auragene Bioscience (Changsha,
China). A cell invasion assay kit was purchased from Merck
Millipore (Darmstadt, Germany).
Tissue collection
All protocols in the present study were approved by
the Ethics Committee of Central South University (Changsha, China).
Written informed consent was obtained from all patients with ccRCC
included in the study. A total of six ccRCC tissues and matched
adjacent normal tissues were collected at the Department of
Nephrology, Xiangya Hospital of Central South University. None of
the patients had received a blood transfusion, radiotherapy or
chemotherapy prior to the surgery. All samples were immediately
snap-frozen in liquid nitrogen (Auragene Bioscience) following
surgical removal and stored at −80°C until further use.
Cell culture
The present study utilized five human ccRCC cell
lines, 786-O, ACHN, SN12C, A704 and TK10, as well as a normal renal
cell line, HEK293, which were obtained from the Cell Bank of
Central South University. Cells were cultured in DMEM with 10% FBS
in a humidified atmosphere containing 5% CO2 at
37°C.
RNA extraction and miRNA expression
assay
miRNAs were isolated from tissues or cells using the
miRNeasy mini kit according to the manufacturer’s instructions. The
miRNA expression was then determined via reverse transcription
quantitative polymerase chain reaction (RT-qPCR) using the TaqMan
MicroRNA assay kit on a 7500 Fast Real Time PCR system (Applied
Biosystems Life Technologies, Foster City, CA, USA), in accordance
with the manufacturer’s instructions. U6 was used as an endogenous
control. For each sample, three independent experiments were
performed. The relative expression levels of mRNA and miRNA were
analyzed using the 2−∆∆Ct method (17).
Measurement of miR-492 promoter CpG
island methylation status using bisulfite genomic sequencing PCR
(BSP)
Genomic DNA was extracted from ccRCC 786-O and ACHN
cells using a genomic DNA extraction kit (Takara, Dalian, China).
Genomic DNA (1 μg) was modified with bisulfite using the
Epitect bisulfite kit (Qiagen) according to the manufacturer’s
instructions, and eluted in a total of 40 ml elution buffer
(Auragene Bioscience). Subsequently, 2 ml modified DNA was used as
the template for the BSP reaction. The primer sequences were as
follows: miR-429 forward, 5′-GTGACCTGGCTCCAGGAAAGGC-3′, and
reverse, 5′-CAGATGGAAAAGATGAAACAATGGG-3′. The PCR cycling
conditions were set at: 94°C for 4 min, followed by 35 cycles at
94°C for 30 sec, 55°C for 30 sec, 72°C for 5 min, and a final step
at 72°C for 5 min. The PCR products were gel purified, cloned into
the pUC18-T plasmid (Sangon Biotech, Shanghai, China) and
subsequently sequenced by BGI (Wuhan, China).
Measurement of miR-492 promoter CpG
island methylation status using methylation specific PCR
(MS-PCR)
Genomic DNA was extracted using a genomic DNA
extraction kit (Takara). Genomic DNA (1 μg) was modified
with bisulfite using the Epitect Bisulfite kit (Qiagen), according
to the manufacturer’s instructions and eluted in a total of 40 ml
elution buffer. MS-PCR was performed on bisulfate-treated DNA. The
primer sequences were as follows: Methylated miR-492 forward,
5′-CGGGGATATTATCGAGGTATATTC-3′ and reverse,
5′-AACTAACACAAACCCTCTACCG-3′ and unmethylated miR-492 forward,
5′-TGGGGATATTATTGAG GTATATTTG-3′ and reverse, 5′-AAACTAACACAAACC
CTCTACCAC-3′. The PCR cycling conditions were set at: 94°C for 4
min, followed by 35 cycles at 94°C for 30 sec, 55°C for 30 sec,
72°C for 5 min, and a final step at 72°C for 5 min.
Western blot analysis
Western blotting was used to examine the protein
expression levels in each group. Cells were lysed in cold
radioimmunoprecipitation buffer (Auragene Bioscience). The BCA
Protein assay kit was used to determine the protein concentration
and was used in accordance with the manufacturer’s instructions.
Subsequently, the proteins were separated by 10% SDS-PAGE (Auragene
Bioscience) and transferred onto a polyvinylidene difluoride (PVDF)
membrane (Auragene Bioscience). The PVDF membrane was blocked in 5%
nonfat dried milk in phosphate-buffered saline (PBS) for 4 h.
Subsequently, the PVDF membrane was incubated with specific primary
antibodies at 37°C for 3 h. Following three washes in PBS, each for
5 min, the PVDF membrane was incubated with the appropriate
secondary antibody at 37°C for 1 h. Following a further three
washes in PBS, each for 5 min, an enhanced chemiluminescence
western blotting kit (Thermo Fisher Scientific, Rockford, IL, USA)
was used to detect the immune complexes on the PVDF membrane.
Epigenetic drug treatment of cells
The human ccRCC cell lines, 786-O and ACHN were
treated with Aza (15.55 nM), or PBA (1.5 nM) or both in
combination, with or without anti-miR-429 (20 μM)
(HmiR-AN0497; Genecopoeia, Guangzhou, China), for 72 h.
Cell counting kit (CCK)-8 cell
proliferation assay
CCK-8 was used to evaluate cell proliferation. A
total of 5×103 cells were seeded in 96-well plates for
24 h, treated with the indicated drugs and further incubated for 0,
24, 48 and 72 h, respectively. At 1 h prior to the completion of
the incubation, 10 μl CCK-8 was added to each well. The
optical density at 450 nm in each well was determined using an
enzyme immunoassay analyzer (Multiskan M3; Thermo Fisher
Scientific).
MTT cell proliferation assay
For all groups, 10×103 cells/well were
plated in a 96-well plate and incubated for 0, 24, 48 and 72 h,
respectively, at 37°C with 5% CO2. To assess cell
proliferation, 50 μl MTT (5 mg/ml; Auragene Bioscience) in
PBS was added and cells were then incubated for 4 h at 37°C and 5%
CO2. Subsequently, the supernatant was removed and 150
μl dimethyl sulfoxide (Auragene Bioscience) was added. The
absorbance was detected at 450 nm with a Microplate Reader (Model
680 XR; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell apoptosis assay
Flow cytometry was used to determine the level of
cell apoptosis. At 24 h post-transfection, cells were ha r vested a
nd washed twice with cold PBS. Subsequently, 10×105
cells were resuspended in 200 μl binding buffer with 10
μl Annexin-V-fluorescein isothiocyanate and 5 μl
propidium iodide-phycoerythrin and incubated in the dark for 30
min. Following this stage, 300 μl binding buffer (Keygentec
Biotech. Co., Ltd., Nanjing, China) was added and cells were
subjected to flow cytometric analysis (Moflo XDP; Beckman Coulter,
Krefeld, Germany).
