Introduction
Retinoic acid (RA), an oxidative metabolite of
vitamin A, is present in the vertebrate embryonic limb bud and is
critical for correct limb development. An excess or a deficiency of
all-trans-retinoic acid (ATRA) may cause congenital malformations,
including limb defects (1).
Delgado-Baeza et al (2)
have established a fetal rat clubfoot model using a single
intragastric dose of RA on day 10 of gestation. Our group
previously demonstrated that inhibition of hindlimb cartilage
development is a crucial factor for ATRA-induced congenital
clubfoot (CCF) in a rat model (3,4).
Limb patterning and growth are carefully regulated through a
complex network of transcription factors and signaling molecules
(5,6). Despite a well-established role of
ATRA in CCF, the essential cellular and molecular targets and the
signaling mechanism whereby ATRA induces CCF remain to be
elucidated.
A key aspect of embryonic development is dependent
upon cell cycle control; p53 is an important mediator of the cell
cycle checkpoint at the G1-S transition (7) and is intricately involved in the
cellular decision-making process (8). p21, as a cyclin-dependent kinase
inhibitor, is a negative regulator of cell cycle progression and is
required for chondrocyte differentiation (9,10).
Induction of p53 is generally characterized by increased
transcription of p21, whose product interacts with the
cyclin-dependent complexes and regulates the cell cycle (11,12).
However, it is becoming increasingly clear that p21 can be induced
in a p53-independent manner (13).
In addition, p53 induces apoptosis or programmed cell death to
remove unwanted cells and to prevent teratogenesis in
preimplantation as well as in the early organogenesis period, which
is essential for normal development (14,15).
However, p53-dependent apoptosis is also responsible
for excessive cell loss in the predigital regions, which may result
in defects of the digits. p53+/+ animals have
been previously reported to be more sensitive to radiation-induced
apoptosis and defects of the digits than p53+/−
and p53−/− mice during the mid-gestational period
and the late gestational period. However, p53−/−
embryonic mice were more sensitive to apoptosis and defects than
were p53+/− and p53+/+ mice
during late organogenesis (16,17).
As p53-knockout (KO) mice developed and were viable,
polydactyly of the hindlimbs and other defects were reported to
occur at a higher incidence in p53-deficient mice (18). The importance of p53 may be more
pronounced in other species, as p53-KO Xenopus laevis
embryos exhibited inhibition of mesodermal differentiation and
severe gastrulation defects (19).
This difference may be explained by the evidence that other p53
family members, p63 and p73, which are expressed during mouse
embryogenesis and may compensate for the absence of p53, are not
expressed during early developmental stages in frogs (20). Similar to that observed in the
embryonic development of frogs, p53 was revealed to be involved in
embryogenesis in zebrafish (21).
In addition, inhibiting p53 expression in salamanders resulted in
inhibition of limb regeneration (22). High levels of p53 are present in
all tissues, including the limb bud until mid-gestation. During
organogenesis, p53 levels decrease until they are scarcely
detectable in terminally-differentiated tissues (23). However, it remains to be elucidated
what specific role p53 has during early embryonic limb bud
development.
A synchronized and tightly coupled mechanism is
hypothesized to be involved in the two p53-associated processes,
with the levels of p53 and cell type being important in bud
patterning. The results of the present study supported the
possibility that p53 or its signaling pathways may be etiologically
responsible for the increased incidence of congenital developmental
abnormalities, including CCF. It was hypothesized that ATRA may
induce CCF by regulating the expression of p21 during early
embryonic development in cartilage-specific molecules, including
Sox9, aggrecan and col2al, which are required for chondrogenesis of
rat embryo hindlimb bud mesenchymal cells (rEHBMCs). In the present
study, mesenchymal cells were collected from embryonic day 12.5
(E12.5) rat embryo hindlimb bud mesenchymal cells (rEHBMCs) and the
mechanisms whereby ATRA affected chondrogenesis were investigated
in vitro.
Materials and methods
Animals
Primiparous female Sprague-Dawley (SD) rats,
attained from the Laboratory Animal Center of Shantou University
Medical College (Shantou, China) were mated with adult males from
the same strain and supplier. The day when sperm was detected in
the vaginal smear was considered to be day 0 of gestation (E0).
Mated females were housed individually in clear polycarbonate cages
with stainless steel wire lids and corncob granules as bedding.
Food pellets and fitered tap water were available ad
libitum. Animals were maintained at 24°C with a humidity of 50%
and a 12-h light/dark cycle. All animal protocols were approved by
the Institutional Animal Care and Use Committee of Shantou
University Medical College (Shantou, China) and the study was
performed in accordance with the established institutional and
national guidelines on the experimental use and care of
animals.
Cell culture
Cell culturing was performed as described by Flint
and Orton (24). Hindlimb bud
mesenchymal cells were isolated from E12.5 female SD rats. Briefly,
pregnant rats were euthanized with CO2 and embryos from
the female rats were separated and placed in calcium- and
magnesium-free Earle’s balanced salt solution (HyClone
Laboratories, Inc., Logan, UT, USA)/fetal bovine serum (Gibco Life
Technologies, Carlsbad, CA, USA) (EBSS-CMF/FBS). The distal
three-quarters of the subridge at the distal tip of the hindlimbs
were dissociated in EBSS-CMF containing 0.1% trypsin (Gibco Life
Technologies), 0.1% EDTA and 50 mM Tris-HCl buffer (Shanghai
Chemical Reagent Co., Ltd., Shanghai, China) (pH 7.4). All cells
were filtered through a pre-moistened 40 μm cell strainer to
remove the ectoderm and cell clumps, which were rendered into
single cell suspensions. Cells were resuspended at a density of
2×107 cells/ml in 50% Dulbecco’s modified Eagle’s medium
(DMEM) and 50% F12 (Gibco Life Technologies) containing 10% FBS
(Gibco Life Technologies), penicillin (100 IU/ml; Sigma-Aldrich,
Beijing, China) and streptomycin (50 mg/ml; Sigma-Aldrich). Cells
at a density of 2×107cells/ml were plated onto 60-mm
dishes with 19 drops of media (25 μl) each or a total of
3×105 cells in 15 ml media placed as micromass in the
center of a 24-well plate. The cells were incubated for 1 h at 37°C
under 5% CO2 to allow attachment prior to being flooded
with the above culture medium. The day of cell seeding was referred
to as culture day 0 (CD 0).
Alcian blue staining
Alcian blue staining of sulfated cartilage
glycosaminoglycans was utilized to examine chondrogenic
differentiation using a previously described method (25). To demonstrate the deposition of
cartilage matrix proteoglycans, representative cultures were
collected on day one and two of incubation and stained with 0.5%
alcian blue 8 GX (Sigma-Aldrich, St. Louis, MO, USA), pH 1.0.
