Introduction
Colon cancer is the third most common type of cancer
worldwide and its incidence is increasing in East Asia and Western
countries (1,2). Systematic chemotherapy is an
important component of colon cancer treatment, particularly for
patients with advanced disease (3). However, advances in chemotherapy for
colon cancer have been limited as the underlying mechanisms causing
chemoresistance remain to be elucidated. Doxorubicin (Dox) is
extensively used as a chemotherapeutic agent in the treatment of
colon cancer (4). Dox exerts its
effect through interference with nucleoside metabolism, and
nucleosides can be incorporated into RNA and DNA, leading to
cytotoxicity and cell death (5).
However, a significant difficulty associated with Dox treatment is
the development of Dox-induced chemoresistance. An improved
understanding of the ways in which resistance arises, and the
molecular alterations that are associated with these processes, may
result in the development of novel therapeutic strategies for the
successful treatment of patients diagnosed with colon cancer.
Recent studies have suggested that
epithelial-mesenchymal transition (EMT), metastasis and
chemoresistance are closely associated with tumor progression
(6,7). During the development of drug
resistance, reversible epigenetic changes indicate alterations in
the differentiation state of the tumor, which are likely to reflect
EMT (8). The transforming growth
factor β (TGFβ) family of cytokines are mediators of embryonic
development and regulators of multiple types of physiological and
pathophysiological EMT (9). RNA
interference (RNAi) allows sequence-specific gene silencing. It
involves small non-coding RNAs, which are associated with
nuclease-containing regulatory complexes that then bind to
complementary messenger RNA targets, thereby preventing the
expression of these mRNAs (10).
The present study assessed whether the inhibition of
TGFβ signaling represents a novel pathway regulating
chemoresistance and the mechanisms underlying this effect.
Materials and methods
Cell culture
The HCT116 human colon cancer cell line was obtained
from the Cell Bank of the Shanghai Institute of Biochemistry and
Cell Biology, Chinese Academy of Sciences (Shanghai, China) and
cultured in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD, USA)
supplemented with 10% heat-inactivated fetal bovine serum, 10 U/ml
penicillin, and 10 μg/ml streptomycin in a humidified
atmosphere containing 5% CO2 at 37°C.
Reagents and antibodies
Dox was purchased from Aladdin Reagent Co., Ltd.
(Shanghai, China). The primary polyclonal rabbit antibodies against
human Smad4, Smad2, Smad3, phospho-Smad2 Ser465 (p-Smad2),
phospho-Smad3 Ser425 (p-Smad3) and MDR p-gp (p-gp) were purchased
from Cell Signaling Technology, Inc. (Beverly, MA, USA) and primary
polyclonal rabbit antibodies against human E-cadherin (sc-7870),
Vimentin (sc-5565), N-cadherin (sc-7939), Snai1 (sc-28199) and Slug
(sc-15391) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). These antibodies were used in western blot
analysis and immunofluorescence staining at a dilution of 1:1,000.
The TGFβ1 enzyme-linked immunosorbent assay (ELISA) kit was
obtained from Invitrogen Life Technologies (Carlsbad, CA, USA).
3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and other materials, including secondary antibodies were purchased
from Beyotime Institute of Biotechnology (Shanghai, China).
Lentiviral vector construction and
transfection
The short hairpin RNA (shRNA)-encoding complementary
single stranded oligonucleotides corresponding to Smad4 were
designed as previously described (11). The RNAi sequence
(5′-GTACTTCATACCATGCCGA-3′) was complementary to the human Smad4
cDNA sequence and was annealed and cloned into the pGCSIL-green
fluorescent protein vector (GeneChem, Shanghai, China). The control
sequence of RNAi (5′-TTCTCCGAACGTGTCACGT-3′) was used as a negative
control. The recombinant virus was packaged using the Lentivector
expression system (GeneChem). HCT116 cells were transfected with
the recombinant lentivirus using Lipofectamine 2000 (Invitrogen
Life Technologies) and transfected cells were screened under 800
μg/ml G418 (Calbiochem, Darmstadt, Germany) for 4 weeks to
generate stable monoclonal cell lines (Smad4 stable downregulation
cell lines, RNAi-Smad4 and control stable cell lines, RNAi-NC). The
expression of Smad4 was confirmed using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis.
Drug treatment
Cells were divided into four groups, termed HCT116
cells, RNAi-NC cells, Smad4-RNAi cells and HCT116 cells untreated
by Dox as a control (the control group). Cells were treated with 50
nmol/l Dox, or an equal volume of phosphate-buffered saline (PBS)
for the controls, for 7 days (the medium was changed every
alternate day, with the addition of the same dosage of Dox on
occasion) for RT-qPCR, western blot analysis, immunofluorescence
staining and ELISA.