Cell invasion assay
Cells were administered the indicated drug
treatments for 72 h, starved in serum-free medium for 24 h and then
resuspended in serum-free medium. The cells were added to the upper
chamber of a transwell (Transwell kit; BD Biosciences, Bedford, MA,
USA), while the lower chamber was filled with base medium
containing 10% FBS. Following incubation for 24 h, cells attached
to the bottom of the chamber were stained with crystal violet
(Auragene Bioscience) for 20 min and then washed and air-dried.
Invasive cells were observed under a microscope (AE31; Motic,
Fujian, China).
Cell adhesion assay
Cells administered with the indicated drug
treatments for 72 h were seeded into a 24-well plate and 500
μl fibronectin (20 μg/ml; Sigma-Aldrich) was added
into each well. The 24-well plate was incubated at 4°C overnight.
Following washing with PBS, 1% bovine serum albumin (Auragene
Bioscience) was added to each well for blocking at 37°C for 2 h.
Subsequently, the blocking solution was removed and the cells were
dried. Cells were then digested and centrifuged at 200 × g for 5
min. Serum-free medium (HyClone, Beijing, China) was used to
produce a single-cell suspension and the cell concentration was
adjusted to 106 cells/ml. The cell suspension (1 ml) was
added into each well with a slide (Auragene Bioscience) and then
incubated at 37°C with 5% CO2 for 2 h. Following the
removal of non-adherent cells using PBS, 4% neutral formalin
(Auragene Bioscience) was used to fix adherent cells for 3 h and
each well was treated with 500 μl crystal violet and
incubated for 2 h. Adherent cells were then observed under a
microscope (AE31; Motic).
Statistical analysis
Data are expressed as the mean ± standard deviation
of three independent experiments and were analyzed using SPSS 17.0
statistical software (SPSS, Inc., Chicago, IL, USA). The
differences between groups were determined using a one-way analysis
of variance. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-492 is markedly
downregulated in ccRCC tissues and cells
The expression levels of miR-492 in ccRCC tissues
and cells, as well as in their matched adjacent tissues and normal
renal cells were examined. As shown in Fig. 1A, the expression of miR-492 was
significantly reduced in ccRCC tissues compared with that of their
matched adjacent tissues. In addition, analogous results were
observed in the ccRCC cell lines, 786-O, ACHN, SN12C, A704 and
TK10, compared with HEK293 cells. The methylation status in the CpG
island of the miR-492 promoter, which may be involved in the
downregulation of miR-492, was further investigated. MS-PCR was
used to determine the methylation status in the CpG island of the
miR-492 promoter in six ccRCC tissues, as well as two normal
adjacent tissues. As shown in Fig.
1B, tumor 1 and tumor 4 exhibited hemimethylation and tumors 2,
3, 5 and 6 exhibited methylation. Conversely, the two normal
adjacent tissues were observed to be unmethylated. These findings
suggested that hypermethylation of the miR-492 promoter may
contribute to the downregulation of miR-492 in ccRCC.
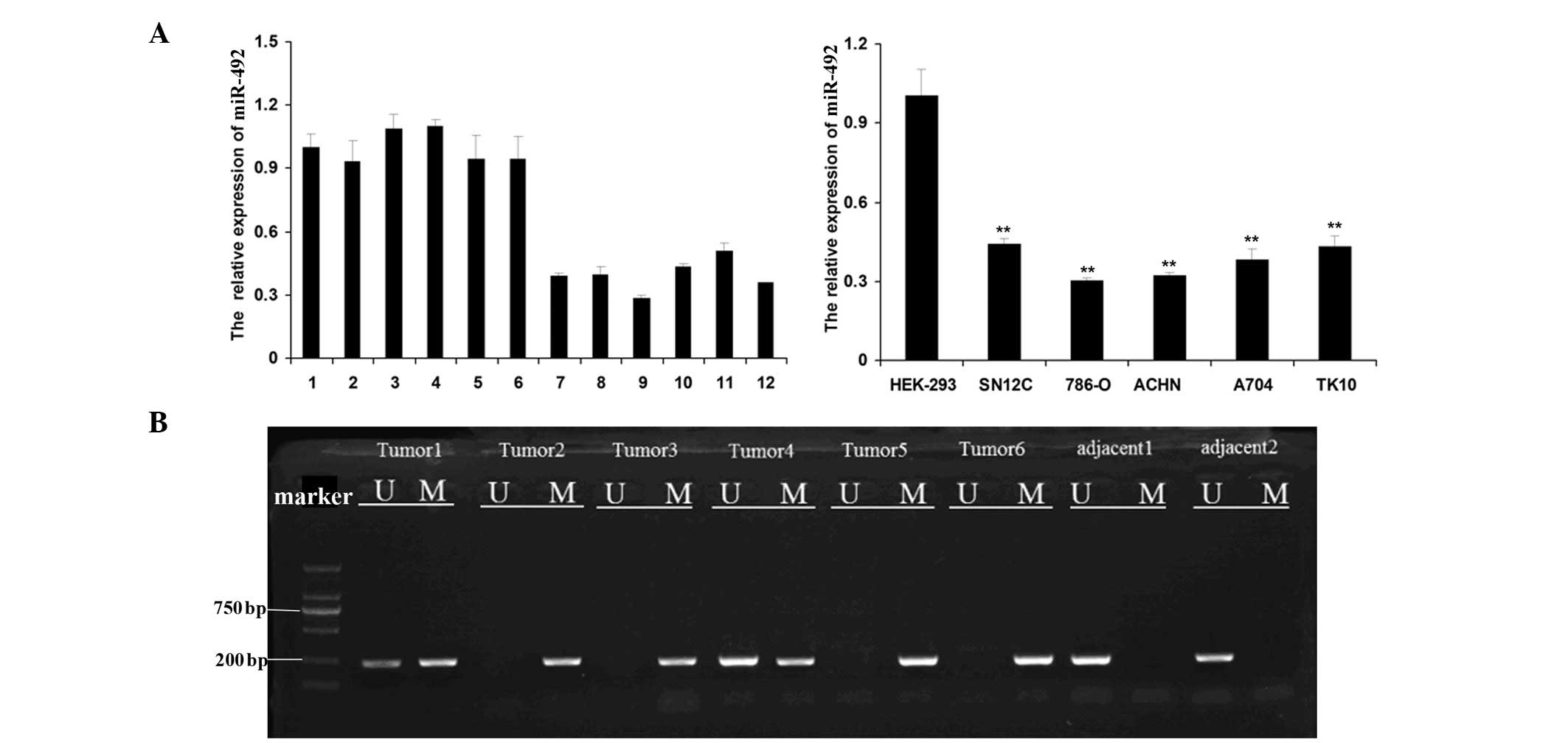 | Figure 1miR-492 expression is downregulated in
ccRCC cells and tissues. (A) Reverse transcription quantitative PCR
was performed to determine the relative expression of miR-492 in
six matched adjacent normal tissues (1–6) and
ccRCC tissues (7–12). The relative expression of miR-492
was also examined in five ccRCC cell lines, 786-O, ACHN, SN12C,
A704 and TK10, as well as a normal renal cell line HEK293.
