Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignant cancer and the third leading cause of
cancer-associated mortality worldwide (1). Despite remarkable achievements in
improving HCC treatment, patient prognosis generally remains very
poor (2). Although numerous
molecules and signaling pathways involved in the development of HCC
have been identified (3–6), the molecular mechanisms underlying
the tumorigenesis and proliferation of HCC have remained
elusive.
Enoyl-coenzyme A (CoA) hydratase short chain 1
(ECHS1) catalyzes the hydration of 2-trans-enoyl-CoA intermediates
to form L-3-hydroxyacyl-CoAs, which constitutes the second step in
the β-oxidation pathway of fatty acid metabolism (7). In addition to its role in fatty acid
metabolism, ECHS1 may also be involved in tumor development. A
proteomics study revealed that ECHS1 was consistently upregulated
in prostate cancer tissues and is therefore considered a candidate
disease biomarker in prostate needle-biopsy specimens (8). ECHS1 was also identified as a
candidate gene in human colorectal carcinogenesis (9,10).
Downregulation of ECHS1 enhances PP2-induced apoptosis in human
breast cancer MCF-7 cells (11).
Chang et al demonstrated that ECHS1 interacts with and
inhibits signal transducer and activator of transcription 3
phosphorylation, transcriptional activity and subsequent target
gene expression (12). The results
of a previous study by our group confirmed ECHS1 as a novel
hepatitis B surface antigen (HB) binding protein that enhances
HepG2 cell apoptosis by reducing the mitochondrial membrane
potential; and explored the potential role of ECHS1 in HCC
(13), although the precise
molecular mechanisms of ECHS1 in HCC progression remain to be
elucidated.
Several studies have demonstrated aberrant
activation of epidermal growth factor receptor (EGFR) in HCC
pathogenesis (14–17). Significant alterations in the
downstream cellular signaling cascades, including phosphoinositide
3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) and
Raf/mitogen activated protein kinase kinase (MEK)/extracellular
signal-regulated kinase (ERK), as well as altered gene expression
result in hepatoma formation via increased proliferation,
cell-cycle progression and apoptotic resistance (14,18–20).
However, the molecular mechanisms underlying these effects have
remained elusive. The present study aimed to investigate the
molecular mechanisms of ECHS1 in HCC cells in vitro and
in vivo.
Materials and methods
Ethics statement
The present study was approved by the Ethics
Committee of Zhongshan Hospital Affiliated to Xiamen University
(no. 20081009; Xiamen, China). All procedures involving
experimental animals were performed in accordance with protocols
approved by the Committee for Animal Research of Xiamen University
(Xiamen, China) and the Guide for the Care and Use of Laboratory
Animals (National Institutes of Health publication no. 86–23,
revised 1985).
Cell cultures and reagents
HepG2 and HuH7 cells were obtained from the American
Type Culture Collection (Manassas, VA, USA) and cultured in
Dulbecco’s modified Eagle’s medium supplemented with 10% fetal
bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin, 2 mM
glutamine, 1 mM sodium pyruvate and 0.1 mM non-essential amino
acids at 37°C with 5% CO2 (All from Gibco Life
Technologies, Carlsbad, CA, USA).
Construction of ECHS1-interfering
vectors
A plasmid encoding a short interfering RNA (siRNA)
targeted to ECHS1 was constructed as described previously (13). Briefly, two siRNA target sequences
(5′-GCCCATATCGTTTCATAGCTT-3′ and 5′-GTAGATGAGATGTGACGAATT-3′) for
the ECHS1 gene (siECHS1–1 and siECHS1–2) were selected using the
RNAi Target Selector algorithm (http://www.clontech.com/GB/Support/Online_Tools), and
the most efficient of the two (siECHS1–2) was selected for use
using the Basic Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi) in the
present study. The oligonucleotides were annealed, to form siRNA
duplexes, and were cloned into the BbsI and XbaI restriction sites
of the pu6 vector (Dharmacon, Inc., Chicago, IL, USA) and named
siECHS1–1 and siECHS1–2. The final clones were verified by DNA
sequencing (Beijing Genomics Institution, Beijing, China).
Establishment of stable ECHS1-knockdown
cell pools
HepG2 and HuH7 cells were transfected with
pu6-siECHS1 using Lipofectamine® 2000 reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA). Cells
transfected with empty pu6 vectors served as controls. Cells were
cultured in medium containing 1 μg/ml puromycin (Invitrogen
Life Technologies) for selection (21,22).
Following two weeks of culture, ECHS1 protein expression was
examined by western blotting (as described in the ‘Western blot
analysis’ section) to validate knockdown efficiency; and these
experiments were repeated three times. The ECHS1-knockdown cell
lines were named HepG2-siECHS1 and HuH7-siECHS1 and the control
cell lines were named HepG2-pu6 and HuH7-pu6.
Cell proliferation assays
Cell proliferation was measured by Cell Counting
kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). Cells were cultured at a density of ~2,500
cells/well in a 96-well plate, in which 10 μl CCK-8 was
added to 100 μl culture medium. The cells were incubated for
1.5 h at 37°C and absorbance was measured at 450 nm using a
Fluorescence Multi-Well Plate Reader CytoFluor 4000–2 (PerSeptive
Bio-systems, Inc., Framingham, MA, USA). Experiments were repeated
three times.