Western blotting
Following drug treatment, cells were washed twice
with PBS and lysed in ice-cold radioimmunoprecipitation assay
buffer (20 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.5% Nonidet P-40; 1
mM EDTA; 50 μg/ml leupeptin; 30 μg/ml aprotinin; 1 mM
Na3VO4; and 1 mM phenylmeth-ylsulfonyl
fluoride). The cell lysate (20 μg) was separated on a sodium
dodecyl sulfate polyacrylamide gel (SDS-PAGE) and then transferred
onto a nitrocellulose membrane (Pall Corporation, Port Washington,
NY, USA) by use of the wet transfer system (Bio-Rad, Hercules, CA,
USA). Nonspecific binding sites were then blocked for 2 h at room
temperature in Tris-buffered saline (TBS, pH 7.4) containing 0.1%
Tween-20 and 10% bovine serum albumin (BSA). The primary antibodies
were diluted (1:1,000) in TBS containing 0.1% Tween-20 and 5% BSA
and incubated overnight at 4°C. The appropriate
peroxidase-conjugated secondary antibodies (immunopure rabbit
anti-goat IgG; goat anti-mouse IgG; and goat anti-rabbit IgG;
Pierce Biotechnology, Inc., Rockford, IL, USA) were used at a
dilution of 1:1,500. Positive antibody reactions were detected
using SuperSignal West Pico Chemiluminescent Substrate (Pierce
Biotechnology, Inc.).
RT-qPCR
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies) according to the manufacturer’s
instructions and cDNA was synthesized from the mRNA obtained by
RT-PCR using the SuperScript first-strand synthesis system
(Fermentas, Vilnius, Lithuania). PCR was performed according to the
standard protocol using a Roche LightCycler (Roche Diagnostics
GmbH, Mannheim, Germany) with SYBR Green supermix (Bio-Rad). The
PCR conditions were as follows: 31 Cycles of denaturation for 5 sec
at 95°C, annealing for 30 sec at 60°C and primer extension for 60
sec at 72°C. cDNA was used as the template for the qPCR reaction
with the following specific primers: Smad4, F
5′-TGTGACAGTGTCTGTGTGA-3′ and R 5′-CCTACCTGAACGTCCATTTC-3′; actin,
F 5′-GTCCACCGCAAATGCTTCTA-3′ and R 5′-TGCTGTCACCTTCACCGTTC-3′. The
human actin gene was amplified as an endogenous control. The gene
expression of mRNA from each sample was calculated by normalizing
against that of the reference gene, β-actin. Primer sequences are
available upon request.
MTT assay
The cell lines (HCT116, RNAi-NC and RNAi-Smad4) were
seeded onto 96-well plates (6.0×103 cells/well) and
allowed to attach overnight. Following cellular adhesion, freshly
prepared Dox at the appropriate concentration (10, 30, 50, 80 or
100 nmol/l), was added for the 24 h treatment, and 50 μmol/l
Dox was added for the 7 day treatment, respectively. The viability
of the cells was evaluated using an MTT assay, according to the
manufacturer’s instructions (Roche Applied Science, Indianapolis,
IN, USA). Briefly, MTT was added at a concentration of 500 mg/l and
the cells were incubated for 4 h at 37°C. The absorbance reading of
each well was determined using a computer-controlled microtiter
plate reader (iMARK; Bio-Rad) at a wavelength of 570 nm. The cell
viability rates were defined as the relative absorbance of treated
vs. untreated cells.
Immunofluorescent staining
Cells were plated on 20 mm circular microscope
coverslips. According to the manufacturer’s (Beyotime Institute of
Biotechnology) instructions, cells were fixed in 4%
paraformaldehyde for 15 min and permeabilized with 0.1% Triton
X-100 for 15 min. Non-specific binding sites were then blocked for
1 h at room temperature in PBS (pH 7.4) containing 0.1% Tween-20
and 5% BSA. Primary antibodies (Snail and Slug, 1:200) in PBS
containing 0.1% Tween-20 and 1% BSA were added overnight at 4°C in
a humidified chamber. Cells were then incubated with the
appropriate fluorescein isothiocyanate-conjugated secondary
antibodies (1:200) for 1 h at room temperature in a humidified
chamber in darkness. The samples were subsequently treated with
4′,6-diamidino-2-phenylindole (10 μg/ml) for 30 sec to
detect the cell nuclei. Images were obtained using fluorescence
microscopy (BX53; Olympus, Tokyo, Japan) at magnification,
×400.
ELISA
HCT116 cells, RNAi-NC cells and Smad4-RNAi cells
were treated with 50 nmol/l Dox for 7 days and HCT116 cells, which
were not subject to treatment with Dox were used as a control
group. The concentration of TGFβ1 in the supernatant of the cells
was measured using ELISA kits (Anogen-Yes Biotech Laboratories
Ltd., Mississauga, ON, Canada) according to the manufacturer’s
instructions.
Statistical analysis
Statistical comparisons between the two groups were
performed using an unpaired t-test. All groups were compared using
a one-way analysis of variance, followed by Tukey’s post hoc test
where appropriate. SPSS software, version 17.0 (SPSS, Inc.,
Chicago, IL, USA) was used for all analyses. P<0.05 was
considered to indicate a statistically significant difference. All
data are expressed as the mean ± standard deviation from at least
three independent experiments.
Results
Dox treatment results in upregulation of
TGFβ1 and phosphorylation of Smad2/3
Dox has been observed to induce expression of
circulating TGFβ in xenograft and transgenic animal models
(12,13). In order to identify whether and how
the TGFβ/Smad4 signaling pathway was affected by Dox
administration, the effects of Dox treatment on TGFβ1 concentration
and p-Smad2/Smad3 expression level in HCT116 cells were examined
using ELISA and western blotting. Following treatment with 50
nmol/l Dox for 7 days, it was shown that the expression of TGFβ1 in
HCT116 cells was significantly higher (P<0.05) than that in the
control (HCT116 cells treated with PBS; Fig. 1A). Similarly, the levels of p-Smad2
and Smad3 in the HCT116 cells increased in comparison with the
control group (Fig. 1B). Thus, the
present results indicated that long-term administration of Dox may
activate the TGFβ/Smad4 pathway in HCT116 cells.