**P<0.01, vs. HEK293. (B) Methylation specific PCR
was used to determine the methylation status in the CpG island of
the miR-492 promoter in six ccRCC tissues (Tumor 1–6) as well as in
two adjacent normal tissues (adjacent 1–2). U, unmethylated; M,
methylated; PCR, polymerase chain reaction; ccRCC, clear cell renal
cell carcinoma; miR, microRNA; bp, basepairs. |
Upregulation of miR-492 is induced by
epigenetic drug treatment in ccRCC cells
Following treatment of the ccRCC cell lines, 786-O
and ACHN, with Aza, PBA or Aza + PBA, the methylation status of the
CpG island of the miR-492 promoter was determined using BSP. As
shown in Fig. 2A, a significantly
higher level of methylation was observed in the 786-O and ACHN
cells following treatment with Aza, PBA or Aza + PBA. In addition,
the expression levels of miR-492 in each group were determined. As
indicated in Fig. 2B, miR-492
expression was significantly upregulated following treatment with
Aza and PBA, particularly following treatment with a combination of
Aza + PBA.
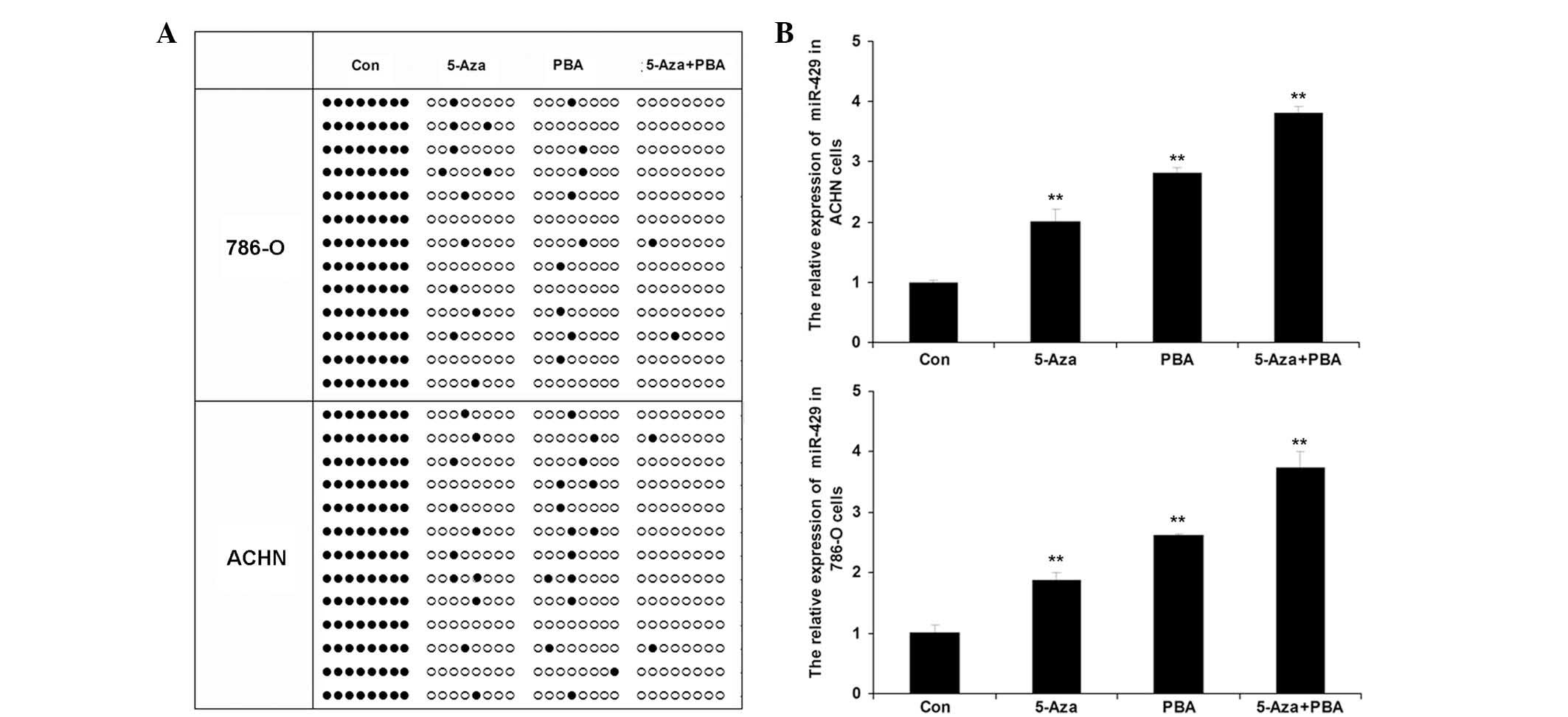 | Figure 2Epigenetic drug treatment induces
upregulation of miR-492 expression in ccRCC cells. (A) Bisulfite
genomic sequencing PCR was used to determine the methylation status
in the CpG island of the miR-492 promoter in two ccRCC cell lines,
786-O and ACHN, treated with Aza (15.55 nM), PBA (1.5 nM) or Aza
(15.55 nM) + PBA (1.5 nM) for 72 h. Black dots indicate
unmethylated sites and white circles indicate methylated sites. (B)
Reverse transcription quantitative PCR was performed to determine
the relative expression of miR-492 in ccRCC 786-O and ACHN cells
treated with Aza (15.55 nM), PBA (1.5 nM) or Aza (15.55 nM) + PBA
(1.5 nM) for 72 h. Con, cells without any treatment.
