Introduction
Osteosarcoma is the most common type of bone
sarcoma. It affects ~560 infants and adolescents annually in the
United States (1). There are two
peaks in incidence, the first is during the pubertal skeletal
growth spurt (15–19 years old) and the second is during old age
(75–79 years old). The latter is attributed to the sarcomatous
transformation of Paget’s disease of bone and other benign bone
lesions (1). Twenty percent of
patients with osteosarcoma (OS) present with clinically detectable
metastases, with micrometastases presumed to be present in a number
of the remaining patients (2).
The management of patients with OS is complicated.
Current treatments typically include preoperative chemotherapy,
surgical resection, postoperative chemotherapy and adjunctive
immunotherapy (2,3). Limb-salvage procedures with wide
surgical margins that aim to completely remove all clinically
detectable tumors surgically are the mainstay of surgical
intervention (1). The use of
intensive chemotherapeutic agents, such as high-dose methotrexate
with leucovorin rescue, adriamycin, cisplatin, ifosfamide and
cyclophosphamide has improved relapse-free survival rates in
patients with localized extremity tumors from <20% with surgery
only, to ~70% (1–3). However, unfortunately the efficacy
and toxicity of these agents limits their use (4). Despite aggressive multi-modal
treatment, patients with synchronous and metachronous metastatic
disease with local relapse and nonresectable primary disease have a
poor 5-year survival rate, which has remained unchanged over the
past two decades (3). This
highlights the urgent requirement for novel, innovative therapeutic
approaches, such as targeted therapies (5,6) and
interferon-mediated immunotherapy (7).
Signal transducers and activators of transcription 3
(STAT3) is a latent transcription factor that participates in the
transcriptional activation of apoptosis and cell cycle progression
(8). STAT3 regulated genes are
involved in invasiveness, proliferation, angiogenesis,
lymphangiogenesis and inflammation. It has been implicated as an
oncogene in a variety of neoplastic diseases as well as OS
(8,9). A study demonstrated that mice lacking
STAT3 undergo incomplete gestation (10). Dysregulation of STAT3 has been
implicated as a key participant in tumor cell survival,
proliferation and metastasis (8).
A constitutively active mutant of STAT3 has been demonstrated to
transform fibroblasts and induce tumor formation in nude mice and
OS transformation (11). STAT3
activation (phosphorylation) has been demonstrated in a subset of
human OS tissues, and human and canine OS cell lines (12,13),
and OS stem like cells (9,14). Gibbs et al (14) showed the existence of a small
subpopulation of self-renewing bone sarcoma cells that are capable
of forming suspended spherical clonal colonies (also termed
sarcospheres) in anchorage-independent serum-starved conditions.
These bone sarcoma cells express activated STAT3 (14). Murase et al (9) demonstrated that a side population in
OS cell lines induce tumorigenesis in nude mice. STAT3 is often
correlated with a malignant tumor phenotype and STAT3 expression in
OS can be used as a prognostic predictor. The high level of STAT3
protein is associated with poor tumor differentiation and
presentation of metastasis (15).
Moreover, the 5-year overall and relapse-free survival rates for
patients with OS with high STAT3 expression are lower than those
for patients with low STAT3 expression (15). In addition, the status of STAT3
protein expression was an independent prognostic factor for
disease-free survival and overall survival (12,16).
Materials and methods
Cell culture
The 143.98.2 OS cell line was obtained from American
Type Culture Collection (ATCC CRL-11226™; Manassas, VA, USA). Cells
were maintained in Dulbecco’s modified Eagle’s medium, 10% fetal
bovine serum and 1% penicillin/streptomycin (GE Healthcare Life
Sciences, Logan, UT, USA) at 37°C with 5% CO2. For drug
treatment, inhibitors were added 2 h after plating cells. FLLL32
was provided by Dr James Fuchs (17). NSC74859 was purchased from
Selleckchem (Houston, TX, USA). The study was approved by the
ethics committee of the Second Affiliated Hospital of Soochow
University (Suzhou, China).
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-
2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay
The MTS assay was performed as described previously
(18). Briefly, OS cells were
plated in quadruplate for each dose of STAT3 inhibitor on 96-well
plates at a density of 500 cells/well in serum-containing growth
medium. Plates were incubated at 37°C and 5% CO2. Two
hours after plating, cells were treated with carrier (0.1% dimethyl
sulfoxide), FLLL32 or NSC 74859 at 1, 10, 100, 1,000 or 10,000 nM.
