Introduction
Partial trisomy 9 is the fourth most common
autosomal trisomy after trisomies 21, 18 and 13. Since Rethoré
et al (1) reported the
first identified case of partial trisomy 9 as a chromosomal
anomaly, >150 cases have been described. In addition to
non-specific psychomotor delay and mental retardation, common
clinical features, including moderately abnormal characteristic
facial features, clinodactyly of the 5th fingers, shortened digits,
hypoplastic nails, abnormal dermatoglyphics and hypoplastic brain
association with Dandy-Walker malformation are observed (2). Trisomy for 9pter-p21 is hypothesized
to be responsible for the majority of these features (3). Intrauterine growth retardation, cleft
lip/palate, skeletal anomalies and heart defects are more common
with trisomic segments extending through 9q22-9q32 (4–7). In
general, partial trisomy 9 leads to variable phenotypes dependent
upon the size and position of the duplicated region (8). However, a precise genotype/phenotype
map has not yet been proposed. The present study describes the case
of a 3-year-old female with a number of the typical features of
trisomy 9p syndrome, as well as distinctive features that include
sensorineural hearing loss and mild body asymmetry. Cytogenetic
results showed the presence of a de novo extra der (9) with 69.5 Mb duplication.
Case report
Case presentation and analysis
A 3-year-old Chinese female was referred to us for
further investigation for mental retardation and hearing loss. The
girl was born full-term with uneventful gestation by elective
cesarean as the first child of nonconsanguineous parents. The
mother and father were 28 and 27 years old, respectively, at her