**P<0.01, vs. Con. PCR, polymerase chain reaction;
ccRCC, clear cell renal cell carcinoma; miR, microRNA; Con,
control; Aza, 5-Aza-2′-deoxycytidine; PBA, 4-phenylbutyric
acid. |
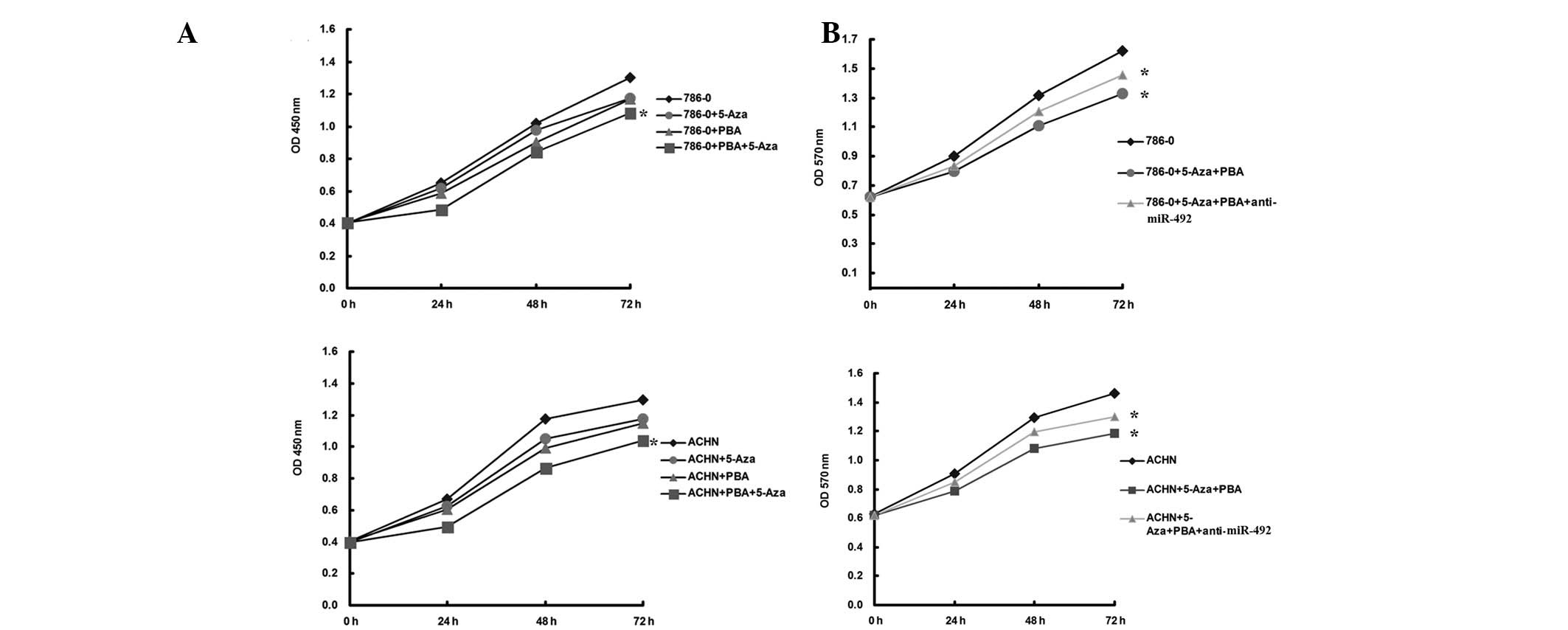
Upregulation of miR-492 induced by
epigenetic drug treatment inhibits proliferation in ccRCC
cells
The effects of miR-492 upregulation on ccRCC cells
were further investigated. As shown in Fig. 3A, the epigenetic drug-induced
upregulation of miR-492 significantly inhibited the proliferation
of ccRCC cells. To further confirm these findings, ccRCC cells were
transfected with anti-miR-492, which is able to reverse the
upregulation of miR-492 induced by treatment with Aza + PBA, and
further MTT investigation revealed that cellular proliferation was
enhanced in the Aza + PBA + anti-miR-492 group, when compared with
that in the Aza + PBA group (Fig.
3B). These data suggested that miR-492 has a suppressive role
in the regulation of ccRCC cell proliferation.