Alcian blue bound to sulfate was extracted with 6 M guanidine-HCl
and quantified by measuring the absorbance of the extracts at 600
nm using a microplate reader (Chromate 4300; Awareness Technology,
Inc., Palm City, FL, USA). Chondrogenic cartilage was expressed in
optical density (OD) value.
Immunofluorescent microscopy
For determination of the distribution of p53 and
p21, cells were fixed in ice-cold 4% (w/v) paraformaldehyde in
phosphate-buffered saline (PBS) for 30 min and permeabilized with
0.2% Triton Χ-100 (Shanghai Chemical Reagent Co., Ltd.) for 10 min
at 40°C. Cells were treated with 3% H2O2 for
20 min and non-specific binding was blocked with 10% normal goat
serum (Wuhan Boster Biological Technology, Ltd., Wuhan, China) for
30 min at 37°C. Following washing with PBS three times, cells were
incubated with mouse monoclonal anti-p53 (1:200; cat. no. AP062;
Beyotime Institute of Biotechnology, Haimen, China) and rabbit
polyclonal p21 antibodies (1:200; cat. no. sc-397; Santa Cruz
Biotechnology, Dallas, TX, USA) overnight at 4°C followed by 1 h
incubation with goat anti-rabbit (1:64; cat. no. BA1105; Boster
Biological Technology, Ltd.) and goat anti-mouse fluorescein
isothiocyanate-conjugated (1:64; cat. no. BA1101; Boster Biological
Technology, Ltd.) antibodies. Following counterstaining with DAPI
(Invitrogen Life Technologies, Carlsbad, CA, USA), cells were
visualized under the Nikon TE2000-S inverted microscope (Nikon,
Tokyo, Japan).
Cell proliferation assay
Proliferation of rEHBMCs was determined by the
direct counting of cells from micromass cultures. Control and
treated cultures were maintained for the indicated periods of time,
trypsinized and counted in triplicate using a hemocytometer
(Invitrogen Life Technologies).
Flow cytometric analysis
Levels of apoptosis were analyzed using flow
cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA).
For detection of propidium iodide staining, cells were excited at
488 nm and emission was detected at 585 nm. The data were analyzed
with the use of WinMDI software (v2.9; Bio-soft Net; http://en.bio-soft.net/other/WinMDI.html).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total cellular RNA was isolated from mesenchymal
cells using an RNA Simple Total RNA kit (Tiangen Biotech, Beijing,
China). The concentration of RNA was determined from the optical
density of the sample, which was measured at 260 nm. Aliquots of
the extracted RNA samples were initially reversely transcribed for
first strand cDNA synthesis. The sequences for specific primers
(Generay Biotech Co., Shanghai, China) for detecting mRNA
transcripts of the rat p53, p21, Sox9, collagen, type II, α 1
(Col2al), aggrean and GAPDH genes were as follows: p53 forward,
5′-CCATCTACAAGAAGTCACAACAC-3′ and reverse,
5′-CCCAGGACAGGCACAAAC-3′; p21 forward, 5′-TGTCCGTCAGAACCCATG-3′ and
reverse, 5′-TGGGAAGGTAGAGCTTGG-3′; Sox9 forward,
5′-TAGCCCTGGTTTCGTTCTCT-3′ and reverse, 5′-TCCTGCTCGTCGGTCATCTT-3′;
Col2al forward, 5′-GGAGCAGCAAGAGCAAGGAGAAGAA-3′ and reverse,
5′-CTCAGTGGACAGTAGACGGAGGAAAGT-3′; aggrecan forward,
5′-GGAGAGGACTGCGTAGTGATGA-3′ and reverse,
5′-AGCCTGTGCTTGTAGGTGTTGG-3′ and GAPDH forward,
5′-CAGTGCCAGCCTCGTCTCAT-3′ and reverse, 5′-AGGGGCCATCCACAGTCTTC-3′.
The PCR conditions were as follows: 35°C for 15 min, followed by 36
cycles of 95°C for 10 sec and 62°C for 30 sec. The RT-PCR products
were resolved using agarose gel electrophoresis. The signal
intensity was quantified using Quantity One Software (v4.5.2;
Bio-Rad, Hercules, CA, USA). The transcript levels were normalized
against GAPDH and the relative mRNA levels were expressed as the
ratio of the density of the detected genes to GAPDH at the same
time-point.
Western blot analysis
Cellular lysates were prepared using
radioimmunoprecipitation assay lysis buffer (Bocai, Shanghai,
China). Western blotting assays were performed as described
previously (26) and the following
antibodies were used for the procedure: rabbit polyclonal anti-p53
(1:1,000; cat. no. 9282; Cell Signaling Technology, Inc., Danvers,
MA, USA), rabbit polyclonal Sox9 (1:2,000; cat. no. ab26414; Abcam,
Cambridge, MA, USA), rabbit polyclonal p21 (1:1,000; cat. no.
BS6561) and rabbit polyclonal Col2a1 (1:1,000; cat. no. BS1071),
and rabbit polyclonal GAPDH (1:1,000; cat. no. BS60630) antibodies
(Bioworld Technology, Nanjing, China). The membranes were then
incubated with goat anti-rabbit fluorescein
isothiocyanate-conjugated secondary antibodies (1:1,000; cat. no.
BA1055; Boster Biological Technology, Ltd.). Densitometric analysis
was performed using Quantity One Software (v4.5.2; Bio-Rad,
Hercules, CA, USA). GAPDH was used as loading control.
Statistical analysis
Values are expressed as the mean ± standard
deviation for three or more independent experiments. Statistical
analyses were conducted using SPSS version 17.0 (SPSS Inc.,
Chicago, IL, USA). Statistical significance was estimated using a
one-way analysis of variance with Bonferroni’s post-hoc test
(multiple comparisons) and the Student-Newman-Keuls test
(comparisons between two groups) was used where appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ATRA inhibits chondrogenesis by
downregulating cartilage-specific proteins in rEHBMCs
Chondrogenic differentiation is regulated at three
stages, which are precartilage condensation, cell proliferation and
cartilage nodule formation (27,28).
To determine whether ATRA affected chondrogenesis, undifferentiated
rEHBMCs were cultured at a density of 2×107 cells/ml and
stimulated with 0 to 10 μM ATRA. Precartilage condensation,
one of the two main stages of chondrogenesis (29,30),
was then assessed using Alcian blue staining for sulfated
proteoglycans and cartilage nodules on days one and two. The
results were expressed as the ratio of the density of the detected
group to that of the control group at the same time-point. It was
identified that ATRA caused a marked dose-dependent reduction in
the quantity of cartilage (10 μM ATRA, 0.280±0.035 vs.
controls, 1.200±0.150; P<0.01). At 1 μM ATRA, Alcian blue
staining uptake was significantly decreased on day two (Fig. 1A and B) and 1 μM ATRA was
therefore used for all subsequent experiments.