Proliferation was quantified four days following the addition of
drug by an MTS assay using a Cell Titer 96 Proliferation kit
(Promega Corporation, Madison, WI, USA). Absorbance was read at 490
nm in a Spectramax M2 plate reader (Molecular Devices, Sunnyvale,
CA, USA).
Immunohistochemistry and histology
Tissue was embedded in paraffin and blocks were cut
to generate 6-μm sections. Deparaffinized sections were
stained with either hematoxylin and eosin or incubated overnight at
4°C with the following antibodies: Polyclonal rabbit anti-human
NCL-Ki67p antibody (1:1,000; #KI67P-CE; Leica Microsystems, Inc.,
Buffalo Grove, IL, USA) or monoclonal rabbit anti-human pSTAT3
(Tyr705) (1:1,000; #9145; Cell Signaling Technology, Inc., Danvers,
MA, USA). The following day, sections were incubated in horseradish
peroxidase (HRP)-labeled secondary antibody and staining was
detected by DAB (DAKO, Herndon, VA, USA). Sections were viewed with
a Axiovert 200 inverted microscope (Carl Zeiss, Thornwood, NY, USA)
equipped with a digital imaging system.
Western blot analysis
Tumor proteins were extracted using extraction
buffer (20 mM NaPO4, 150 mM NaCl, 2 mM MgCl2,
0.1% Nonidet P-40, 10% glycerol, 10 mM sodium fluoride, 0.1 mM
sodium orthovanadate, 10 mM sodium pyrophosphate, 100 μM
phenylasine oxide, 10 nM okadaic acid, 1 mM dithiothreitol, 10
μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml
pepstatin, 10 μg/ml tosyl-l-phenylalanine chloromethyl
ketone and 10 μg/ml Nα-tosyl-L-lysine
chloromethyl ketone). Protein concentration was estimated using
Coomassie® Plus Protein Assay Reagent (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Proteins (40
μg/lane) were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis on 4–20% or 16%
tris-glycine gel (Invitrogen Life Technologies, Carlsbad, CA, USA)
and electrotransferred to polyvinylidene diflouride membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
non-fat milk+0.1% Tris-buffered saline with Tween-20 to minimize
nonspecific binding. The pSTAT3 antibody; monoclonal rabbit
anti-human STAT3 (1:1,000; #4904) and cleaved caspase 3 (Asp175)
(1:2,000; #9664) antibodies; and polyclonal rabbit anti-human
β-actin antibody (1:10,000; #4967) (Cell Signaling Technology,
Inc.) were used. Binding of the HRP-conjugated secondary antibody
to the membrane was visualized using a chemiluminescent detection
system (Amersham, Arlington Heights, IL, USA). Anti-β-actin was
used as a loading control. At least three different tumor lysates
were analyzed for each antigen.
Lentiviral infection
143.98.2 OS cells were infected at 60% confluence
with human shSTAT3 (TRCN0000353630) or non-target control
(pLKO.1-puro-non-target control, cat. no. SHC016; Sigma-Aldrich,
St. Louis, MO, USA). Lentiviral particles were incubated with OS
cells in the presence of polybrene (8 μg/ml; Sigma-Aldrich)
daily for 2 days, followed by selection in puromycin (2
μg/ml; Sigma-Aldrich) for at least five days. Surviving
cells were collected for xenograft injection.
Mouse xenograft
Female athymic nude (nu/nu) mice (Harlan
Laboratories, Harlan, IN, USA) were maintained in a pathogen-free
environment with free access to food and water, 12 h on/off light
cycle, 22°C temperature, and 21–22% oxygen. A total of 143.98.2 OS
cells (2×106) were injected in a total volume of 150
μl 30% Matrigel (BD Biosciences, San Jose, CA, USA) into the
flanks of 5- to 6-week-old mice; right flanks were injected with
shSTAT3 cells and left with non-target control cells. Tumor volume
was calculated as L × W2 (π/6), where L is the longest
diameter and W is the width. For the drug treatment xenograft,
143.98.2 OS cells were injected only into the right flanks of nude
mice. Tumors were dissected 1 h after the final dose was
administered and immediately flash frozen in liquid nitrogen for
biochemical analysis or fixed in 4% paraformaldehyde for
histological analysis.