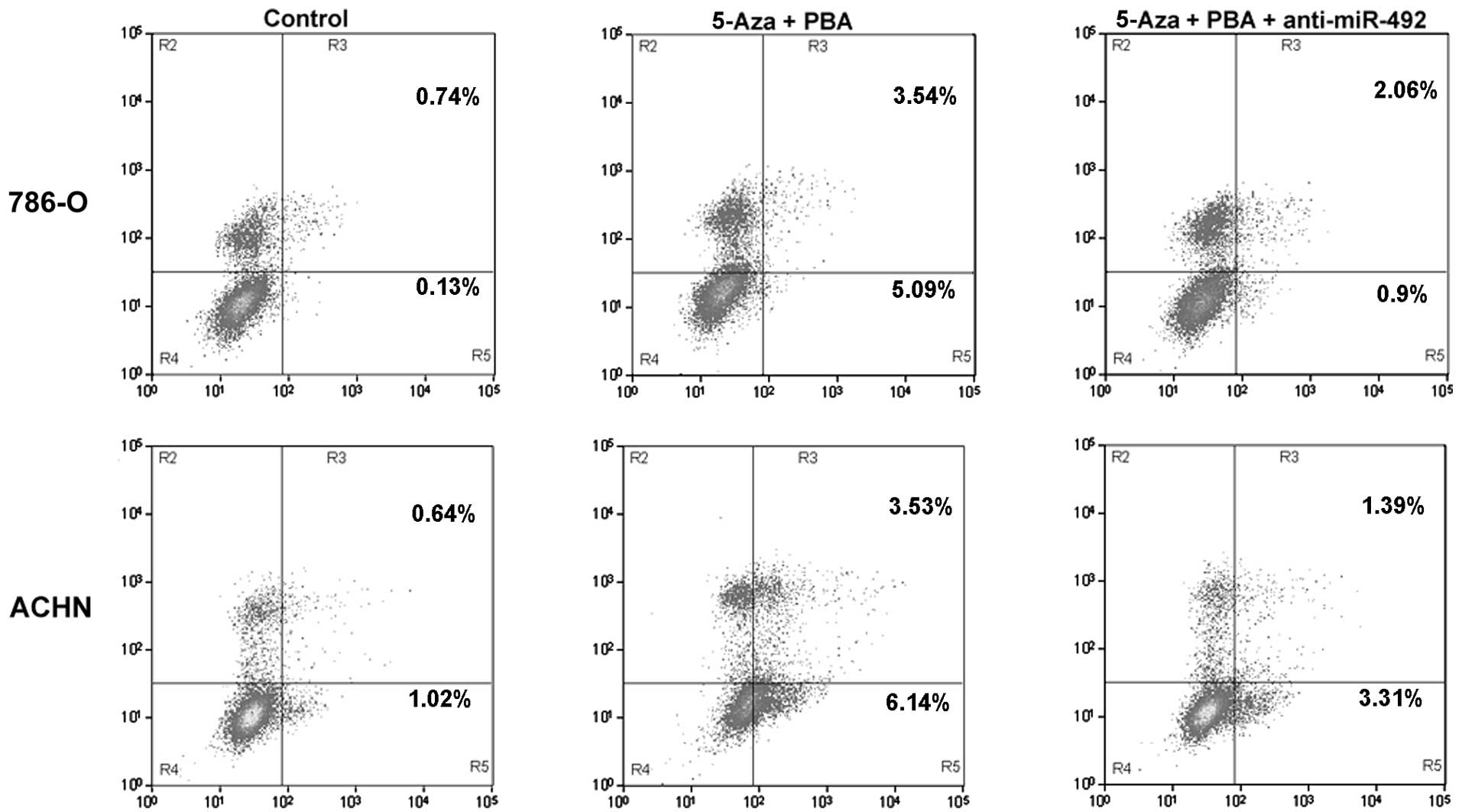
Upregulation of miR-492 induced by
epigenetic drug treatment promotes apoptosis in ccRCC cells
The effect of miR-492 on apoptosis was determined in
the ccRCC cell lines, 786-O and ACHN. As shown in Fig. 4, the upregulation of miR-492
induced by treatment with Aza + PBA significantly promoted
apoptosis in the ccRCC cell lines, 786-O and ACHN. However,
transfection with anti-miR-492 markedly attenuated this effect.
These findings suggested that miR-492 upregulation was able to
induce ccRCC cell apoptosis.
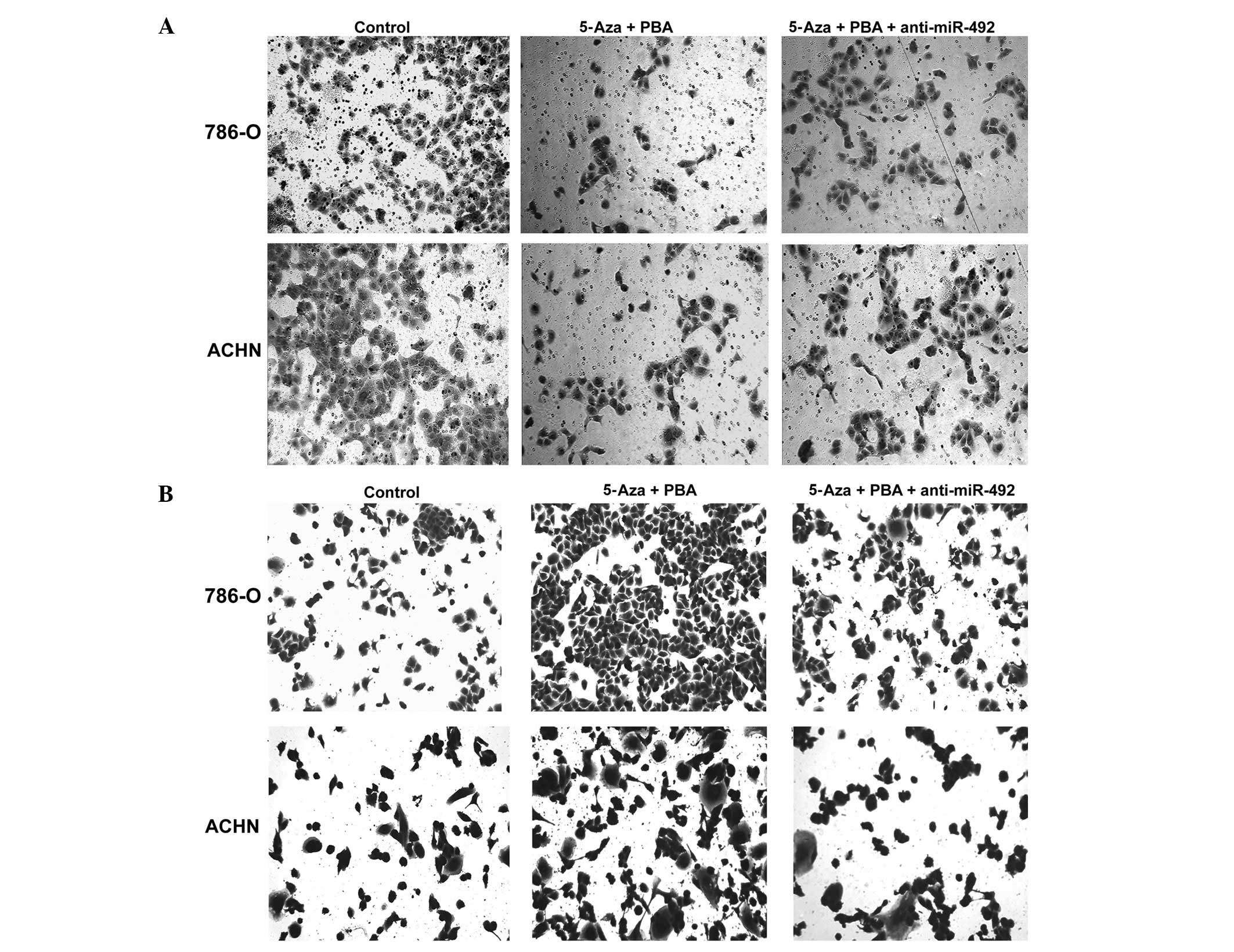
Upregulation of miR-492 induced by
epigenetic drug treatment inhibits invasion while promoting
adhesion in ccRCC cells
As shown in Fig.