The 5-bromo-2-deoxyuridine (BrdU) assay was
performed with the BrdU Cell Proliferation Assay kit according to
the manufacturer’s instructions (Cell Signaling Technology, Inc.,
Danvers, MA, USA). Briefly, cells were seeded into three wells of a
96-well plate at 0.5×104, 1×104 and
2×104 cells/well and incubated at room temperature for
24 h. BrdU (10 μl) was added to each well and incubated at
room temperature for an additional 4 h. Following removal of the
medium, 100 μl/well Fixing/Denaturing solution was added and
incubated at room temperature for 30 min, followed by antibody
detection solution for 1 h. The plate was washed three times and
the anti-mouse horseradish peroxidase-conjugated secondary
immunoglobulin G (IgG) antibody was added and incubated for 30 min,
followed by 100 μl TMB Substrate for 30 min. The reaction
was terminated with STOP solution and absorbance was measured at
450 nm on the PerSeptive Bio-systems, Inc. microplate reader.
Enzymatic activity of ECHS1
ECHS1 activity was assayed according to the
manufacturer’s instructions of the Cellular Short Chain ENOYL COA
HYDRATASE Activity Assay kit (Genmed Scientifics, Inc., Arlington,
MA, USA). Activity was measured by determining absorbance at 263 nm
with the PerSeptive Bio-systems, Inc. microplate reader. Protein
concentrations were determined by the Bradford method (23) with Quick Start Bradford Protein
Assay kit 1 assay solution and bovine serum albumin (BSA) was used
as the standard (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Xenograft assay
To determine the oncogenicity of ECHS1, ten 4–6
month-old male nude mice were randomly divided into two groups and
administered 6×106 HepG2-siECHS1 or HepG2-pu6
cells/mouse via injection to the gluteal region. HepG2 cells were
selected for use, due to Huh7 cells exhibiting poor tumorigenesis
effects in preliminary experiements. Tumor size was assessed every
three days using calipers and the tumor volume was calculated using
the formula: [length (mm) × width (mm)2]/2.
Western blot analysis
The total protein was extracted from cells and
tissue specimens using Mammalian Cell Lysis reagent (Fermentas;
Thermo Fisher Scientific, Pittsburgh, PA, USA) according to the
manufacturer’s instructions. The proteins were resolved using 10,
12 or 15% SDS-PAGE and detected with the appropriate antibodies.
The following primary antibodies were all purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA), apart from those
against ECHS1 and EGFR which were purchased from ProteinTech Group,
Inc. (Chicago, IL, USA): Mouse monoclonal proliferating cell
nuclear antigen (PCNA; 1:1,000; #2586), rabbit monoclonal AKT
(1:1,000; #4691), rabbit monoclonal phosphorylated (p-)Akt(Ser473)
(1:1,000; #4060), ERK1/2 (1:1,000; #4695), rabbit monoclonal
p-ERK1/2(Thr202/Tyr204) (1:1,000; #4370), rabbit polyclonal nuclear
factor (NF)-κB (1:1,000; #4882), rabbit polyclonal p-NF-κB
(1:1,000; #4810), rabbit monoclonal p-EGFR(Tyr1068) (1:1,000;
#11862), rabbit monoclonal glycogen synthase kinase (GSK)-3β
(1:1,000; #12456), rabbit monoclonal p-GSK-3β(Ser9) (1:1,000;
#5558), rabbit monoclonal C-Myc (1:1,000; #13987), rabbit
monoclonal β-catenin (1:1,000; #8480), rabbit monoclonal cyclin
dependent kinase (CDK)4 (1:1,000; #12790), rabbit monoclonal CDK6
(1:1,000; #13331), rabbit monoclonal cyclin D1 (1:1,000; #2978),
mouse monoclonal cyclin D3 (1:1,000; #2936), rabbit polyclonal p15
(1:1,000; #4822), rabbit poly-clonal p16 (1:1,000; #4824), mouse
monoclonal p21 (1:1,000; #2946), rabbit monoclonal p27 (1:1,000;
#3686), rabbit polyclonal ECHS1 (1:2,000; 11305–1-AP) and rabbit
polyclonal EGFR (1:3,000; 18986–1-AP). The antibodies were diluted
in 5% w/v BSA, 1X Tris-buffered saline and 0.1% Tween-20 (Amresco,
Solon, OH, USA) at 4°C with gentle shaking, overnight.
Subsequently, the membranes were washed and incubated with the
following secondary antibodies from Cell Signaling Technology,
Inc.: Anti-mouse IgG (1:2,500; #7076) and anti-rabbit IgG (1:2500;
#7074) conjugated to horseradish peroxidase.
Immunoreactivity was detected with an
Enhanced Chemiluminescence kit (GE Healthcare Life Sciences,
Chalfont, UK) and quantified using densitometry with the ImageQuant
LAS 4000 mini system (GE Healthcare Life Sciences)
Tubulin (Cell Signaling Technology, Inc.) was used
as a loading control.
Statistical analysis
Experimental data are presented as the mean ±
standard deviation of at least three independent experiments, as
calculated using SPSS, version 13.0 (SPSS, Inc., Chicago, IL, USA).
The independent-samples and paired-samples t-tests were used to
compare data. P<0.05 was considered to indicate a statistically
significant difference. Statistical graphs with error bars
representing the standard deviation of the mean were created using
GraphPad Prism 5 Software (GraphPad Software, Inc., San Diego, CA,
USA).