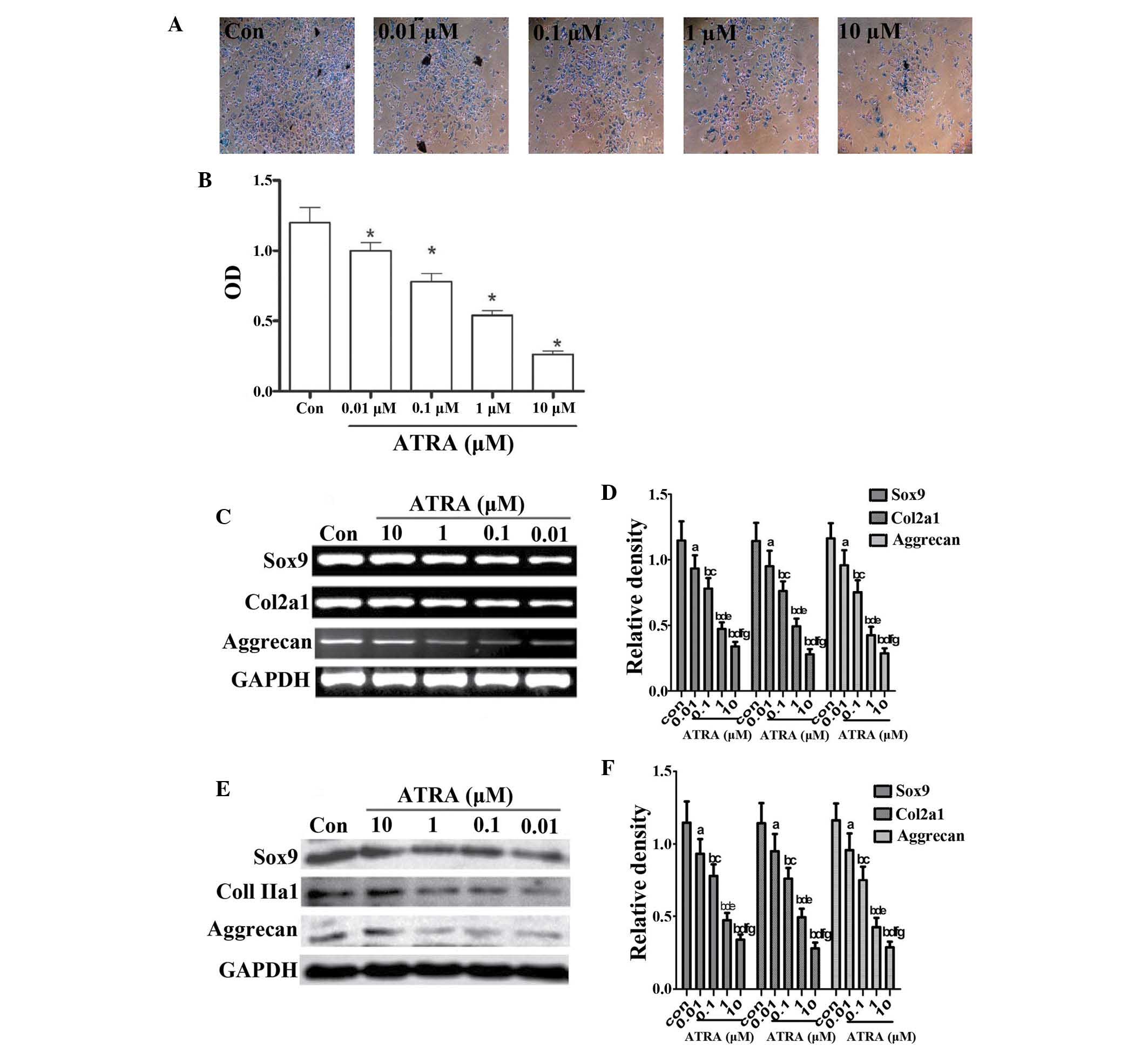 | Figure 1ATRA suppresses chondrogenesis. (A)
Alcian blue staining of rEHBMCs grown in micromass cultures in the
presence of varying concentrations of ATRA on day two of culture
(magnification, ×100). (B) Quantification of chondrogenesis was
determined by measuring the absorbance of bound Alcian blue at 600
nm. *P<0.05, compared with control cells. (C) and (E)
Changes in the levels of Sox9, aggrecan and Col2a1 in control and
ATRA-treated cultures were determined by reverse transcription
polymerase chain reaction and western blotting on day two of
culture. Results shown are representative of at least three
independent experiments. GAPDH was used as a loading control. (D)
and (F) Densitometric quantification of Sox9, Col2a1 and aggrecan
was performed. (D) aP>0.05; bP<0.05,
versus control group; cP>0.05; dP<0.05,
versus 0.01 μM ATRA group; eP>0.05;
fP<0.05, versus 0.1 μM ATRA group;
gP>0.05, versus 1 μM ATRA group. (F)
aP>0.05; bP<0.05 versus control group;
cP>0.05; dP<0.05, versus 0.01 μM
ATRA group; eP>0.05; fP<0.05, versus
0.1 μM ATRA group; gP>0.05, versus 1 μM
ATRA group. ATRA, all-trans-retinoic acid; rEHBMCs, rat embryo
hindlimb bud mesenchymal cells; Col2a1, collagen, type II, α 1; OD,
optical density. |
It was further investigated whether ATRA inhibited
chondrogenesis by downregulating the expression of
cartilage-specific molecules, including aggrecan, Sox9 and Col2al.
The expression of aggrecan, Sox9 and Col2al on day one was not
detected (data not shown). The RT-PCR assays revealed that ATRA
dose-dependently reduced the mRNA transcript levels of Sox9,
aggrecan and Col2a1 on day two (Fig.
1C and D) with a 40–50% reduction in the mRNA transcript levels
of Sox9, aggrecan and Col2a1 in cells treated with 1 μM
ATRA. Consistently, a dose-dependent reduction in the protein
levels of aggrecan, Sox9 and Col2al (Fig. 1E and 1F) were also observed. These results
demonstrated that ATRA dose-dependently inhibited sulfated
proteoglycan accumulation and cartilage nodule formation during
chondrogenesis by downregulating cartilage-specific proteins,
suggesting that ATRA inhibited chondrogenesis by decreasing
precartilage condensation of rEHBMCs.