5A, the upregulation of miR-492 induced by epigenetic drug
treatment significantly inhibited the invasion of the ccRCC cell
lines, 786-O and ACHN, which was attenuated by transfection with
anti-miR-492. Furthermore, miR-492 upregulation induced by
treatment with Aza + PBA markedly promoted cell adhesion in the
ccRCC cell lines, 786-O and ACHN, which was also inhibited by
transfection with anti-miR-492 (Fig.
5B). These findings suggested that miR-492 upregulation was
able to inhibit invasion while promoting adhesion in ccRCC
cells.
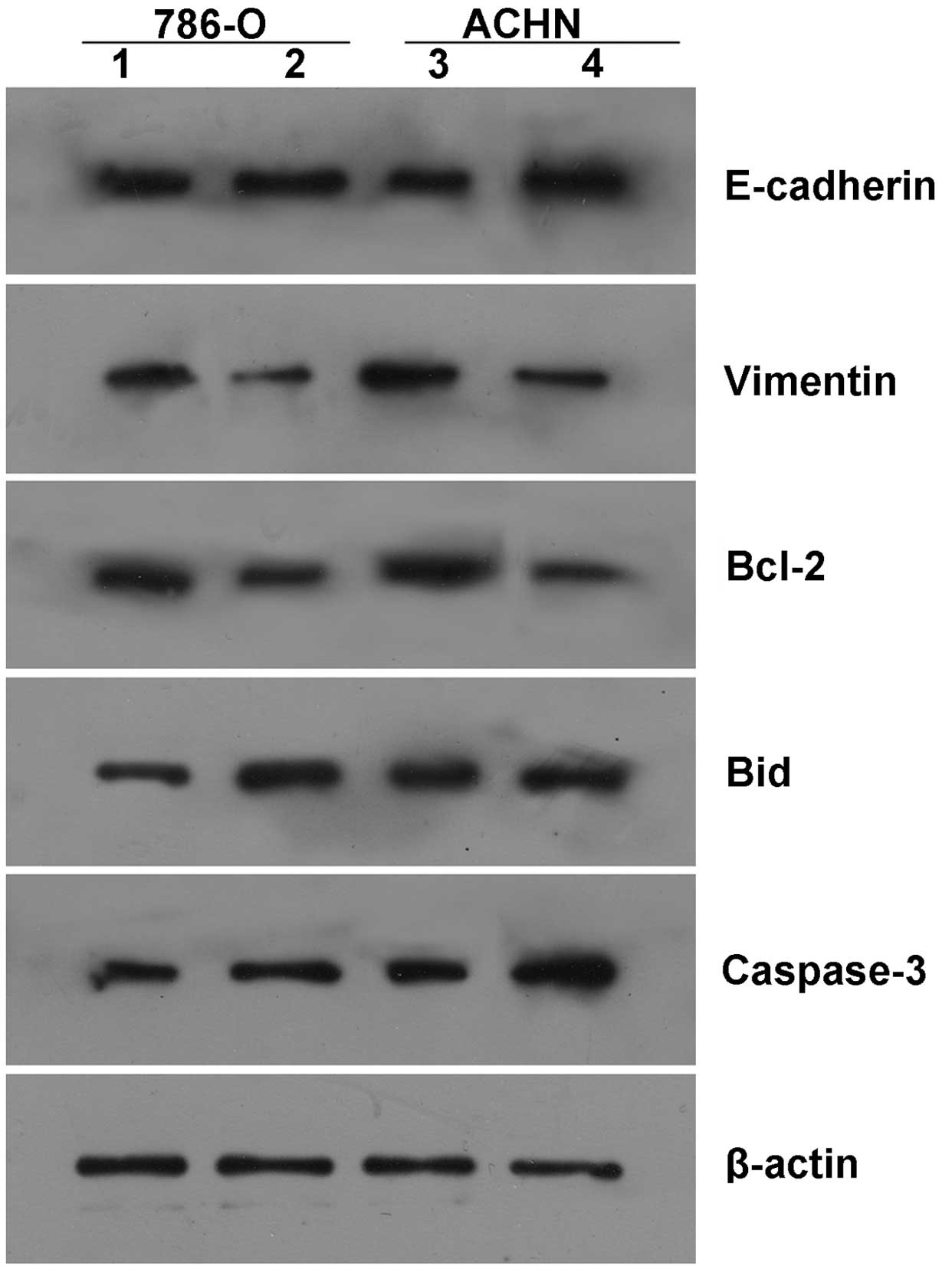
Changes in gene expression following
upregulation of miR-492 in ccRCC cells
To further investigate the molecular mechanism
underlying the effects of miR-492 in ccRCC cells, the expression of
several key factors associated with cell survival and adhesion in
ccRCC cells were examined following treatment with epigenetic
drugs. As shown in Fig. 6, the
upregulation of miR-492 expression induced by treatment with Aza +
PBA promoted the expression of pro-adhesive E-cadherin, and
inhibited the expression of anti-adhesive Vimentin, consistent with
the aforementioned data demonstrating that miR-492 upregulation
promoted adhesion in ccRCC cells. In addition, upregulation of
miR-492 also inhibited the expression of the anti-apoptotic protein
Bcl-2, and enhanced the expression of pro-apoptotic Bid and Caspase
3, which was consistent with the findings that miR-492 upregulation
promoted ccRCC cell apoptosis.
Discussion
In the present study, it was demonstrated that the
expression levels of miR-492 were significantly downregulated in
ccRCC tissues and cells, when compared with those of normal renal
tissues and cells. In addition, this downregulation was accompanied
by hypermethylation of the CpG island of the miR-492 promoter.