birth. Family history was negative, meaning the other families in
this pedigree exhibited no similar ilness. The girl had a birth
weight of 3,900 g (95th centile), length of 50 cm (50th centile)
and head circumference was 35 cm (50th centile), as well as 1 min
and 5 min Apgar scores were of 10, respectively. Her psychomotor
development was substantially delayed with severe speech
retardation. The patient spoke at the age of 3-years and walked
without assistance at the age of 2-years. On examination, the girl
had a height of 98.5 cm (77th centile) and weight of 16.5 kg (80th
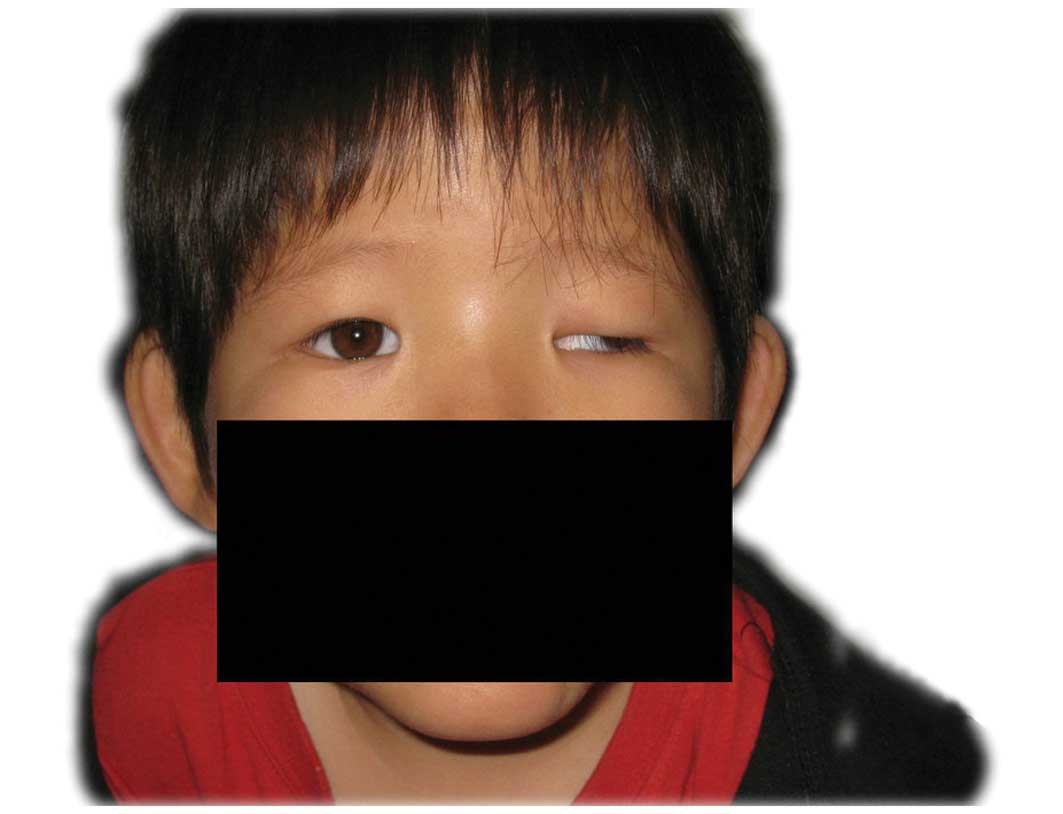
centile). The patient presented with a characteristic face with an
antimongoloid slant of palpebral fissures, a broad and prominent
nasal bridge, low-set and forwardly-rotated auricles, large poorly
lobulated ears and downturned corners of the mouth (Fig. 1). A short neck, clinodactyly of
both of the 5th fingers, a bilateral simian crease, joint
hyperlaxity and hypoplasia of the toenails were also observed. In
addition to the phenotypes of typical trisomy 9p, the patient
presented with distinctive features, including the left side of the
body slightly smaller than the right with ptosis and strabismus of
left eye and sensorineural hearing loss (left ear at 100 decibels,
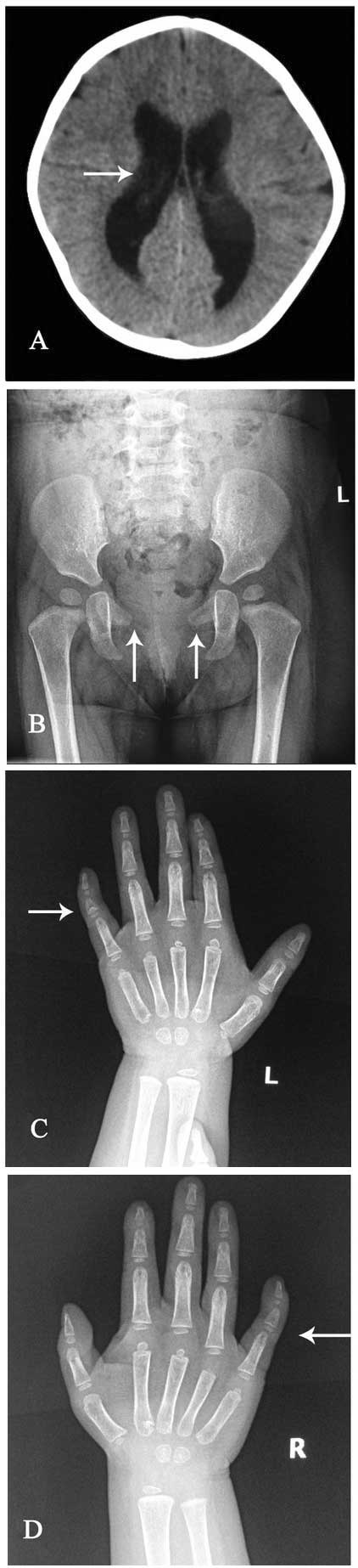
right at 40 decibels). Cerebral computerized tomography showed
enlargement of the lateral ventricles (Fig. 2A), 3rd, 4th ventricles and basal
cistern, with a mild agenesis of the cerebellar tonsil.
Roentgenograms of the skeleton demonstrated hypoplastic pubic bones
(Fig. 2B), and bilateral
hypoplastic distal phalanges of the feet, pes valgus and bilateral
clinodactyly of both 5th fingers (Fig.