Results
Establishment of stable ECHS1-knockdown
cell pools using siRNA in human HepG2 and HuH7 cells
HepG2 and HuH7 cells were transfected with ECHS1
siRNA or the empty pu6 vector (control) to stably establish
HepG2-siECHS1 and HuH7-siECHS1 ECSH1 knocked-down cell pools and
HepG2-pu6 and HuH7-pu6 control cells. Western blot analysis
revealed that ECHS1 expression was downregulated in the
HepG2-siECHS1 and HuH7-siECHS1 cells compared with that of the
HepG2-pu6 and HuH7-pu6 cells, respectively (Fig. 1A). As expected, the enzymatic
activity of ECHS1 was attenuated in the HepG2-siECHS1 and
HuH7-siECHS1 cells compared with that of HepG2-pu6 and HuH7-pu6
cells (Fig. 1B).
 | Figure 1Silencing of ECHS1 attenuates HCC
cell proliferation in vitro. (A) Western blot analysis
confirmed siRNA silencing of ECHS1 and proliferation marker PCNA
expression. Cells transfected with empty vectors were used as
controls and tubulin was used as an internal control. (B) Relative
enzymatic activity of ECHS1 in HepG2-siECHS1, Huh7-siECHS1 and
control cells, *P<0.01. (C and D) CCK8 assay of cell
proliferation in HepG2-siECHS1, Huh7-siECHS1 and control cells,
performed every 12 h for 72 h; *P<0.05. (E and F)
BrdU assay of cell proliferation in HepG2-siECHS1, Huh7-siECHS1 and
control cells following 24 h incubation at 0.5×104,
1×104 and 2×104 cells/well, respectively;
*P<0.05. ESCHS1, enoyl-coenzyme A hydratase short
chain 1; HCC, hepatocellular carcinoma; siRNA, short interfering
RNA; PCNA, proliferating cell nuclear antigen; CCK8, cell counting
kit-8; BrdU, 5-bromo-2-deoxyuridine; pu6, control cells; siE, ECHS1
siRNA-transfected cells; SiECHS1, ESCHS1 siRNA-transfected
cells. |
Silencing of ECHS1 attenuates HCC cell
proliferation in vitro
A reduction in the proliferation rate of
HepG2-siECHS1 and HuH7-siECHS1 cells compared with that of the
control cells was identified by CCK8 assay following 24, 36, 48 and
72 h of incubation (Fig. 1C and
D). The BrdU staining results at 0.5×104,
1×104 and 2×104 cells/well following 24 h of
incubation were consistent with those of the CCK-8 assay (Fig. 1E and F). PCNA, a proliferation
marker, was also downregulated in HepG2-siECHS1 and HuH7-siECHS1
cells (Fig. 1A). Therefore,
silencing of ECHS1 inhibited the proliferation of HepG2 and HuH7
cells.
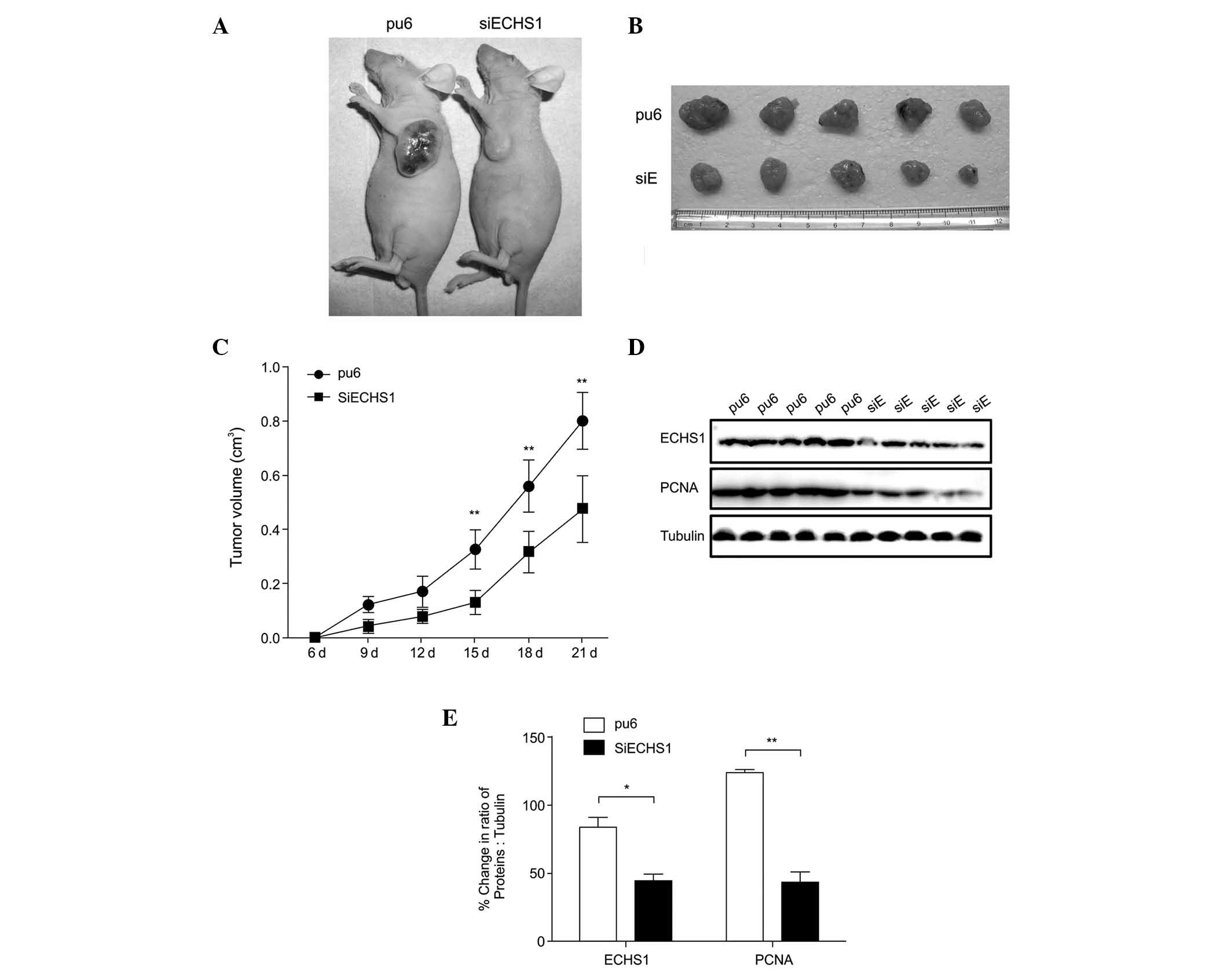