Furthermore, treatment with epigenetic drugs markedly promoted the
expression of miR-492 in ccRCC cells. Further investigation
demonstrated that the upregulation of miR-492 induced by epigenetic
drug treatment markedly inhibited proliferation and invasion,
whilst promoting apop-tosis and adhesion in ccRCC cells, suggesting
that miR-492 has a suppressive role in the regulation of malignant
phenotypes in ccRCC cells.
The detailed role of miR-492 in cancer has remained
elusive. The deregulation of miR-492 has been previously reported
in retinoblastoma; Zhao et al (7) revealed that miR-492 was highly
expressed in retinoblastoma, suggesting that miR-492 may be
involved in the tumorigenesis of retinoblastoma. Subsequently, von
Frowein et al (8) reported
that miR-492 was markedly upregulated in metastatic hepatoblastoma,
suggesting that miR-492 may promote certain processes involved in
the regulation of hepatoblastoma metastasis. Additionally, miR-492
was also reported to be associated with non-small cell lung cancer,
pancreatic cancer and oropharyngeal carcinoma (11–13).
In the present study, it was revealed that miR-492 was
significantly downregulated in ccRCC tissues and five ccRCC cell
lines. Gaedcke et al (10)
also observed a decrease in miR-492 expression in rectal cancer
tissues. In addition, Wu et al (14) revealed that miR-492 expression was
downregulated in ccRCC tissues. However, details of the specific
mechanisms underlying the involvement of miR-492 in ccRCC remains
to be elucidated.
Similarly to protein-coding genes, epigenetic
mechanisms, including DNA methylation and histone acetylation have
also been revealed to be involved in the regulation of miRNA
transcription (16). It has been
well-established that DNA methylation in the CpG island of the gene
promoter is the most common epigenetic modification observed in
eukaryotic genomes (18) and
hypermethylation may lead to decreased gene transcription (19). However, to the best of our
knowledge, the epigenetic mechanisms by which the expression of
miR-492 is mediated have not previously been investigated with
regards to cancer. In the present study, the methylation level in
the CpG island of the miR-492 promoter was significantly
upregulated in ccRCC tissues. However, the normal renal tissues
were observed to be unmethylated. As hypermethylation in the gene
promoter has an inhibitory role in the regulation of gene
transcription, it was hypothesized that hypermethylation of the
miR-492 promoter may contribute to the downregulation of miR-492,
as well as the development and progression of ccRCC. To further
confirm the involvement of an epigenetic mechanism in the
downregulation of miR-492, the ccRCC cell lines, 786-O and ACHN,
were treated with two common epigenetic drugs, Aza and PBA. Aza is
a DNA methyltransferase inhibitor, which is able to induce DNA
demethylation, while PBA is a histone deacetylase inhibitor, which
may induce histone acetylation (20,21).
DNA demethylation and histone acetylation promote gene
transcription (22,23). The present data revealed that
treatment with these epigenetic drugs significantly promoted the
expression of miR-492, particularly when administered in
combination, indicating that the expression levels of miR-492 in
ccRCC were tightly regulated by epigenetic modulations, including
DNA methylation and histone acetylation.
In addition, the upregulation of miR-492 induced in
ccRCC cells treated with Aza and PBA resulted in a significant
decrease in the proliferation of ccRCC cells. Accordingly, it was
hypothesized that miR-492 may have an inhibitory role in the
regulation of ccRCC cell proliferation. To verify this hypothesis,
anti-miR-492 was applied, and the results demonstrated that
anti-miR-492 reversed the inhibitory effects of Aza and PBA on
ccRCC cell proliferation. Subsequently, the effects of miR-492
upregulation on ccRCC cell apoptosis, invasion and adhesion were
further examined. The present data demonstrated that miR-492
upregulation significantly promoted cell apoptosis and adhesion,
while suppressing cell invasion in ccRCC cells.
Furthermore, upregulation of apoptosis was
accompanied by an increase in the expression of Caspase 3 and Bid,
as well as a decrease in the expression of Bcl-2. Bcl-2 and Bid are
two key members of the Bcl-2 family, which is involved in the
regulation of cell survival. However, Bcl-2 and Bid have opposing
effects on cell survival rate, since Bcl-2 is a key anti-apoptotic
factor, whereas Bid functions as a death agonist able to promote
cell apoptosis through heterodimerization with Bcl-2 (24,25).
In addition, sequential activation of the caspases is crucial in
the execution phase of cell apoptosis, and Caspase 3 is a key
executor (26,27). Accordingly, the present data
suggested that these three key apoptosis-associated proteins may
act as downstream effectors of miR-492 in ccRCC cells. In addition,
the upregulation of adhesion and inhibition of invasion observed
were consistent with the increased expression of E-cadherin as well
as the reduced expression of Vimentin. E-cadherin is a cell-cell
adhesion molecule and its increased expression may lead to
upregulation of adhesion as well as inhibition of cell motility
(28,29). By contrast, Vimentin functions as a
cytoskeletal linker protein and is critical in the regulation of
cell motility (30,31). In accordance with these results, it
was suggested that miR-492 upregulation promoted adhesion while
suppressing invasion in ccRCC cells, partially at least, through
promoting the expression of E-cadherin and inhibiting the
expression of Vimentin.
In conclusion, the present study revealed that
miR-492 was markedly downregulated in ccRCC cells due to the
hyper-methylated status of the miR-492 promoter. In addition, the
upregulation of miR-492 induced by epigenetic drug treatment
inhibited cell proliferation and invasion, while it promoted cell
apoptosis and adhesion in ccRCC cells. Based on these results, it
was hypothesized that miR-492 has an inhibitory role in ccRCC, and
may be a novel diagnostic or therapeutic target for ccRCC.
Acknowledgments
The present study was supported by funding from
National Natural Science Foundation of China (grant no. 81201672),
Research Fund for the Doctoral Program of Guangdong Medical College
(grant no. XB1331) and Research Fund for the Doctoral Program of
Affiliated Hospital of Guangdong Medical College (grant no.
BK201208).