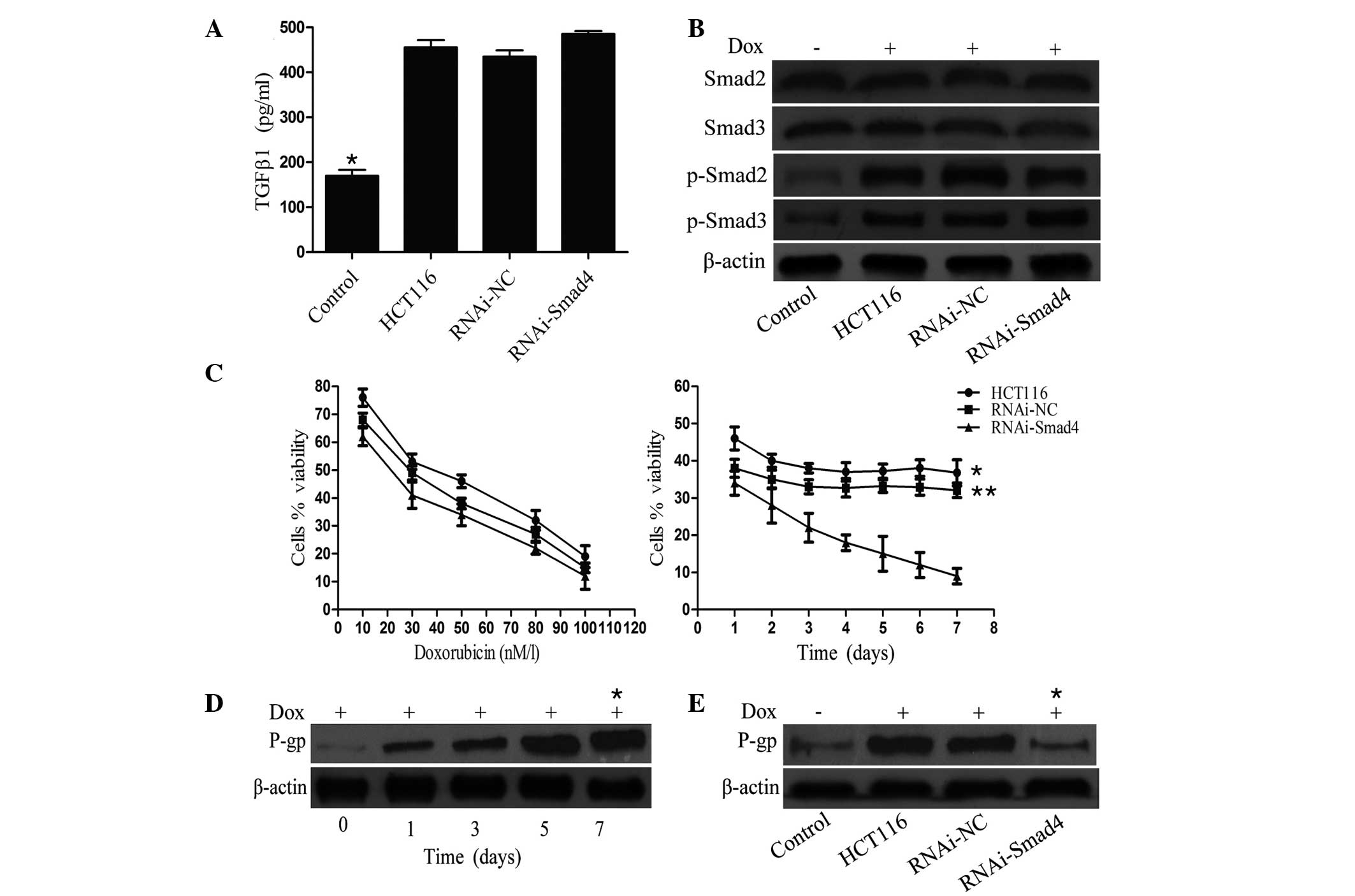 | Figure 1Changes in expression of molecules
involved in the TGFβ/Smad4 signaling pathway, the cell viability
ratio and p-gp expression, following Dox or Smad4 RNAi treatment.