Silencing of ECHS1 suppresses xenograft
tumor growth
ECHS1 knockdown suppressed HCC cell proliferation
in vitro; therefore, the role of ECHS1 in HCC development
in vivo was subsequently evaluated. Nude mice were
subcutaneously injected with HepG2-siECHS1 and HepG2-pu6 cells to
induce xenograft tumors (Fig. 2A and
B), which developed over time (Fig. 2C). Slower growth was observed in
tumors derived from HepG2-siECHS1 cells than in those derived from
HepG2-pu6 cells (Fig. 2A–C).
Western blotting verified that ECHS1 knockdown was maintained in
the transplanted tumors, and PCNA expression remained lower in mice
injected with HepG2-siECHS1 cells than those of the controls
(Fig. 2D and E). Therefore it was
concluded that the absence of ECHS1 was responsible for the
inhibition of tumor growth.
ECHS1 regulates HCC cell proliferation
via the EGFR signaling pathway
To define the signaling pathways involved in
ECHS1-mediated regulation of HCC cell proliferation, western
blotting was used to detect the phosphorylation levels of EGFR and
its downstream effectors ERK and AKT, which mediate proliferation,
survival, apoptosis and cell cycle progression. The expression
levels of p-EGFR, p-ERK1/2 (Thr202/Tyr204) and p-AKT (Ser473) were
markedly decreased in HepG2-siECHS1 and HuH7-siECHS1 cells compared
with those of the control cells (Fig.
3A and B). EGFR, ERK1/2 and AKT phosphorylation were also
analyzed in xenograft tumors derived from ECHS1 knock-down cells.
Consistent with the results of the in vitro study, the
phosphorylation of EGFR, ERK1/2 and AKT was down-regulated in ECHS1
knock-down tumor tissues at the end of the study period (Fig. 3C–F). The expression of GSK-3β,
which is a major inhibitor of the Wnt/β-catenin pathway, was also
assessed. Phosphorylation of GSK-3β at serine-9 was markedly
reduced in HepG2-siECHS1 and Huh7-siECHS1 cells compared with that
of the control cells (Fig. 4A),
thereby increasing the negative regulatory effect of GSK-3β on the
Wnt/β-catenin pathway. Concurrently, C-Myc and β-catenin expression
were also downregulated in HepG2- and HuH7-siECHS1 cells (Fig. 4B). However, no significant
differences in NF-κB phosphorylation were observed in these cells
(data not shown). Therefore, it was hypothesized that ECHS1 may
regulate cancer cell proliferation in vitro and in
vivo via an EGFR-mediated signaling pathway.