ATRA stimulates apoptotic cell death of
rEHBMCs in vitro
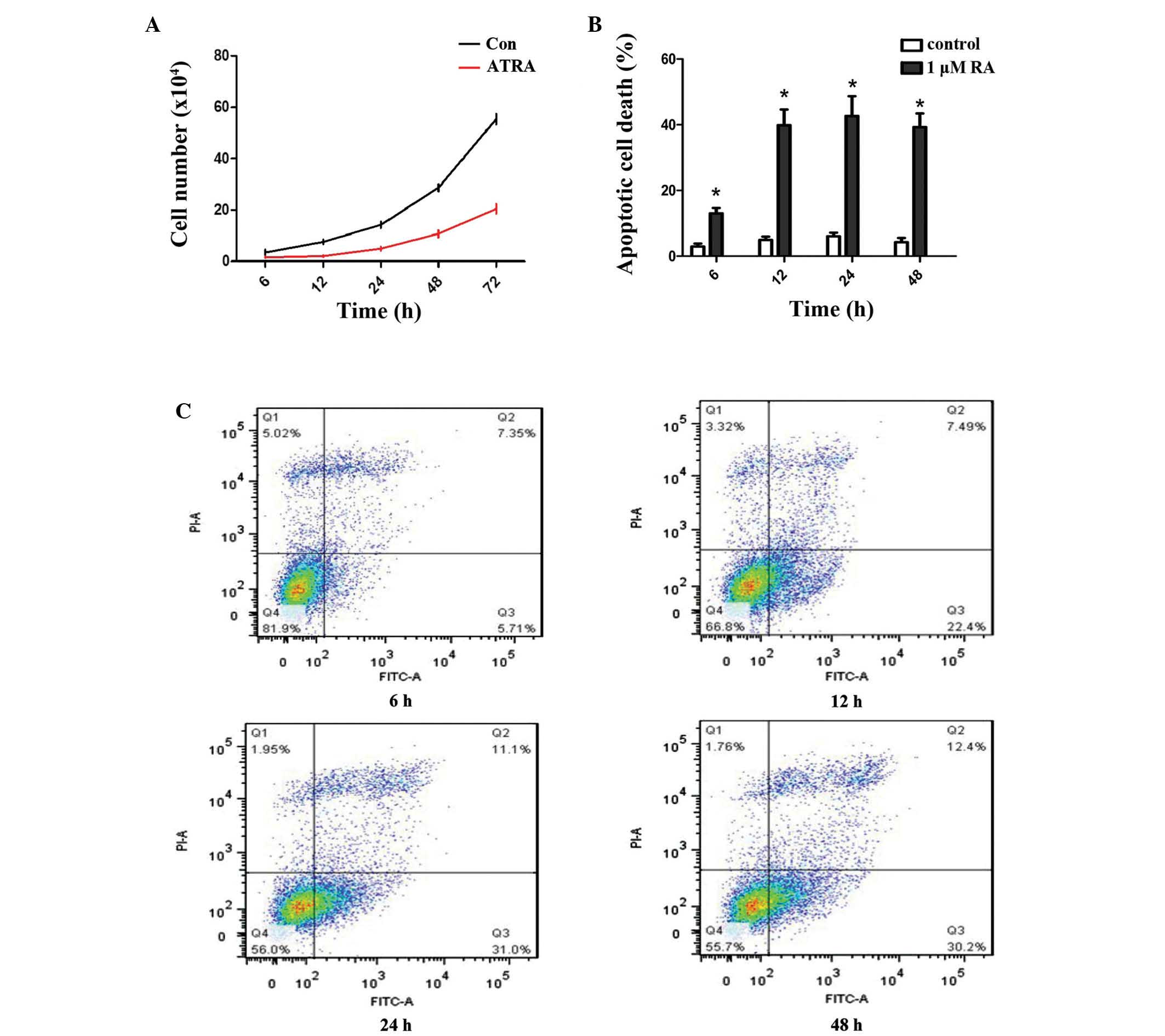
It was subsequently examined whether ATRA-inhibited
chondrogenesis occurred as a result of changes in the proliferation
of undifferentiated rEHBMCs. It was identified that, compared with
the controls, ATRA markedly reduced the viability of rEHBMCs at 6–7
h post-treatment (Fig. 2A). Flow
cytometric analysis revealed that ATRA caused a marked increase in
the rate of apoptotic cell death of rEHBMCs. At 48 h
post-treatment, the apoptotic rate was 42% for rEHBMCs compared
with 2% for the controls (P<0.01; Fig. 2B and C).
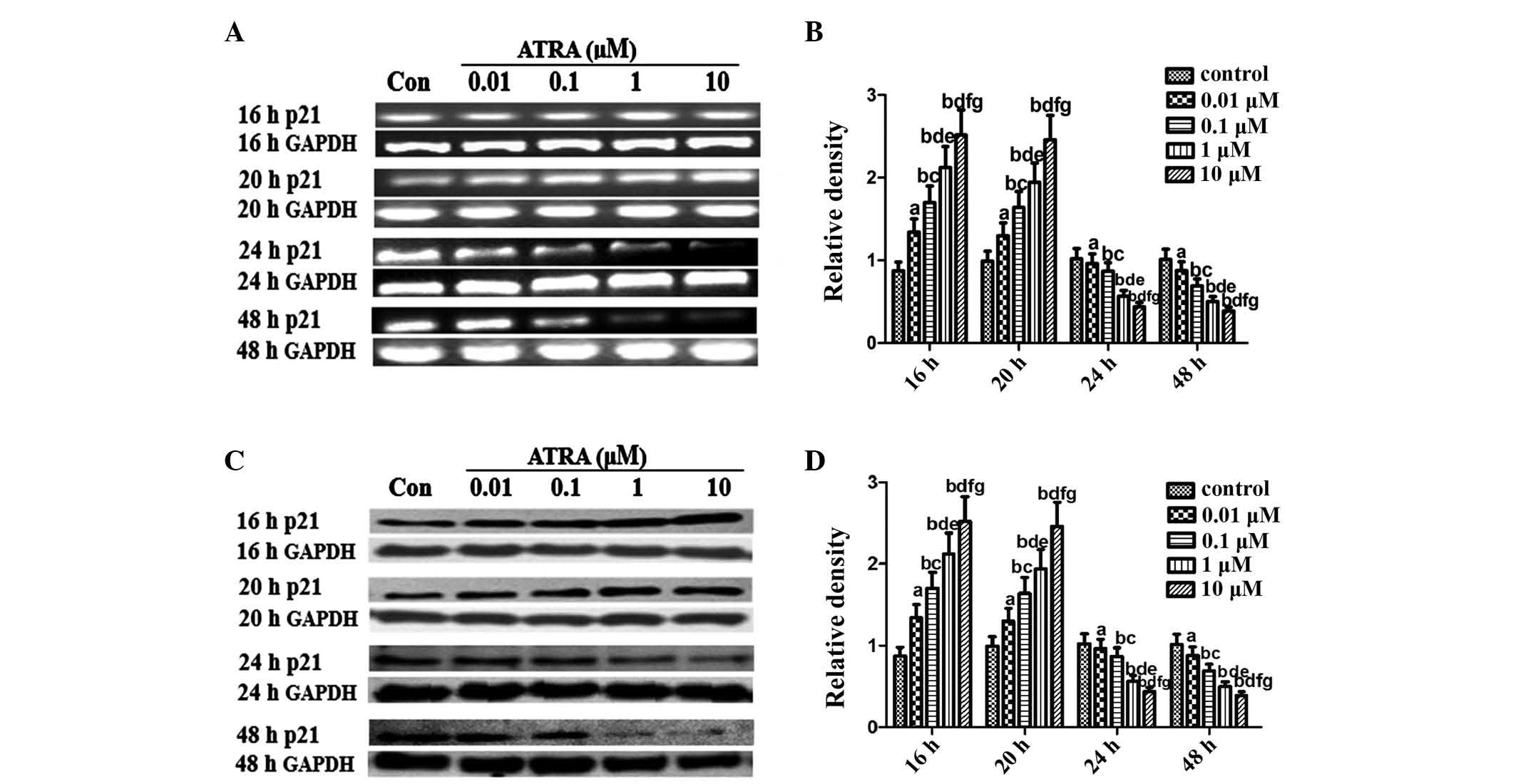
ATRA modulates p53 and p21 expression in
rEHBMCs during chondrogenesis
An important objective of the present study was to
elucidate the pattern of p53 expression during differentiation in
rEHBMCs. RT-PCR assays revealed that undifferentiated rEHBMCs
expressed high mRNA transcription levels of p53. The mRNA
transcript levels of p53 markedly increased dose-dependently at 16
and 20 h after treatment with ATRA, which then underwent a rapid
decline 24 and 48 h after ATRA treatment (Fig. 3A and B). A similar pattern of
changes in the levels of p53 protein was observed in rEHBMCs
following treatment with ATRA (Fig. 3C
and D).