Expression of (A) TGFβ1, (B) Smad2/3 and p-Smad2/3 were examined
using ELISA and western blotting. *P<0.05 vs. HCT116
cells (P=0.022), RNAi-NC cells (P=0.018) and Smad4-RNAi cells
(P=0.013). (C) Cell viability ratio was assessed using an MTT
assay. Data are presented as a percentage of the control cell
viability. (D) and (E) p-gp level was investigated using a western
blot assay, with significant differences observed between p-gp
expression levels at 0 and 7 days (*P=0.029), and
between RNAi-Smad4 vs. HCT116 cells (*P=0.004) and
RNAi-NC cells (*P=0.007). β-actin expression was used as
a reference gene. TGF, transforming growth factor; Dox,
doxorubicin; p-gp, multi-drug resistant plasma membrane
glycoprotein; RNAi, RNA interference; NC, normal control. |
HCT116 cells exhibit resistance following
long-term administration of Dox
To further examine the effects of Dox on HCT116
cells, the cell viability and p-gp levels were assessed using an
MTT assay and western blotting. HCT116 cells were treated with 10,
30, 50, 80 and 100 nmol/l Dox for 24 h, and with 50 nmol/l Dox for
1, 2, 3, 4, 5, 6 and 7 days, respectively. Cells treated with PBS
alone were used as a control. The results from the MTT assay
indicated that the cell viability of HCT116 cells decreased in a
dose-dependent manner (Fig. 1C). A
concentration of 50 nmol/l was determined to be appropriate for
long-term Dox administration for the MTT assay. It was observed
that the viability of HCT116 cells decreased between 1 and 3 days
in a time-dependent manner. However, it did not change markedly
between 3 and 7 days, following treatment (Fig. 1C). Following administration of 50
nmol/l Dox, the p-gp expression of HCT116 cells increased in a
time-dependent manner (Fig. 1D).
These data demonstrated that HCT116 cells acquired resistance to
Dox following long-term treatment at a low concentration.
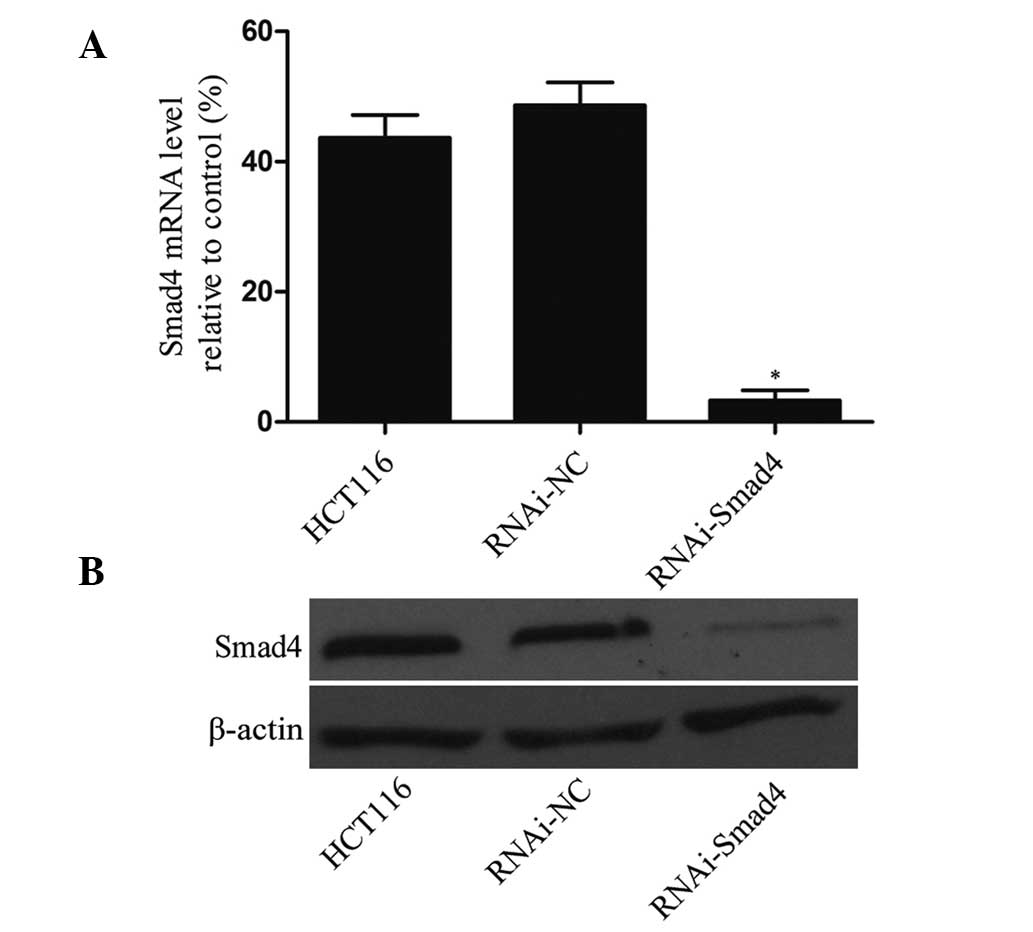
Expression of Smad4 is significantly
downregulated using a lentivirus vector
In order to assess the knockdown efficiency of the
lentiviral vector, the mRNA and protein expression of Smad4 in
HCT116 cells, RNAi-NC cells and Smad4-RNAi cells was assessed. The
results of the RT-qPCR and western blotting experiments revealed
that mRNA and protein levels of Smad4 in Smad4-RNAi cells were
significantly lower than that in HCT116 cells or RNAi-NC cells
(P<0.01), while no significant difference was observed between
HCT116 cells and RNAi-NC cells (P>0.05; Fig. 2).