2C and D). Cardiac and renal ultrasound findings were normal.
This study was approved by the ethics committee of Jinling
Hospital, Nanjing University School of Medicine (Nanjing, China),
and written informed consent was obtained from the parents.
Chromosome analysis
Karyotype analysis
Karyotyping was performed on peripheral blood
lymphocytes from the patient and her parents. Peripheral blood
lymphocyte cultures were cultivated using RPMI media supplemented
with 10% fetal calf serum (Lai Fu institute of biotechnology, Qing
Dao, China). Metaphase chromosomes were GTG-banded using standard
procedures.
Multiplex fluorescence in situ
hybridization (M-FISH) analysis
M-FISH was performed on the metaphase spreads using
Spectra Vysion WCP probe (Vysis, Inc., Downers Grove, IL, USA)
according to manufacturer’s procedures. Images were captured with
Olympus BX51 microscope (Olympus, Tokyo, Japan) and analyzed with
the Cytovision 3.0 (Applied Imaging, Sunderland, UK) image analyses
software.
OaCGH analysis
In order to investigate the extent of duplication on
molecular level, analysis of using a genomic-wide high density
oligo array (OaCGH244 K) was conducted according to Agilent
manufacturer’s procedures and statistical algorithms (www.agilent.com.chem/gocgh) (9).
Chromosomal analysis. showed a female non-mosaic
karyotype with an extra chromosome in all metaphases analyzed
(Fig. 3A). M-FISH analysis using
the Spectra Vysion WCP Probe (Vysis, Downers Grove, IL, USA)
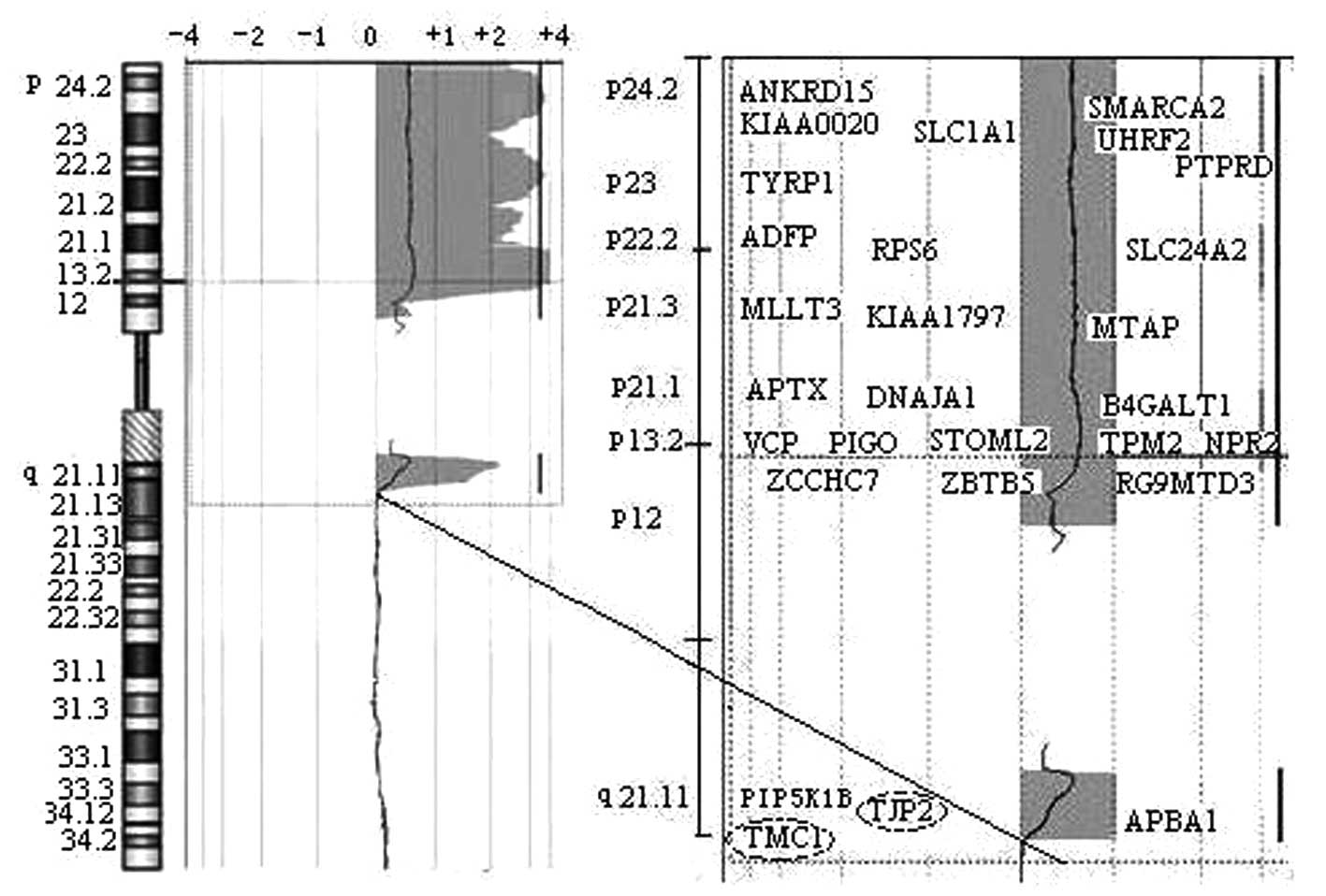
confirmed the extra chromosome from chromosome 9 (Fig. 3B). A 69.5 Mb duplication segment at
genomic position 273,048 bp →72,521,148 bp in the 9pter→q21.12
region was confirmed (Fig. 4). The
final karyotype was interpreted to be 47, XX, +mar.ish der