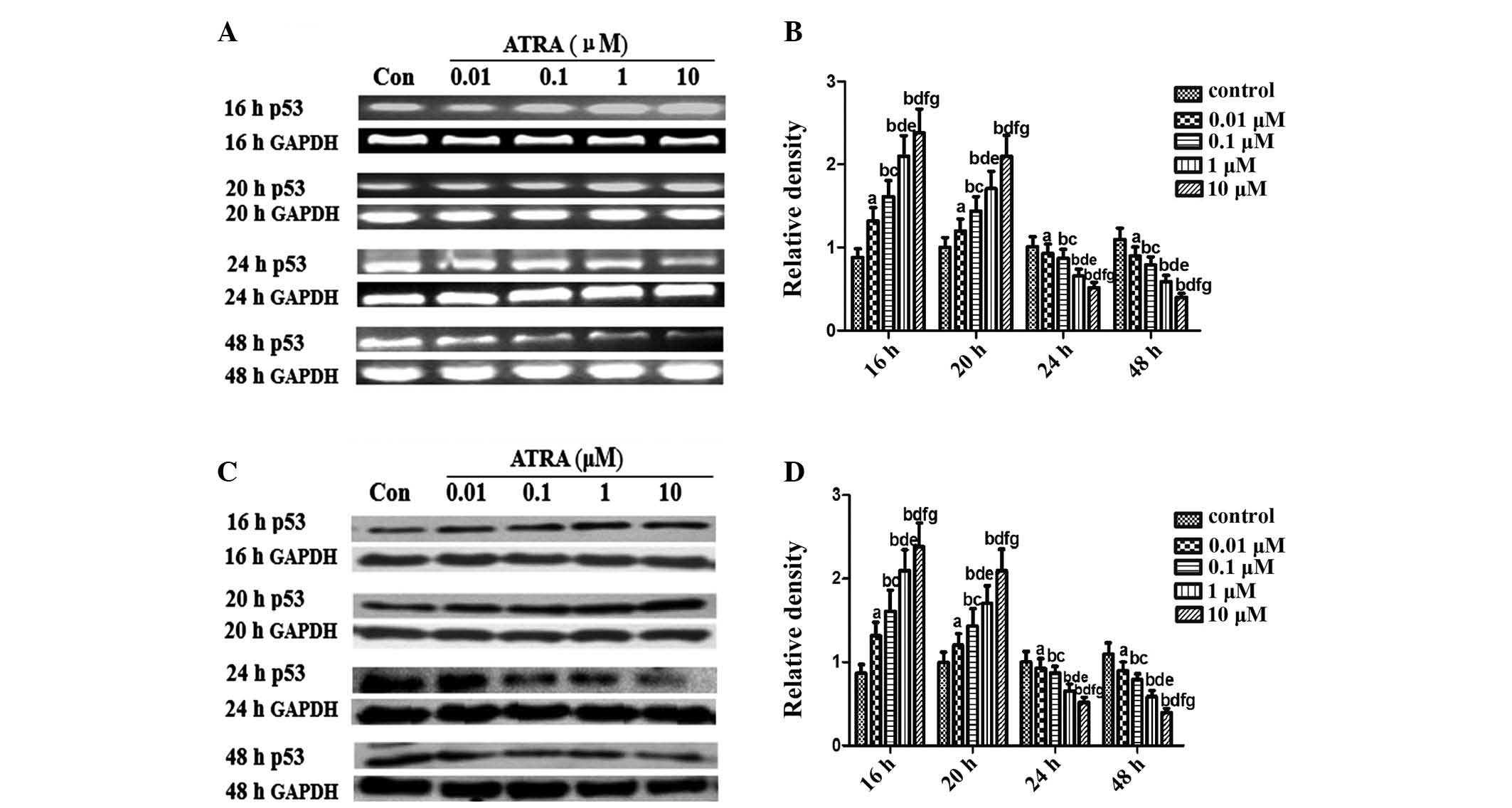 | Figure 3Expression of p53 in spontaneously
differentiating rEHBMCs for a period of two days. Undifferentiated
mesenchymal cells express high levels of p53 and a decline was
noted during differentiation. (A) and (C) Changes in the levels of
p53 in control and ATRA-treated cultures were determined by RT-PCR
and western blotting at the indicated time-points. Values shown are
representative of at least three independent experiments. GAPDH was
used as a loading control. (B) and (D) Densitometric quantification
of p53 mRNA and p53 protein were performed. (B) p53 mRNA was
subjected to RT-PCR in the exponential growth phase and normalized
to GAPDH. aP>0.05, bP<0.05, versus
control group; cP>0.05, dP<0.05, versus
0.01 μM ATRA group; eP >0.05,
fP<0.05, versus 0.1 μM ATRA group;
gP>0.05, versus 1 μM ATRA group. (D) p53
Protein was subjected to western blot analysis in the exponential
growth phase and normalized to GAPDH. aP>0.05;
bP<0.05, versus control group; cP>0.05;
dP<0.05 versus, 0.01 μM ATRA group;
eP>0.05, fP<0.05, versus 0.1 μM
ATRA group; gP>0.05, versus 1 μM ATRA group.