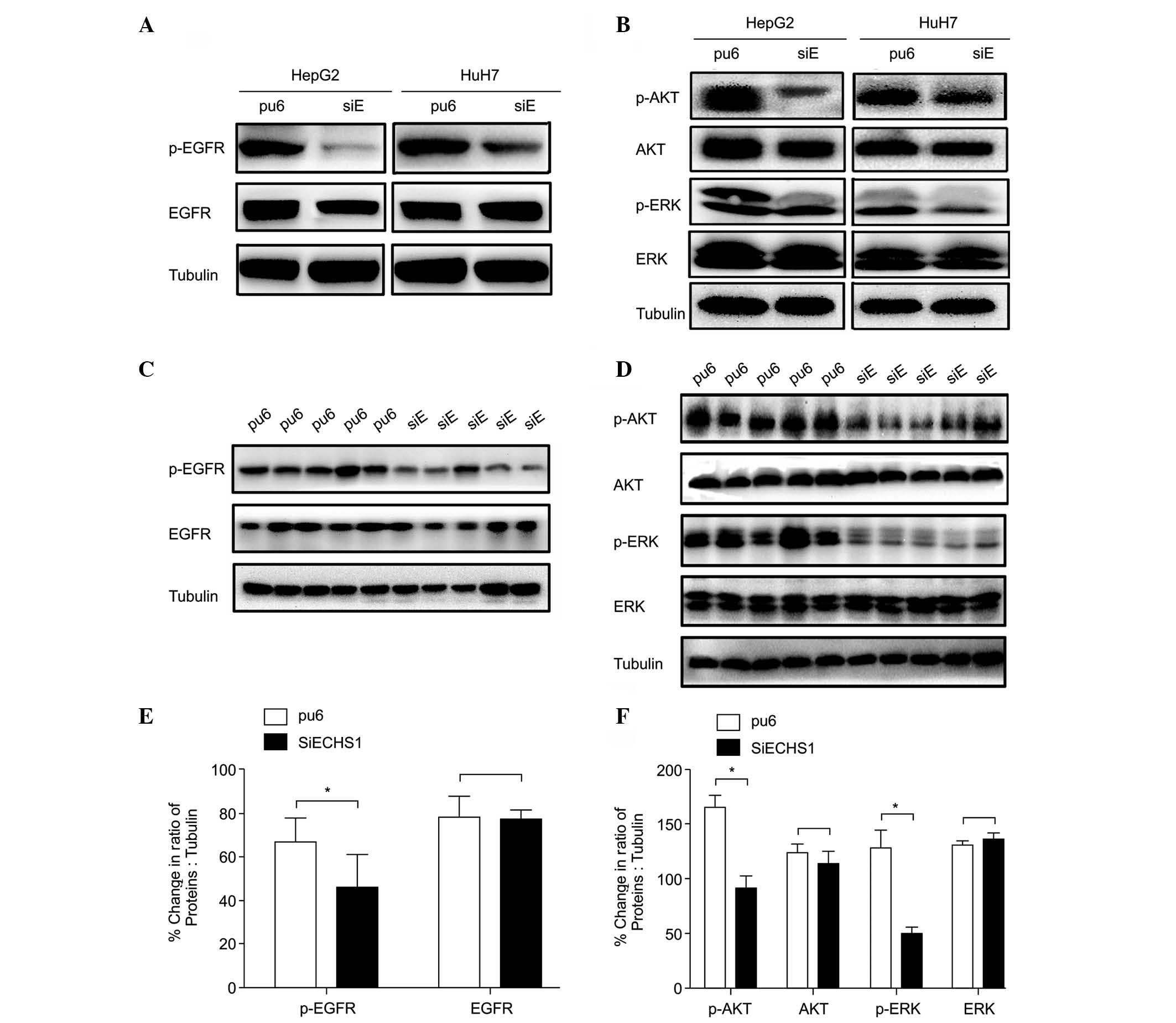 | Figure 3Silencing of ECHS1 downregulates
hepatocellular carcinoma cell proliferation by inhibiting EGFR
signaling in vitro and vivo. Western blot analysis of
EGFR, AKT, and ERK phosphorylation in (A and B) HepG2- and
Huh7-siECHS1 and control cells and (C and D) tumors derived from
ECHS1 knock-down cells and controls. Bands were quantified by
optical density scanning of (E) EGFR, (F) AKT and ERK in
vivo (as indicated in C and D). The percent changes in the
ratio of the indicated proteins to tubulin are presented as the
mean ± standard deviation; *P<0.05,
**P<0.01. ESCHS1, enoyl-coenzyme A hydratase short
chain 1; siRNA, short interfering RNA; pu6, control cells; siE,
ECHS1 siRNA-transfected cells; SiECHS1, ESCHS1 siRNA-transfected
cells; EGFR, epidermal growth factor receptor; ERK, extracellular
signal-regulated kinase; p-, phosphorylated. |
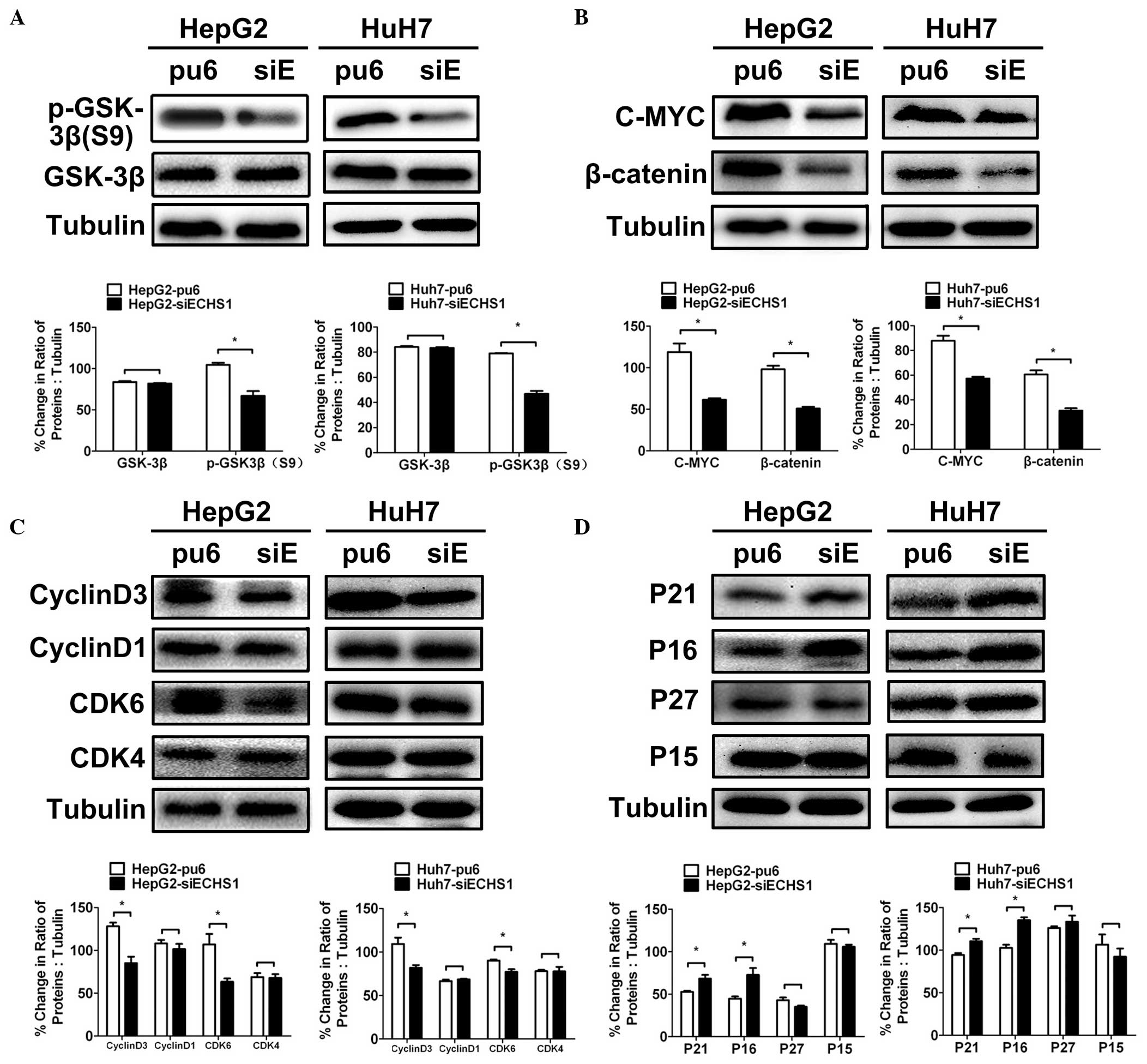 | Figure 4Silencing of ECHS1 alters the
expression of proteins involved in Wnt/β-catenin signaling and cell
cycle regulation. (A and B) Western blot analysis and
quantification of GSK-3β, p-GSK-3β(Ser9), β-catenin and C-MYC in
HepG2- and Huh7-siECHS1 and control cells. (C and D) Western blot
analysis and quantification of cell cycle-associated proteins in
HepG2-siECHS1, Huh7-siECHS1 and control cells. Data are presented
as the mean ± standard deviation (*P<0.05,
**P<0.01 vs. control). ESCHS1, enoyl-coenzyme A
hydratase short chain 1; siRNA, short interfering RNA; pu6, control
cells; siE, ECHS1 siRNA-transfected cells; SiECHS1, ESCHS1
siRNA-transfected cells; GSK-3β, glycogen synthase kinase 3β; CDK,
cyclin dependent kinase; p-, phosphorylated. |
ECHS1 modulates the expression of cell
cycle regulators
The expression levels of various cell cycle
regulators that are directly associated with cellular proliferation
were evaluated. The expression of cyclin D3 and CDK6 was reduced in
HepG2- and Huh7-siECHS1 cells (Fig.
4C), whereas expression of the CDK inhibitors p16 and p21 was
enhanced (Fig. 4D). Expression