(9) (wcp9+). arr cgh 9pterq21.12
(DOCK8→LOC138225)×3. The duplicate region spanned 148 annotated
genes in which 28 genes are expressed in the cochlea (Fig. 4). Chromosome analysis of the
parents showed normal karyotype, indicating a de novo extra
chromosome.
Discussion
To date, 65 genes for non-syndromic hearing loss
have been identified (http://hereditaryhearingloss.org/) (10). However, to the best of our
knowledge, hearing loss with isolated partial trisomy 9
(9pter→q21.12) has not been previously reported. The functions of
the 28 genes identified in the chromosomal analysis, which are
expressed in the cochlea, are mostly unknown. Reviewing the
literature, cases of two males with partial trisomy 9, including
duplication of 9per→q21 was reported by Morrissette et al
(11) and Centerwall et al
(12), respectively; however, the
patients succumbed to the disease at four weeks following birth and
thus it was uncertain whether or not hearing loss occurred.
Comparing our case with other cases in the literature (2–8,13–18)
it was found that the patients without hearing loss have
overlapping regions of 9pter→9q13 or 9q22-9q32. On the basis of
these data, it was hypothesized that 9q13-q21 may be a critical
region for hearing. Recently, mutations of two genes in the region
of 9q13-9q21.1 were confirmed to be responsible for deafness. For
example, transmembrane channel-like gene 1 (TMC1, MIM 606706,
GenBank ID NT_023935 position 4301249-4615799), mutations are
identified by Kurima et al (19) as a cause of autosomal dominant
(#MIM 606705) and autosomal recessive non-syndromic hearing loss
(#MIM 600974). The association between mutations in the gene with
hearing loss were further confirmed in other studies (20–23).