ATRA, all-trans-retinoic acid; rEHBMCs, rat embryo hindlimb bud
mesenchymal cells; RT-PCR, reverse transcription polymerase chain
reaction. |
p21 inhibits proliferation in vivo and in
vitro
Considering the prominent role of p53/p21 in
chondrogenesis (19,20), an aim of the present study was to
investigate whether ATRA inhibited chondrogenesis of rEHBMCs by
modulating the expression of p21. The expression of p21 was
examined between 6 and 48 h after the induction of chondrogenic
differentiation of rEHBMCs. The levels of p21 at 6 and 12 h
remained low or undetectable (data not shown). The levels of p21
markedly increased at 16–20 h after ATRA treatment; however, they
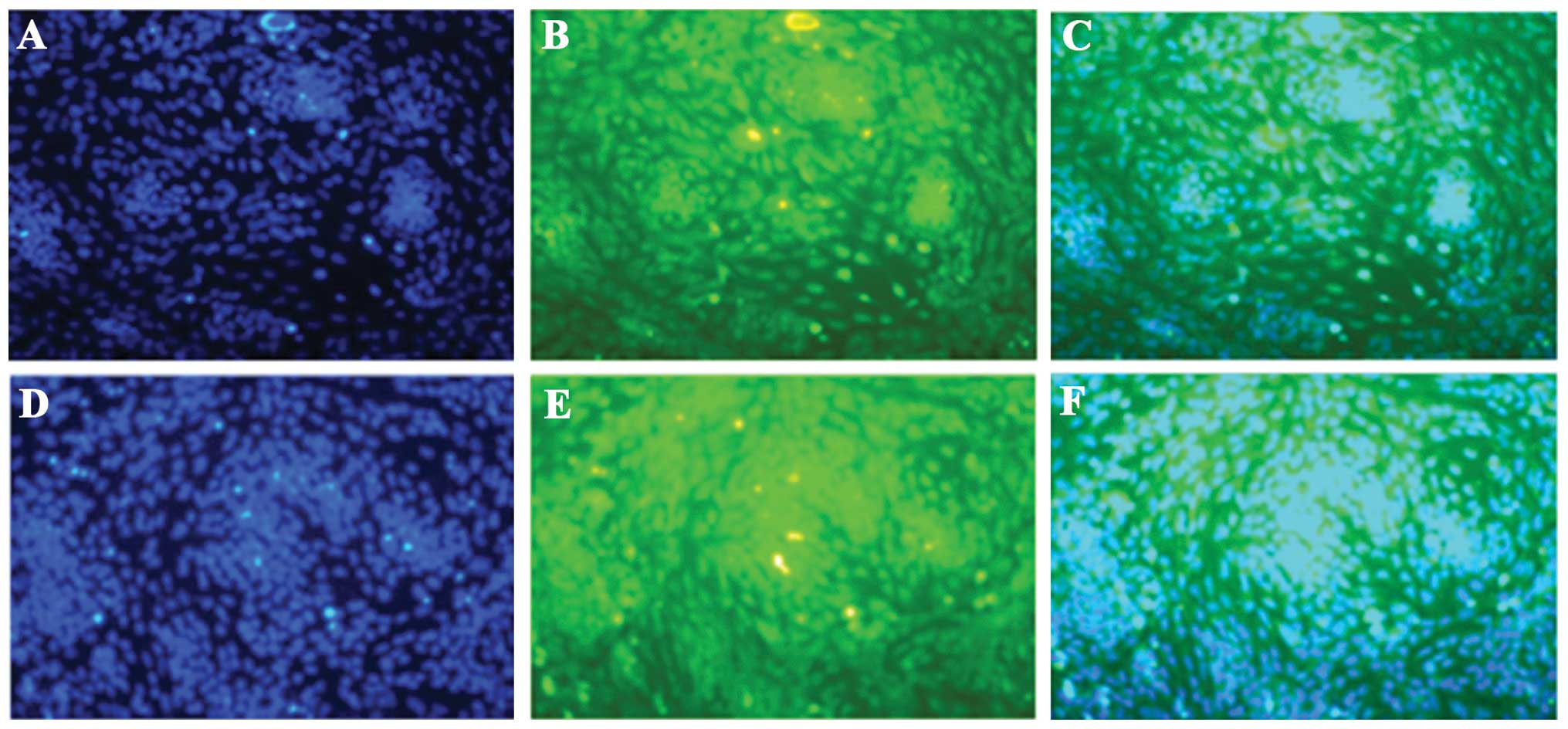
rapidly decreased 24–48 h after treatment (Fig. 4). The immunofluorescent microscopy
data further revealed that p53 and p21 were predominantly expressed
in the cartilage nodules, mainly in the nucleus (Fig. 5A–D). These findings indicated that
ATRA suppressed chondrogenesis of rEHBMCs by modulating the
expression of p53/p21 at the transcriptional and translational
level.
Discussion
During early embryonic development, the expression
of RA in a specific region of the embryo enables the determination
of the position along the embryonic anterior/posterior axis by
serving as an intercellular signaling molecule guiding the
development of the posterior portion of the embryo (31). Appropriate RA expression in the
vertebrate embryo is essential for the proper development and
patterning of the organism. It has previously been reported that an
excess or a deficiency of RA causes congenital malformations,
including limb defects (1) that
result from the failure of chondrogenesis in the embryonic hindlimb
bud mesenchymal cells (32–34).
Additionally, it has been proposed that the teratogenic effects of
ATRA, leading to limb defects, are based on increased cellular
apoptosis in the developing limb (33,34).
Previous studies by our group have demonstrated that a rat embryo
CCF-like model may be established via a single intragastric dose of
ATRA, which exhibits hindlimb congenital malformations, impaired
chondrogenesis and increased cellular apoptosis (3,4,35).
These findings suggested that defective chondrogenesis of the
primary hindlimb bud mesenchymal cells may contribute to CCF.
Formation of cartilage nodules is a morphological
characteristic of the condensation stage of chondrogenic
development (36). In the present
study, it was demonstrated that ATRA dose-dependently reduced the
number of cartilage nodules and the area of cartilage nodules in
rat hindlimb buds in vitro, suggesting that ATRA inhibited
the condensation of hindlimb bud cells. There is a requirement for
a minimal number of cells prior to prechondrogenic cells being able
to differentiate; if the number of cells is too low, this prevents
a skeletal element from forming, which may also reduce the number
of skeletal elements formed (37).
A reduction in precartilaginous condensations below a critical size
is a developmental mechanism for the evolutionary loss of skeletal
elements, including chondrocytes. It was identified that ATRA
initiated apoptotic cell death of rEHBMCs in the initial 6 h of
ATRA exposure, which decreased the proliferation of rEHBMCs,
suggesting that ATRA inhibited the condensation of rEHBMCs by
promoting the apoptotic cell death of these cells.