levels of CDK4, cyclin D1, p15 and p27 were not significantly
altered in HepG2- and Huh7-siECHS1 cells compared with those of the
control cells (Fig. 4C and D). It
was therefore confirmed that ECHS1 knockdown in HCC cells
suppressed cyclin D3 and CDK6 expression and enhanced the
expression of p16 and p21.
Discussion
Late diagnosis and high recurrence are the leading
causes of poor survival amongst patients with HCC (24). Therefore, elucidation of the
molecular mechanisms that mediate HCC progression and the
identification of efficient therapeutic targets are urgently
required. A previous study by our group reported that ECHS1 binds
HBs to enhance HepG2 cell apoptosis. It was also demonstrated that
knockdown of endogenous ECHS1 attenuated the cell growth,
proliferation and migration of HepG2 cells. Furthermore, the
involvement of pro-apoptotic and pro-survival proteins and Akt
phosphorylation levels in HepG2 cells was defined, suggesting that
ECHS1 is a significant regulator of HCC progression (13). Zhu et al (25) confirmed that ECHS1 knockdown
inhibited HCC cell proliferation via suppression of AKT activity in
HepG2 cells. However, to the best of our knowledge, the upstream
and downstream regulators of this signaling cascade have not
previously been investigated, and the mechanisms underlying the
involvement of ECHS1 in HCC proliferation have remained
elusive.
In the present study, the high expression levels of
ECHS1 in human HepG2 and Huh-7 cells were verified. Subsequently,
the suppression of proliferation in HepG2 and Huh-7 cells by stable
ECHS1 knockdown was observed. Tumors derived from transplanted
HepG2-siECHS1 cells grew more slowly than tumors derived from
HepG2-pu6 cells. The downregulation of proliferation marker PCNA
(26) in ECHS1 knockdown cells was
also observed in vitro and in vivo. These results
suggested that ECHS1 mediates HCC proliferation; however,
elucidation of the mechanisms underlying this process require
further investigation.
The EGFR-induced mitogenic signaling pathway is
activated in various malignancies, including HCC (14–17).