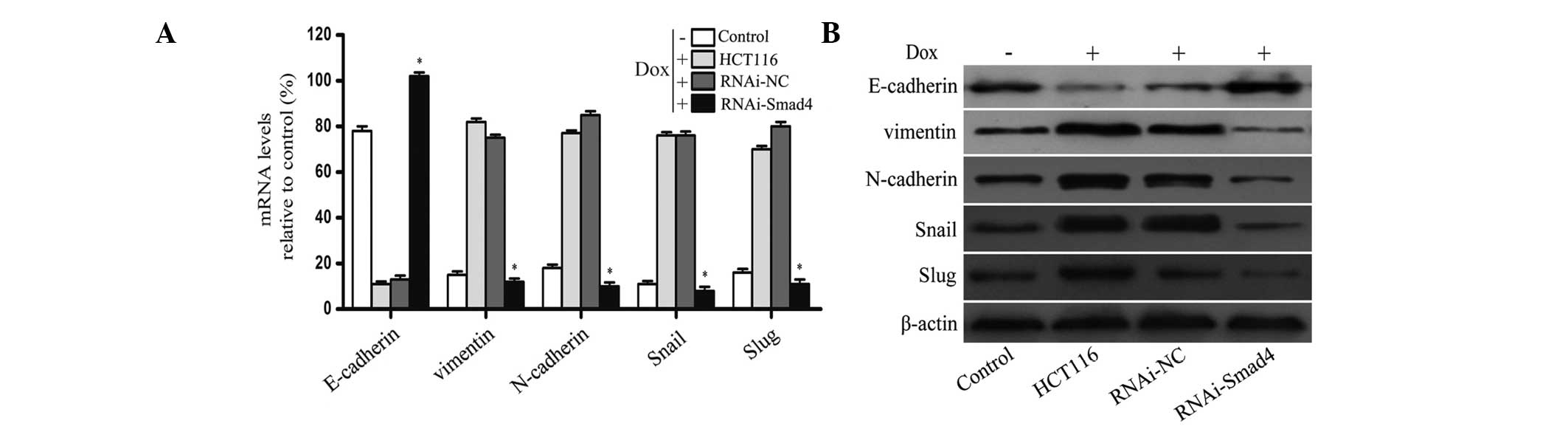
HCT116 cells exhibit molecular changes
consistent with EMT following Dox treatment
To determine whether the acquisition of Dox
resistance was associated with specific molecular changes that are
consistent with EMT, the expression of epithelial and mesenchymal
phenotypic markers, following 50 nmol/l Dox administration, were
examined using RT-qPCR and western blotting. From the PCR data, a
significant reduction in the expression of E-cadherin, and an
upregulation of Vimentin and N-cadherin, in HCT116 cells was
observed compared with that in the control group. The expression of
the transcription factors, Snail and Slug, were significantly
increased in resistant cells and the western blotting results were
in accordance with the PCR data (Fig.
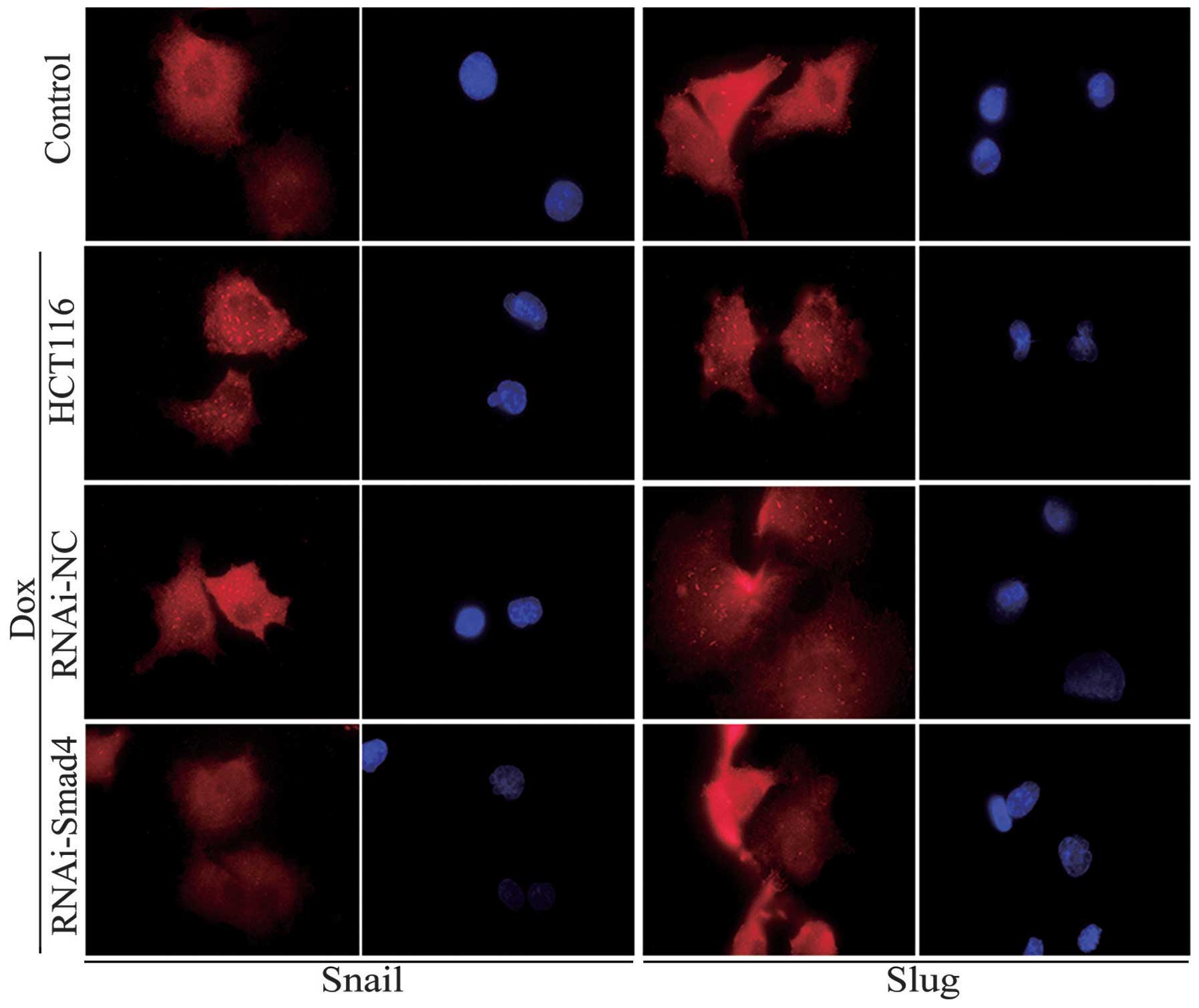
3A and B). Furthermore, immunofluorescence staining revealed
that Snail and Slug were more localized in the cytoplasm of the
HCT116 cells than it was in the control group (Fig. 4). These results suggested that EMT
occurred in HCT116 cells following treatment with Dox.