Between 2002 and 2008, a total of 2 dominant and 18 recessive TMC1
mutations were reported as the cause of hearing loss in 34 families
(24). Additionally, Hilgert et
al (24) found the other six
families with non-syndromic hearing loss were associated with
mutations in DFNA36 and DFNB7/11, rather than mutations in TMC1,
which implied at least one additional deafness-causing gene at loci
DFNA36 and DFNB7/11. Another candidate gene, tight junction protein
2 (TJP2, MIM 607709), was considered a good candidate due to its
function as a tight junction protein and its expression in the
cochlea. Hilgert et al (24) reported a Guatemalan family with
autosomal dominant nonsyndromic hearing loss. In exon 19 of the
gene, a novel sequence variant, The mutation, c.2971A>T, was
identified in the girl with the hearing loss phenotype, and this
lead to an amino acid change from proline to valine at codon 924
(P924V). This aspartic acid residue is a member of a conserved
acidic domain of the protein. The mutation was predicted to cause
decreased stability by bioinformatic analysis. However, our
hypothesis remains to be proven.
In addition to the typical clinical features of
partial trisomy 9, the present case presented a group of
distinctive phenotypes: The left side of the body was slightly
smaller than the right one; left hearing loss was more severe than
right; ptosis and strabismus of the left eye, all of which were not
previously associated with partial trisomy 9. Body asymmetry is a
complex developmental malformation and has already been described
in syndromes, such as Beckwith-Wiedemann Syndrome (MIM 147470),
Silver-Russell Syndrome (MIM 180860), Proteus syndrome (MIM 176920)
and Klippel-Trenaunay-Weber syndrome (MIM 149000). Reviewing the
literature, only one case of mosaic tetrasomy 9p with this anomaly
was found (25). Considering the
malformations are rare, it is uncertain whether the distinctive
features were associated with partial trisomy 9 or not. However,
the unusual clinical features with a detailed molecular karyotyping
may provide information on this phenotype and expand existing
knowledge.
In conclusion, the patient carrying a segmental
duplication of 9pter-q21.12 exhibits distinctive phenotypes, such
as sensorineural hearing loss. Although the molecular mechanism
underlying the hearing loss is not clear, it was proposed that the
region of 9q13→9q21 may be critical for hearing.
Acknowledgments
This study was supported by the Key foundation of
Jiangsu Science and Technology Bureau (grant no. BM2013058), the
National Natural Science Foundation of China (grant no. 30901652)
and the Foundation of Jiangsu province (grant no. BK2011660). The
authors would like to thank all members of the family for their
cooperation in the study.
References
|
1
|
Rethoré MO, Larget-Piet L, Abonyi D, et
al: 4 cases of trisomy for the short arm of chromosome 9.
Individualization of a new morbid entity. Ann Genet. 13:217–232.
1970.
|
|
2
|
Tonni G, Lituania M, Chitayat D, Bonasoni
MP, Keating S, Thompson M and Shannon P: Complete trisomy 9 with
unusual phenotypic associations:Dandy-Walker malformation, cleft
lip and cleft palate, cardiovascular abnormalities. Taiwan J Obstet
Gynecol. 53:592–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lewandowski RC Jr, Yunis JJ, Lehrke R,
O’Leary J, Swaiman KF and Sanchez O: Trisomy for the distal half of
the short arm of chromosome 9. A variant of the trisomy 9p
syndrome. Am J Dis Child. 130:663–667. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hou JW and Wang TR: Molecular c1993netic
studies of duplication 9q32->q34.3 inserted into 9q13. Clin
Genet. 48:148–150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Naritomi K, Izumikawa Y, Goya Y, Gushiken
M, Shiroma N and Hirayama K: Trisomy 9q3 syndrome: a case report
and review of the literature. Clin Genet. 35:293–298. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kleczkowska A, Fryns JP, Lemay P and Van
den Berghe H: The characteristic phenotype of distal 9q3 trisomy is
due to duplication of band 9q32. Genet Couns. 4:217–221.