By contrast to the condensation stage, where the
formation of cartilage nodules occurs, the differentiation stage is
characterized by the expression of a number of specific molecular
markers. Sox9, which is expressed where cartilage formation takes
place, is a key transcription factor required for chondrogenesis
(38,39). It is essential for mesenchymal
condensation prior to chondrogenesis and chondrocyte
differentiation fails in the absence of Sox9. Mutations or loss of
Sox9 lead to severe malformations of the endochondral skeleton,
while homozygous deletion of Sox9 completely inhibits
chondrogenesis (40). Of note,
Sox9-deficient cells are excluded from cartilage condensation
(38), indicating that Sox9 is
required to establish the chondrogenic lineage. Sox9-null
embryonic stem cells are inhibited in their differentiation to
chondrocytes and persist as mesenchymal cells (38). In the present study, it was
identified that ATRA dose-dependently suppressed the expression of
Sox9 at the transcriptional and translation level, suggesting that
ATRA may inhibit chondrogenesis of the hindlimb bud mesenchymal
cells through its inhibitory effect on Sox9.
p21 is required for chondrocytic differentiation
(9,10) and its expression is noticeably
increased during the differentiation of chondrogenic cells
(41). Additionally, p21 is
upregulated in maturing and differentiated chondrocytes in
vivo (41). Sox9-transfected
cells were observed to accumulate in the G0/G1 phase, which was
associated with an increase in the expression and promoter activity
of p21, suggesting that Sox9 inhibits cell cycle progression to
facilitate its pro-differentiating function. These studies
demonstrated that Sox9 alters the rate of cell cycle progression of
chondrocytes and their differentiation by enhancing or inhibiting
the expression of p21. It was revealed in the present study that
ATRA downregulated the mRNA and protein expression of p21 in
primary hindlimb bud mesenchymal cells in a dose-dependent manner.
Combined with the evidence that p21 is predominantly expressed in
cartilage cells, these findings indicated that ATRA suppresses
chondrogenesis by modulating the expression of Sox9 and its
downstream target p21 in primary hindlimb bud mesenchymal cells.
Col2a1 is the principal constituent of the extracellular cartilage
matrix (42). Sox9 has also been
observed to be capable of regulating the expression of
cartilage-specific molecules, including Col2a1 and aggrecan
(38,43,44).
The present study demonstrated that ATRA downregulated the mRNA and
protein expression of Col2al and aggrecan in primary hindlimb bud
mesenchymal cells in a dose-dependent manner. These findings
indicated that ATRA suppresses chondrogenesis by modulating the
expression of Sox9 and its downstream target Col2a1 in primary
hindlimb bud mesenchymal cells. In addition, the effects occurred
early and at a stage prior to the onset of chondrocyte
hypertrophy.
Numerous studies have demonstrated that expression
of p53 in undifferentiated cells can induce differentiation
(43,45). Correspondingly, reduction of p53
expression in human stem cells prevents them from spontaneous
apoptosis and differentiation (46). p53 has a central role in cartilage
development by regulating key genes for chondrogenesis, including
Sox9 and Col2a1 (47). It was
identified that undifferentiated rEHBMCs express a high level of
p53 mRNA and protein. The p53 mRNA transcript levels decreased
during early spontaneous differentiation of undifferentiated
rEHBMCs in the absence of any inducing agents, which was
accompanied by a decrease in p53 protein, suggesting a regulatory
mechanism, which is affected not only by protein stability, but
also by a decrease in mRNA transcript levels. The levels of p53
were markedly increased from 16–20 h after treatment with ATRA in a
dose-dependent manner. However, the levels then decreased from
24–48 h after treatment with ATRA in a dose-dependent manner. The
results demonstrated that ATRA modulated the expression of p53 in
the rat hindlimb bud mesenchymal cells during the entire process of
chondrogenesis in a dose- and stage-dependent manner. Of note, ATRA
induced apoptosis 10 h prior to the upregulation of p53. These
results demonstrated that ATRA-mediated apoptosis promoted changes
in p53, suggesting that p53 upregulation did not initiate
ATRA-induced apoptosis during cell proliferation and precartilage
condensation of chondrogenic competent cells. ATRA-induced
apoptosis of undifferentiated rat hindlimb bud mesenchymal cells
may occur independently of p53 function. The levels of p53 were
downregulated in parallel with the expression of Sox9, Col2a1 and
aggrecan in the primary hindlimb bud mesenchymal cells treated with
ATRA. Combined with the evidence that p53 protein is mainly
expressed in cartilage cells, these findings suggested that ATRA
suppressed the differentiation of undifferentiated rat hindlimb bud
mesenchymal cells in association with p53. These findings
demonstrated that ATRA inhibited chondrogenesis of primary hindlimb
bud mesenchymal cells by downregulating the expression of p53/p21,
Sox9, aggrecan and Col2al. Although further investigations are
required, it is hypothesized that the effects of ATRA on these
chondrogenesis-associated genes may be mediated through modulating
the p53/p21 signaling pathways. The p53/p21 signaling pathway may
be important for chondrogenesis and the disruption of this process
may contribute to a significant proportion of ATRA-induced CCF
cases.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81271947, 81341103
and 31240077) and the Administration of Traditional Chinese
Medicine of Guangdong Province, China (grant no. 20131248 and
20142084). All experiments were performed at the Laboratory of
Molecular Cardiology, the First Affiliated Hospital of Shantou
University Medical College (Shantou, China). The authors would like
to acknowledge Dr Tao-gen Shang (Xinsteel Centre Hospital, Jiangxi,
China) for the time and effort provided to the experiments provided
in this study and the support of Professor Yong-gang Zhang, Mr.
Li-biao Wu and Mr. Bo-zhi Cai for their helpful advice and
collaboration.
References
|
1
|
Lee GS, Kochhar DM and Collins MD:
Retinoid-induced limb malformations. Curr Pharm Design.
10:2657–2699. 2004. View Article : Google Scholar
|
|
2
|
Delgado-Baeza E, Santos-Alvarez I and
Martos-Rodriguez A: Retinoic acid-induced clubfoot-like deformity:
pathoanatomy in rat fetuses. J Pediatr Orthoped B. 8:12–18.
1999.
|
|
3
|
Zhong YS, Zheng C, Jia Y, et al:
Quantitative evaluation in vivo of the degree of differentiation of
hindlimb cartilage in a rat clubfoot model. Toxicol Mech Methods.
19:292–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu ZY, Li XD, Chen B, et al: Retinoic
acid retards fetal and hindlimb skeletal development asymmetrically
in a retinoic acid-induced clubfoot model. Exp Toxicol Pathol.
62:663–670. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zelzer E and Olsen BR: The genetic basis
for skeletal diseases. Nature. 423:343–348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tickle C: Patterning systems - from one
end of the limb to the other. Dev Cel. 4:449–458. 2003. View Article : Google Scholar
|
|
7
|
Kastan MB, Zhan Q, El-Deiry WS, et al: A
mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is
defective in ataxia-telangiectasia. Cell. 71:587–597. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sarkar SA and Sharma RP:
All-trans-retinoic acid-mediated modulation of p53 during neural
differentiation in murine embryonic stem cells. Cell Biol Toxicol.
18:243–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beier F, Leask TA, Haque S, et al: Cell
cycle genes in chondrocyte proliferation and differentiation.