Downregulation of Smad4 reverses
Dox-induced EMT
Following successful silencing of the Smad4 gene
using a lentiviral vector, Smad4-specific RNAi was used to
investigate the effect of inhibiting the TGFβ/Smad4 signaling
pathway on the process of EMT, which was induced following Dox
treatment. RNAi-NC cells and Smad4-RNAi cells were also treated
with 50 nmol/l Dox for 7 days, and the expression of EMT markers
and transcription factors were examined using RT-qPCR and western
blotting. The PCR data demonstrated that the expression of
E-cadherin in Smad4-RNAi cells was higher than that observed in the
RNAi-NC cells (P<0.05), and that the expression levels of
Vimentin, N-cadherin, Snail and Slug in Smad4-RNAi cells was
significantly lower than that detected in the RNAi-NC cells
(P<0.05). The western blotting results were in accordance with
the PCR data (Fig. 3A and B).
Furthermore, immunofluorescence staining revealed that in the
RNAi-NC cells, Snail and Slug were more localized to the cytoplasm
than they were in the Smad4-RNAi cells (Fig. 4). These results suggested that
downregulation of Smad4 reverses EMT, which is induced following
Dox treatment.
Downregulation of Smad4 enhances and
increases sensitivity of HCT116 cells to Dox
In order to examine the effect of the downregulation
of Smad4 on HCT116 cell sensitivity to Dox, the cell viability and
MDR p-gp levels were also investigated using an MTT assay and
western blot analysis, in RNAi-NC cells and Smad4-RNAi cells.
Following administration of the same treatment to HCT116 cells, the
MTT assay results indicated that the cell viability ratio of
RNAi-NC and Smad4-RNAi cells decreased in a dose-dependent manner
(Fig. 1C). Concerning the MTT data
from the long-term administration of 50 nmol/l Dox, it was shown
that the cell viability ratio of RNAi-NC cells decreased between 1
and 3 days following treatment, in a time-dependent manner, whilst
it did not alter markedly between 3 and 7 days following treatment.
This result was similar to that obtained in the HCT116 cells.
However, the cell viability ratio of Smad4-RNAi cells decreased in
a time-dependent manner for the duration of the experiment
(Fig. 1C). Following 7 days of 50
nmol/l Dox administration, the p-gp expression of RNAi-NC cells was
higher than that in the control and Smad4-RNAi cells (Fig. 1E). These data suggested that
downregulation of Smad4 increases HCT116 cell sensitivity to Dox
following long-term treatment at a low concentration.
Discussion
There are two primary forms of drug resistance that
are associated with chemotherapy used in cancer. Patients who are
initially refractory to therapy exhibit intrinsic drug resistance,
while patients who relapse following an initial response to the
therapy, do so as a result of acquired drug resistance (14). Previous studies have demonstrated
that resistance to Dox treatment may be due to the activation of
expression of the MDR p-gp gene (15) and activation of intracellular
signaling pathways (16,17). Despite advances in treatment,
chemoresistance remains a significant impediment to the treatment
of colon cancer. An improved understanding of the mechanisms by
which residual tumor cells survive following chemotherapy may
ultimately provide novel, effective chemotherapeutic strategies. In
the present study, HCT116 human colon cancer cells were used to
investigate the molecular mechanisms underlying Dox resistance and
the associated cellular behaviors.
It was shown that HCT116 cells underwent changes
associated with EMT, following long-term Dox treatment. This was
demonstrated by changes in the expression of molecular markers and
proteins, including decreased expression of E-cadherin, and
increased levels of Vimentin and N-cadherin, as elucidated using
RT-qPCR and western blot analysis. These changes correlated with a
significant increase in the expression of transcription factors
Snail and Slug as elucidated using RT-qPCR, western blot analysis
and immunofluorescence staining. In accordance with the present
findings, the induction of EMT has also been reported in acquired
resistance to other chemotherapeutic agents (18,19).