1993.PubMed/NCBI
|
|
7
|
Temtamy SA, Kamel AK, Ismail S, et al:
Phenotypic and cytogenetic spectrum of 9p trisomy. Genet Couns.
18:29–48. 2007.PubMed/NCBI
|
|
8
|
Wilson GN, Raj A and Baker D: The
phenotypic and cytogenetic spectrum of partial trisomy 9. Am J Med
Genet. 20:277–282. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fan YS, Jayakar P, Zhu H, et al: Detection
of pathogenic gene copy number variations in patients with mental
retardation by genomewide oligonucleotide array comparative genomic
hybridization. Hum Mutat. 28:1124–1132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ganapathy A, Pandey N, Srisailapathy CR,
et al: Non-syndromic hearing impairment in India: High allelic
heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23 and
TMIE. PLoS One. 9:e847732014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morrissette JJ, Laufer-Cahana A, Medne L,
et al: Patient with trisomy 9p and a hypoplastic left heart with a
tricentric chromosome 9. Am J Med Genet A. 123A:279–284. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Centerwall WR, Mayeski CA and Cha CC:
Trisomy 9q-. a variant of the 9p trisomy syndrome. Humangenetik.
29:91–98. 1975.PubMed/NCBI
|
|
13
|
Haddad BR, Lin AE, Wyandt H and Milunsky
A: Molecular cytogenetic characterisation of the first familial
case of partial 9p duplication (p22p24). J Med Genet. 33:1045–1047.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanlaville D, Baumann C, Lapierre JM, et
al: De novo inverted duplication 9p21pter involving telomeric
repeated sequences. Am J Med Genet. 83:125–131. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Centerwall WR, Miller KS and Reeves LM:
Familial ‘partial 9p’ trisomy: six cases and four carriers in three
generations. J Med Genet. 13:57–61. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sutherland GR, Carter RF and Morris LL:
Partial and complete trisomy 9: delineation of a trisomy 9
syndrome. Hum Genet. 32:133–140. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smart RD, Viljoen DL and Fraser B: Partial
trisomy 9-further delineation of the phenotype. Am J Med Genet.
31:947–951. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teraoka M, Narahara K, Yokoyama Y,
Ninomiya S, Mizuta S, Une T and Seino Y: Maternal origin of a
unique extra chromosome, der (9)(pter->q13::q13->q12:) in a
girl with typical trisomy 9p syndrome. Am J Med Genet. 102:25–28.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurima K, Peters LM, Yang Y, et al:
Dominant and recessive deafness caused by mutations of a novel
gene, TMC1, required for cochlear hair-cell function. Nat Genet.
30:277–284. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meyer CG, Gasmelseed NM, Mergani A, et al:
Novel TMC1 structural and splice variants associated with
congenital nonsyndromic deafness in a Sudanese pedigree. Hum Mutat.
25:1002005. View Article : Google Scholar
|
|
21
|
Santos RL, Wajid M, Khan MN, et al: Novel
sequence variants in the TMC1 gene in Pakistani families with
autosomal recessive hearing impairment. Hum Mutat. 26:3962005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kitajiri SI, McNamara R, Makishima T, et
al: Identities, frequencies and origins of TMC1 mutations causing
DFNB7/B11 deafness in Pakistan. Clin Genet. 72:546–550. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tlili A, Rebeh IB, Aifa-Hmani M, et al:
TMC1 but not TMC2 is responsible for autosomal recessive
nonsyndromic hearing impairment in Tunisian families. Audiol
Neurootol. 13:213–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hilgert N, Alasti F, Dieltjens N, et al:
Mutation analysis of TMC1 identifies four new mutations and
suggests an additional deafness gene at loci DFNA36 and DFNB7/11.
Clin Genet. 74:223–232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cazorla Calleja MR, Verdú A and Félix V:
Dandy-Walker malformation in an infant with tetrasomy 9p. Brain
Dev. 25:220–223. 2003. View Article : Google Scholar : PubMed/NCBI
|