References
|
1
|
Diamond E, Riches J, Faltas B, Tagawa ST
and Nanus DM: Immunologics and chemotherapeutics for renal cell
carcinoma. Semin Intervent Radiol. 31:91–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rydzanicz M, Wrzesiński T, Bluyssen HA and
Wesoly J: Genomics and epigenomics of clear cell renal cell
carcinoma: recent developments and potential applications. Cancer
Lett. 341:111–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chowdhury S, Matrana MR, Tsang C, Atkinson
B, Choueiri TK and Tannir NM: systemic therapy for metastatic
non-clear-cell renal cell carcinoma: recent progress and future
directions. Hematol Oncol Clin North Am. 25:853–869. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garg D and Cohen SM: miRNAs and aging: A
genetic perspective. Ageing Res Rev. 2014:3–8. 2014. View Article : Google Scholar
|
|
5
|
Choi JD and Lee JS: Interplay between
epigenetics and genetics in cancer. Genomics Inf. 11:164–173. 2013.
View Article : Google Scholar
|
|
6
|
Watanabe K and Takai D: Disruption of the
expression and function of microRNAs in lung cancer as a result of
epigenetic changes. Front Genet. 4:2752013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao JJ, Yang J, Lin J, et al:
Identification of miRNAs associated with tumorigenesis of
retinoblastoma by miRNA microarray analysis. Childs Nerv Syst.
25:13–20. 2009. View Article : Google Scholar
|
|
8
|
von Frowein J, Pagel P, Kappler R, von
Schweinitz D, Roscher A and Schmid I: MicroRNA-492 is processed
from the keratin 19 gene and up-regulated in metastatic
hepatoblastoma. Hepatology. 53:833–842. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoon KA, Yoon H, Park S, et al: The
prognostic impact of microRNA sequence polymorphisms on the
recurrence of patients with completely resected non-small cell lung
cancer. J Thorac Cardiovasc Surg. 144:794–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gaedcke J, Grade M, Camps J, et al: The
rectal cancer microRNAome - microRNA expression in rectal cancer
and matched normal mucosa. Clin Cancer Res. 18:4919–4930. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schultz NA, Werner J, Willenbrock H, et
al: MicroRNA expression profiles associated with pancreatic
adenocarcinoma and ampullary adenocarcinoma. Mod Pathol.
25:1609–1622. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hui AB, Lin A, Xu W, et al: Potentially
prognostic miRNAs in HPV-associated oropharyngeal carcinoma. Clin
Cancer Res. 19:2154–2162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kupcinskas J, Wex T, Link A, et al: Gene
polymorphisms of micrornas in Helicobacter pylori-induced high risk
atrophic gastritis and gastric cancer. PLoS One. 9:e874672014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu X, Weng L, Li X, et al: Identification
of a 4-microRNA signature for clear cell renal cell carcinoma
metastasis and prognosis. PLoS One. 7:e356612012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JG, Kim TO, Bae JH, et al:
Epigenetically regulated MIR941 and MIR1247 target gastric cancer
cell growth and migration. Epigenetics. 9:1018–1030. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suzuki H, Maruyama R, Yamamoto E and Kai
M: Epigenetic alteration and microRNA dysregulation in cancer.
Front Genet. 4:2582013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu Y, Liu J, Jiang B, et al: Mir-199a-5p
loss up-regulated DDR1 aggravated colorectal cancer by activating
epithelial-to-mesenchymal transition related signaling. Dig Dis
Sci. 59:2163–2172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shapiro JA: Epigenetic control of mobile
DNA as an interface between experience and genome change. Front
Genet. 5:872014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Chen Y, Tang J and Xie X:
Frequent loss expression of and promotor hypermethylation in human
cancers: a meta-analysis and systematic review. Pak J Med Sci.
30:432–437. 2014.PubMed/NCBI
|
|
20
|
Izutsu N, Maesawa C, Shibazaki M, et al:
Epigenetic modification is involved in aberrant expression of class
III β-tubulin, TUBB3, in ovarian cancer cells. Int J Oncol.
32:1227–1235. 2008.PubMed/NCBI
|
|
21
|
Song K, Han C, Zhang J, et al: Epigenetic
regulation of MicroRNA-122 by peroxisome proliferator activated
receptor-gamma and hepatitis b virus X protein in hepatocellular
carcinoma cells. Hepatology. 58:1681–1692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fetahu IS, Höbaus J, Aggarwal A, et al:
Calcium-sensing receptor silencing in colorectal cancer is
associated with promoter hyper-methylation and loss of acetylation
on histone 3. Int J Cancer. 135:2014–2023. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Z, Li H and Jin P: Epigenetics-based
therapeutics for neuro-degenerative disorders. Curr Transl Geriatr
Exp Gerontol Rep. 1:229–236. 2012. View Article : Google Scholar
|
|
24
|
Wang Y and Tjandra N: Structural insights
of tBid, the caspase-8-activated Bid, and its BH3 domain. J Biol
Chem. 288:35840–35851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Renault TT and Chipuk JE: Getting away
with murder: how does the BCL-2 family of proteins kill with
immunity? Ann NY Acad Sci. 1285:59–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maurya SK, Tewari M, Sharma B and Shukla
HS: Expression of procaspase 3 and activated caspase 3 and its
relevance in hormone-responsive gallbladder carcinoma chemotherapy.
Korean J Intern Med. 28:573–578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Connolly PF, Jäger R and Fearnhead HO: New
roles for old enzymes: killer caspases as the engine of cell
behavior changes. Front Physiol. 5:1492014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nilsson GM, Akhtar N, Kannius-Janson M and
Baeckström D: Loss of E-cadherin expression is not a prerequisite
for c-erbB2-induced epithelial-mesenchymal transition. Int J Oncol.
45:82–94. 2014.PubMed/NCBI
|
|
29
|
Ponce E, Louie MC and Sevigny MB: Acute
and chronic cadmium exposure promotes E-cadherin degradation in
MCF7 breast cancer cells. Mol Carcinog. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sutoh Yoneyama M, Hatakeyama S, Habuchi T,
et al: Vimentin intermediate filament and plectin provide a
scaffold for inva-dopodia, facilitating cancer cell invasion and
extravasation for metastasis. Eur J Cell Biol. 93:157–169. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fu CH, Lin RJ, Yu J, et al: A novel
oncogenic role of inositol phosphatase SHIP2 in ER-negative breast
cancer stem cells: involvement of JNK/Vimentin activation. Stem
Cells. 32:2048–2060. 2014. View Article : Google Scholar : PubMed/NCBI
|