EGFR phosphorylation leads to activation of the PI3K/Akt and
MEK/ERK signaling cascades, which are involved in cell
proliferation, survival, motility, differentiation and angiogenesis
(27–29). EGFR induces activation of the
Ras/Raf/MEK/ERK pathway via Grb2 or Shc adaptor proteins, and the
PI3K/AKT/mTOR pathway via recruitment of the p85 regulatory subunit
to the activated receptors (30).
In the present study, EGFR phosphor-ylation levels were reduced in
ECHS1 knock-down HepG2 and Huh7 cells; and the expression of
downstream effectors p-AKT and p-ERK were also reduced in
vitro and in xenograft tumors. p-ERK1/2 and p-Akt(ser473) are
highly expressed in HCC tissues, and activation of the ERK and AKT
pathways is indicative of poor prognosis in patients with HCC
(31). Certain studies have
focused on evaluating the synergy between the PI3K/Akt and MEK/ERK
pathways in HCC (31,32). It was therefore hypothesized that
ECHS1 may function via PI3K/Akt and MEK/ERK signaling to mediate
liver tumor development in an EGFR-dependent manner.
Akt-mediated phosphorylation of GSK-3β also
influences the Wnt/β-catenin pathway and the epithelial mesenchymal
transition (33). As expected,
serine-9 phosphorylation of GSK-3β was reduced in HepG2- and
Huh7-siECHS1 cells compared with that of the controls, which may
thereby increase the negative effect of GSK-3β on the Wnt/β-catenin
pathway, leading to downregulation of β-catenin. Upon activation of
the Wnt pathway, β-catenin forms a complex with B-cell lymphoma-9,
Pygo, plakoglobulin and T-cell factor/lymphoid enhancing factor,
which results in the transcription of critical genes including
cyclin D1, c-Myc, SALL4 and PPARδ (33). In the present study, β-catenin,
c-Myc and cyclin D3 expression were decreased in response to
p-GSK3β downregulation in ECHS1-knockdown HepG2 and Huh7 cells. It
was therefore hypothesized that ECHS1 may regulate the PI3K/Akt and
MEK/ERK pathways by targeting EGFR, leading to activation of the
downstream signaling cascade and promoting HCC progression.
However, the specific mechanisms underlying this function require
further investigation.
Previous studies have demonstrated that EGFR
regulates the cell cycle via the PI3K/Akt and MEK/ERK signaling
pathways (34,35). Akt regulates cell cycle progression
by inducing GSK-3β inhibition (36), cyclin D1 degradation and p21
upregulation (37). Cell cycle
regulators were therefore evaluated and it was revealed that
knockdown of ECHS1 suppressed cyclin D-CDK4/6 complex formation and
enhanced expression of the negative upstream regulators p16 and
p21.
In conclusion, ECHS1 promoted the development of
human HCC by regulating cell proliferation and cell cycle
progression. Consistent with a previous study by our group, which
indicated that ECHS1 may be involved in the progression of HCC
(13), the results of the present
study revealed a mechanistic link between ECHS1 and EGFR signal
activation through PI3K/Akt and c-Raf/MEK/ERK. Moreover, ECHS1
modulated the Wnt/β-catenin pathway through p-GSK-3β(ser9) and the
expression of cell cycle markers, including cyclin D1/3, CDK4/6,
p16 and p21. These results provide an insight into the critical
role of ECHS1 in the intricate EGFR signal transduction network,
which may result in the development of novel interventions using
EGFR-targeted therapies for the treatment of HCC. However, the
precise mechanism by which ECHS1 regulates EGFR requires further
evaluation.
Acknowledgments
The present study was supported by grants from the
National Key Basic Research Program of China (no. 2013CB933904),
National Key Sci-Tech Special Project of China (no.
2012ZX10002–011–005), Projects of Xiamen Science and Technology
Program (no. 3502Z20130030) and the National Natural Science
Foundation of China (no. 81171976).
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
ECHS1
|
enoyl-coenzyme A hydratase short chain
1
|
|
EGFR
|
epidermal growth factor receptor
|
|
CCK-8
|
cell counting kit-8
|
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsim NC, Frampton AE, Habib NA and Jiao
LR: Surgical treatment for liver cancer. World J Gastroenterol.
16:927–933. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guégan JP, Ezan F, Théret N, Langouët S
and Baffet G: MAPK signaling in cisplatin-induced death:
predominant role of ERK1 over ERK2 in human hepatocellular
carcinoma cells. Carcinogenesis. 34:38–47. 2013. View Article : Google Scholar
|
|
4
|
Tsao CM, Yan MD, Shih YL, et al: SOX1
functions as a tumor suppressor by antagonizing the WNT/β-catenin
signaling pathway in hepatocellular carcinoma. Hepatology.
56:2277–2287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Cai X, Lu W, Hu C, Xu X, Yu Q and
Cao P: Evodiamine inhibits STAT3 signaling by inducing phosphatase
shatterproof 1 in hepatocellular carcinoma cells. Cancer Lett.
328:243–251. 2013. View Article : Google Scholar
|
|
6
|
Yang P, Li QJ, Feng Y, et al:
TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment
promotes venous metastases of HBV-positive hepatocellular
carcinoma. Cancer Cell. 22:291–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Agnihotri G and Liu HW: Enoyl-CoA
hydratase reaction, mechanism, and inhibition. Bioorg Med Chem.