Matrix Biol. 18:109–120. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beier F, Taylor AC and LuValle P: The
Raf-1/MEK/ERK pathway regulates the expression of the p21Cip1/Waf1
gene in chondrocytes. J Biol Chem. 274:30273–30279. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wade J, Adami GR, Wei N, Keyomarsi K and
Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar
|
|
12
|
El-Deiry WS, Tokino T, Velculescu VE, et
al: WAF1, a potential mediator of p53 tumor suppression. Cell.
75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gartel AL and Tyner AL: Transcriptional
regulation of the p21 [(WAF1/CIP1)] gene. Exp Cell Res.
246:280–289. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Oca Luna RM, Wagner DS and Lozano G:
Rescue of early embryonic lethality in mdm2-deficient mice by
deletion of p53. Nature. 378:203–206. 1995. View Article : Google Scholar
|
|
15
|
Uren AG and Vaux DL: Molecular and
clinical aspects of apoptosis. Pharmacol Ther. 72:37–50. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nomoto S, Ootsuyama A, Shioyama Y, Katsuki
M, Kondo S and Norimura T: The high susceptibility of heterozygous
p53(+/−) mice to malformation after foetal irradiation is related
to sub-competent apoptosis. Int J Radiat Biol. 74:419–429. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Westphal CH, Hoyes KP, Canman CE, et al:
Loss of atm radiosensitizes multiple p53 null tissues. Cancer Res.
58:5637–5639. 1998.PubMed/NCBI
|
|
18
|
Armstrong JF, Kaufman MH, Harrison DJ and
Clarke AR: High-frequency developmental abnormalities in
p53-deficient mice. Curr Biol. 5:931–936. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cordenonsi M, Dupont S, Maretto S, Insinga
A, Imbriano C and Piccolo S: Links between tumor suppressors: p53
is required for TGF-β gene responses by cooperating with Smads.
Cell. 113:301–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Danilova N, Sakamoto KM and Lin S: p53
family in development. Mech Dev. 125:919–931. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campbell WA, Yang H, Zetterberg H, et al:
Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen-2)
demonstrate excessive p53-dependent apoptosis and neuronal loss. J
Neurochem. 96:1423–1440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Villiard É, Brinkmann H, Moiseeva O,
Mallette FA, Ferbeyre G and Roy S: Urodele p53 tolerates amino acid
changes found in p53 variants linked to human cancer. BMC Evol
Biol. 7:1802007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schmid P, Lorenz A, Hameister H and
Montenarh M: Expression of p53 during mouse embryogenesis.
Development. 113:857–865. 1991.PubMed/NCBI
|
|
24
|
Flint O and Orton T: An in vitro assay for
teratogens with cultures of rat embryo midbrain and limb bud cells.
Toxicol Appl Pharmacol. 76:383–395. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lev R and Spicer S: Specific staining of
sulphate groups with alcian blue at low pH. J Histochem Cytochem.
12:309. 1964. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Jiang Y, Wan Y, et al:
Medroxyprogestogen enhances apoptosis of SKOV-3 cells via
inhibition of the PI3K/Akt signaling pathway. J Biomed Res.
27:43–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Solursh M: Differentiation of cartilage
and bone. Curr Opin Cell Biol. 1:989–994. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ruberte E, Dolle P, Chambon P and
Morriss-Kay G: Retinoic acid receptors and cellular retinoid
binding proteins. II. Their differential pattern of transcription
during early morphogenesis in mouse embryos. Development.
111:45–60. 1991.PubMed/NCBI
|
|
29
|
Thorogood P and Hinchliffe J: An analysis
of the condensation process during chondrogenesis in the embryonic
chick hindlimb. J Embryol Exp Morphol. 33:581–606. 1975.PubMed/NCBI
|
|
30
|
Ahrens PB, Solursh M and Reiter RS:
Stage-related capacity for limb chondrogenesis in cell culture. Dev
Biol. 60:69–82. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duester G: Retinoic acid synthesis and
signaling during early organogenesis. Cell. 134:921–931. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Galdones E and Hales BF: Retinoic acid
receptor gamma-induced misregulation of chondrogenesis in the
murine limb bud in vitro. Toxicol Sci. 106:223–232. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kochlar D: Cellular basis of congenital
limb deformity induced in mice by vitamin A. Birth Defects Orig
Artic Ser. 13:111–154. 1977.PubMed/NCBI
|
|
34
|
Paulsen DF, Chen WD, Pang L, Johnson B and
Okello D: Stage- and region-dependent chondrogenesis and growth of
chick wing-bud mesenchyme in serum-containing and defined tissue
culture media. Dev Dyn. 200:39–52. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jia YL, Zhong YS, Chen B, et al: The
primary research of posterior buds in quantificated congenital
clubfoot at pathological and molecule levels. Chin J Exp Surg.
6:726–728. 2009.
|
|
36
|
Hall BK and Miyake T: All for one and one
for all: condensations and the initiation of skeletal development.
Bioessays. 22:138–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hall B and Miyake T: The membranous
skeleton: the role of cell condensations in vertebrate
skeletogenesis. Anat Embryol (Berl). 186:107–124. 1992. View Article : Google Scholar
|
|
38
|
Bi W, Deng JM, Zhang Z, Behringer RR and
de Crombrugghe B: Sox9 is required for cartilage formation. Nat
Genet. 22:85–89. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bi W, Huang W, Whitworth DJ, et al:
Haploinsufficiency of Sox9 results in defective cartilage primordia
and premature skeletal mineralization. Proc Natl Acad Sci USA.
98:6698–6703. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Goldring MB, Tsuchimochi K and Ijiri K:
The control of chondrogenesis. J Cell Biochem. 97:33–44. 2006.
View Article : Google Scholar
|
|
41
|
Stewart M, Farnum C and MacLeod J:
Expression of p21CIP1/WAF1 in chondrocytes. Calcif Tissue Int.
61:199–204. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
de Crombrugghe B, Lefebvre V and Nakashima
K: Regulatory mechanisms in the pathways of cartilage and bone
formation. Curr Opin Cell Biol. 13:721–727. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Almog N and Rotter V: Involvement of p53
in cell differentiation and development. Biochim Biophys Acta.
1333:F1–F27. 1997.PubMed/NCBI
|
|
44
|
Ng LJ, Wheatley S, Muscat GE, et al: SOX9
binds DNA, activates transcription and coexpresses with type II
collagen during chondrogenesis in the mouse. Dev Biol. 183:108–121.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Meletis K, Wirta V, Hede S-M, Nistér M,
Lundeberg J and Frisén J: p53 suppresses the self-renewal of adult
neural stem cells. Development. 133:363–369. 2006. View Article : Google Scholar
|
|
46
|
Qin H, Yu T, Qing T, et al: Regulation of
apoptosis and differentiation by p53 in human embryonic stem cells.
J Biol Chem. 282:5842–5852. 2007. View Article : Google Scholar
|
|
47
|
Ikeda T, Kawaguchi H, Saito T, et al: p63
plays a central role in cartilage development by directly
regulating key genes for chondrogenesis. J Bone Miner Res.
22:s52–s101. 2007.
|