From the MTT and western blotting data in the present study, it was
observed that HCT116 cells exhibited induced resistance to Dox, as
shown by the reduction in the cell viability ratio and the increase
in p-gp expression, in association with EMT. It was hypothesized
that this may be associated with the upregulation of TGFβ1
expression, and the phosphorylation of Smad2 and Smad3, which was
triggered by treatment with Dox.
TGFβ is a ubiquitous, pleiotropic growth factor,
which regulates numerous cellular processes, including cell
proliferation, differentiation and apoptosis (20). There are three TGFβ isoforms,
TGFβ1, TGFβ2 and TGFβ3, which exert their effects by binding to
cell surface receptors; the type I (TβRI), type II (TβRII) and type
III (TβRIII) TGFβ receptors. TβRII is a constitutively active
serine/threonine kinase that, upon ligand binding, recruits and
phosphorylates TβRI, thereby stimulating TβRII serine/threonine
kinase activity. TβRI then phosphorylates and activates the
transcription factors Smad2 or Smad3, which form a complex with
Smad4. This complex translocates to the nucleus and initiates the
transcription of TGFβ target genes in a cell-specific manner
(21). Thus, silencing the Smad4
gene may block TGFβ signaling and inhibit its molecular and
biological effects. Lim et al (22) utilized knockdown of Smad4 in order
to block TGFβ signaling in lung cells. A previous study confirmed
that the TGFβ/Smad4 pathway has an important role in the
chemoresistance of colon cancer cells to Dox-induced cell death
(23). EMT is a latent process,
important during embryonic development, by which certain cells
within a tumor may reactivate mesenchymal traits to disperse and
form metastases in the process of cancer progression (24). Induction of EMT by TGFβ has been
observed to increase motility and chemoresistance via the
disassembly of cell-to-cell contacts, loss of cell polarity and
significant cytoskeletal reorganization in normal and malignant
mammary epithelial cell types (25,26).
In accordance with previous studies (27,28),
the present data indicated that downregulation of Smad4 may be a
crucial event in the reversal of Dox-induced EMT. The current
results demonstrated that Smad4 RNAi reversed the changes in the
expression of the EMT markers E-cadherin, Vimentin and N-cadherin,
and that of the EMT transcription factors, Snail and Slug, which
were induced by Dox, indicating EMT reversal. Concomitantly, Smad4
RNAi resulted in a persistent reduction in the viability and MDR
p-gp expression of cells treated with Dox. Although further studies
are required to elucidate the mechanisms underlying
chemoresistance, the use of Smad4 shRNA during chemotherapy may be
a potential therapeutic approach with which to improve treatment
efficacy. As hypothesized, knockdown of Smad4 did not significantly
affect the expression of TGFβ1, Smad2 or Smad3, or the
phosphorylation of Smad2/3, as they are located upstream of the
TGFβ/Smad4 pathway, in contrast to Smad4.
In conclusion, the present study demonstrated that
low concentration, long-term administration of Dox may promote
resistance in HCT116 colon cancer cells, in part via the activation
of TGFβ signaling. In turn, this triggered Vimentin, N-cadherin,
Snail and Slug expression, indicative of the occurrence of EMT. The
present study also suggested that knockdown of Smad4 to inhibit the
TGFβ signal during chemotherapy may sensitize cancer cells to
chemotherapy, in part through the inhibition of MDR p-gp expression
and reversal of the EMT process. This may result in increased
therapeutic efficacy and the need for lower doses of
chemotherapeutic agents. Therefore, downregulation of Smad4, or
treatment with inhibitors to inhibit TGFβ signaling or Smad2 and/or
Smad3 phosphorylation, combined with Dox may be a potential novel
strategy with which to treat colon cancer.
Acknowledgments
The present study was sponsored by a grant from the
National Natural Science Foundation of China (grant no. 81272693).
The authors would like to thank Mr Hong Xia and Mrs Xiaoqing Cai
for their administrative support and technical assistance.
References
|
1
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, et al: Cancer incidence and mortality patterns
in Europe: Estimates for 40 countries in 2012. Eur J Cancer.
49:1374–1403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cui R, Okada Y, Jang SG, et al: Common
variant in 6q26-q27 is associated with distal colon cancer in an
Asian population. Gut. 60:799–805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jonker DJ, Spithoff K, Maroun J, et al:
Adjuvant systemic chemotherapy for stage II and III colon cancer
after complete resection: An updated practice guideline. Clin Oncol
(R Coll Radiol). 23:314–322. 2011. View Article : Google Scholar
|
|
4
|
Colombo V, Lupi M, Falcetta F, Forestieri
D, D Incalci M and Ubezio P: Chemotherapeutic activity of silymarin
combined with doxorubicin or paclitaxel in sensitive and
multidrug-resistant colon cancer cells. Cancer Chemoth Pharm.
67:369–379. 2011. View Article : Google Scholar
|
|
5
|
Arafa el-Sa, Zhu Q, Shah ZI, et al:
Thymoquinone up-regulates PTEN expression and induces apoptosis in
doxorubicin-resistant human breast cancer cells. Mutat Res.