11:9–20. 2003. View Article : Google Scholar
|
|
8
|
Lin JF, Xu J, Tian HY, et al:
Identification of candidate prostate cancer biomarkers in prostate
needle biopsy specimens using proteomic analysis. Int J Cancer.
121:2596–2605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Komori T, Takemasa I, Higuchi H, et al:
Identification of differentially expressed genes involved in
colorectal carcinogenesis using a cDNA microarray. J Exp Clin
Cancer Res. 23:521–527. 2004.PubMed/NCBI
|
|
10
|
Yeh CS, Wang JY, Cheng TL, Juan CH, Wu CH
and Lin SR: Fatty acid metabolism pathway play an important role in
carcinogenesis of human colorectal cancers by
Microarray-Bioinformatics analysis. Cancer Lett. 233:297–308. 2006.
View Article : Google Scholar
|
|
11
|
Liu X, Feng R and Du L: The role of
enoyl-CoA hydratase short chain 1 and peroxiredoxin 3 in
PP2-induced apoptosis in human breast cancer MCF-7 cells. FEBS
Lett. 584:3185–3192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang Y, Wang SX, Wang YB, et al: ECHS1
interacts with STAT3 and negatively regulates STAT3 signaling. FEBS
Lett. 587:607–613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiao CX, Yang XN, Huang QW, et al: ECHS1
acts as a novel HBsAg-binding protein enhancing apoptosis through
the mitochondrial pathway in HepG2 cells. Cancer Lett. 330:67–73.
2013. View Article : Google Scholar
|
|
14
|
Kira S, Nakanishi T, Suemori S, Kitamoto
M, Watanabe Y and Kajiyama G: Expression of transforming growth
factor alpha and epidermal growth factor receptor in human
hepatocellular carcinoma. Liver. 17:177–182. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ito Y, Takeda T, Sakon M, et al:
Expression and clinical significance of erb-B receptor family in
hepatocellular carcinoma. Br J Cancer. 84:1377–1383. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Prigent SA and Lemoine NR: The type 1
(EGFR-related) family of growth factor receptors and their ligands.
Prog Growth Factor Res. 4:1–24. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barnard JA, Beauchamp RD, Russell WE,
Dubois RN and Coffey RJ: Epidermal growth factor-related peptides
and their relevance to gastrointestinal pathophysiology.
Gastroenterology. 108:564–580. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
DeCicco LA, Kong J and Ringer DP:
Carcinogen-induced alteration in liver epidermal growth factor
receptor distribution during the promotion stage of
hepatocarcinogenesis in rat. Cancer Lett. 111:149–156. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jazag A, Ijichi H, Kanai F, et al: Smad4
silencing in pancreatic cancer cell lines using stable RNA
interference and gene expression profiles induced by transforming
growth factor-beta. Oncogene. 24:662–671. 2005. View Article : Google Scholar
|
|
22
|
Guleng B, Tateishi K, Ohta M, et al:
Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates
in vivo tumor growth by inhibiting angiogenesis in a vascular
endothelial growth factor-independent manner. Cancer Res.
65:5864–5871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu XS, Dai YC, Chen ZX, Xie JP, Zeng W,
Lin YY and Tan QH: Knockdown of ECHS1 protein expression inhibits
hepatocellular carcinoma cell proliferation via suppression of Akt
activity. Crit Rev Eukaryot Gene Expr. 23:275–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kelman Z and O’Donnell M: Structural and
functional similarities of prokaryotic and eukaryotic DNA
polymerase sliding clamps. Nucleic Acids Res. 23:3613–3620. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carpenter G: Receptors for epidermal
growth factor and other polypeptide mitogens. Annu Rev Biochem.
56:881–914. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soltoff SP, Carraway KR III, Prigent SA,
Gullick WG and Cantley LC: ErbB3 is involved in activation of
phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell
Biol. 14:3550–3558. 1994.PubMed/NCBI
|
|
31
|
Schmitz KJ, Wohlschlaeger J, Lang H, et
al: Activation of the ERK and AKT signalling pathway predicts poor
prognosis in hepatocellular carcinoma and ERK activation in cancer
tissue is associated with hepatitis C virus infection. J Hepatol.
48:83–90. 2008. View Article : Google Scholar
|
|
32
|
Saxena NK, Sharma D, Ding X, Lin S, Marra
F, Merlin D and Anania FA: Concomitant activation of the JAK/STAT,
PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma
cells. Cancer Res. 67:2497–2507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Steelman LS, Chappell WH, Abrams SL, et
al: Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in
controlling growth and sensitivity to therapy-implications for
cancer and aging. Aging (Albany NY). 3:192–222. 2011.
|
|
34
|
Bill HM, Knudsen B, Moores SL, Muthuswamy
SK, Rao VR, Brugge JS and Miranti CK: Epidermal growth factor
receptor-dependent regulation of integrin-mediated signaling and
cell cycle entry in epithelial cells. Mol Cell Biol. 24:8586–8599.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lui VW and Grandis JR: EGFR-mediated cell
cycle regulation. Anticancer Res. 22:1–11. 2002.PubMed/NCBI
|
|
36
|
Horn S, Endl E, Fehse B, Weck MM, Mayr GW
and Jücker M: Restoration of SHIP activity in a human leukemia cell
line downregulates constitutively activated phosphatidylinositol
3-kinase/Akt/GSK-3beta signaling and leads to an increased transit
time through the G1 phase of the cell cycle. Leukemia.
18:1839–1849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|