706:28–35. 2011. View Article : Google Scholar :
|
|
6
|
Rosanò L, Cianfrocca R, Spinella F, et al:
Acquisition of chemoresistance and EMT phenotype is linked with
activation of the endothelin a receptor pathway in ovarian
carcinoma cells. Clin Cancer Res. 17:2350–2360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bastid J: EMT in carcinoma progression and
dissemination: Facts, unanswered questions, and clinical
considerations. Cancer Metast Rev. 31:277–283. 2012. View Article : Google Scholar
|
|
8
|
Canino C, Mori F, Cambria A, et al: SASP
mediates chemoresistance and tumor-initiating-activity of
mesothelioma cells. Oncogene. 31:3148–3163. 2012. View Article : Google Scholar
|
|
9
|
Borthwick LA, Gardner A, De Soyza A, Mann
DA and Fisher AJ: Transforming growth factor-β1 (TGF-β1) driven
epithelial to mesenchymal transition (EMT) is accentuated by tumour
necrosis factor α (TNFα) via crosstalk between the SMAD and NF-κB
pathways. Cancer Microenviron. 5:45–57. 2012. View Article : Google Scholar
|
|
10
|
Davidson BL and McCray PB: Current
prospects for RNA interference-based therapies. Nat Rev Genet.
12:329–340. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang X, Huang S, Zhang F, et al:
Lentiviral-mediated Smad4 RNAi promotes SMMC-7721 cell migration by
regulation of MMP-2, VEGF and MAPK signaling. Mol Med Rep.
3:295–299. 2010.
|
|
12
|
Lindner D: Animal models and the tumor
microenvironment: Studies of tumor-host symbiosis. Semin Oncol.
41:146–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakasone ES, Askautrud HA, Kees T, et al:
Imaging tumor-stroma interactions during chemotherapy reveals
contributions of the microenvironment to resistance. Cancer Cell.
21:488–503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong XB and Lavasanifar A: Traceable
multifunctional micellar nanocarriers for cancer-targeted
co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 5:5202–5213.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghosh J, Das J, Manna P and Sil PC: The
protective role of arjunolic acid against doxorubicin induced
intracellular ROS dependent JNK-p38 and p53-mediated cardiac
apoptosis. Biomaterials. 32:4857–4866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sims JT, Ganguly SS, Bennett H, Friend JW,
Tepe J and Plattner R: Imatinib reverses doxorubicin resistance by
affecting activation of STAT3-Dependent NF-κB and HSP27/p38/AKT
pathways and by inhibiting ABCB1. PloS one. 8:e555092013.
View Article : Google Scholar
|
|
18
|
Li QQ, Chen ZQ, Cao XX, et al: Involvement
of NF-κB/miR-448 regulatory feedback loop in chemotherapy-induced
epithelial-mesenchymal transition of breast cancer cells. Cell
Death Differ. 18:16–25. 2011. View Article : Google Scholar :
|
|
19
|
Sun L, Yao Y, Liu B, et al: MiR-200b and
miR-15b regulate chemotherapy-induced epithelial-mesenchymal
transition in human tongue cancer cells by targeting BMI1.
Oncogene. 31:432–445. 2012. View Article : Google Scholar
|
|
20
|
Tian M, Neil JR and Schiemann WP:
Transforming growth factor-β and the hallmarks of cancer. Cell
Signal. 23:951–962. 2011. View Article : Google Scholar :
|
|
21
|
Javelaud D, Alexaki VI, Dennler S,
Mohammad KS, Guise TA and Mauviel A: TGF-β/SMAD/GLI2 signaling axis
in cancer progression and metastasis. Cancer Res. 71:5606–5610.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lim MJ, Lin T and Jakowlew SB: Signaling
mechanisms of transforming growth factor-β (TGF-β) in cancer: TGF-β
induces apoptosis in lung cells by a Smad-dependent mechanism.
Tumor Suppressor Genes. Cheng Y: 1st. InTech; Rijeka, Croatia: pp.
1452012
|
|
23
|
Kang Y, Park M, Heo S, et al: The
radio-sensitizing effect of xanthohumol is mediated by STAT3 and
EGFR suppression in doxorubicin-resistant MCF-7 human breast cancer
cells. Biochim Biophys Acta. 1830:2638–2648. 2013. View Article : Google Scholar
|
|
24
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: Parallels between
normal development and tumor progression. J Mammary Gland Biol.
15:117–134. 2010. View Article : Google Scholar
|
|
25
|
Smith AL, Iwanaga R, Drasin DJ, et al: The
miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and
induces EMT and tumor initiating cell characteristics downstream of
Six1 in human breast cancer. Oncogene. 31:5162–5171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fuxe J, Vincent T and de Herreros AG:
Transcriptional crosstalk between TGFβ and stem cell pathways in
tumor cell invasion: Role of EMT promoting Smad complexes. Cell
Cycle. 9:2363–2374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hesling C, Fattet L, Teyre G, et al:
Antagonistic regulation of EMT by TIF1γ and Smad4 in mammary
epithelial cells. Embo Rep. 12:665–672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Li W, Zang X, et al:
MicroRNA-204-5p regulates epithelial-to-mesenchymal transition
during human posterior capsule opacification by targeting SMAD4.
Invest Ophth Vis Sci. 54:323–332. 2013. View Article : Google Scholar
|