In vivo drug treatment
FLLL32 was dissolved in 12.5% alcohol+12.5%
cremaphor (Sigma-Aldrich) by boiling for 15 min. FLLL32 was
prepared fresh every other day. FLLL32 (200 mg/kg) or vehicle
(12.5% alcohol+12.5% cremaphor) were administered to tumor bearing
nude mice daily for 3 weeks by intraperitoneal injection in 200–300
μl based on mouse weight. Mice were weighed twice a week in
order to monitor potential side effects of weight loss. The treated
mice were sacrificed with 100% CO2 for 5 min and
cervical dislocation subsequent to 15 consecutive days of
treatment, when the tumor size reached >10% of the mouse body
weight.
Statistical analysis
All statistical analyses were performed by unpaired,
two-tailed Student’s t-test using Microsoft Excel 2000 software
(Microsoft Corporation, Redmond, WA, USA). P<0.05 was considered
to indicate a significant difference.
Results
JAK2/STAT3 inhibition prevents OS cell
proliferation in vitro
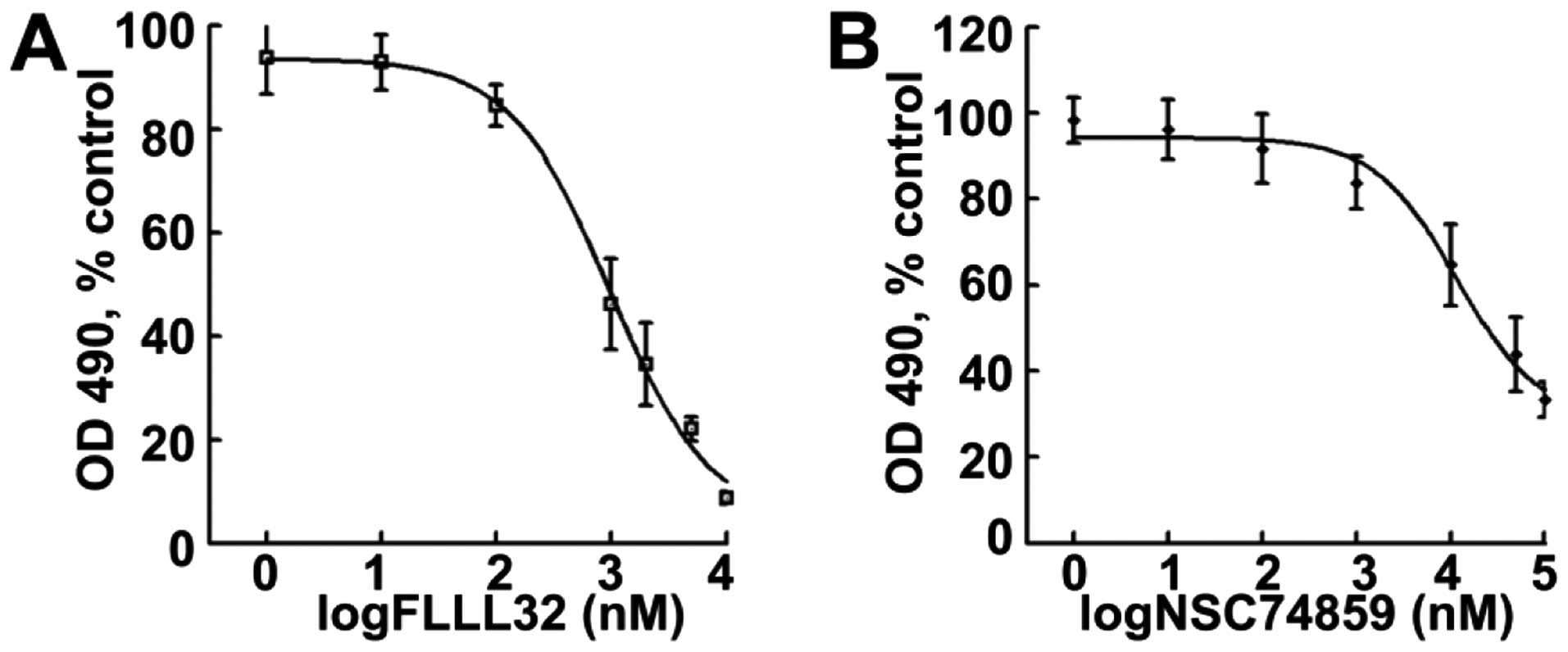
To determine whether the JAK2/STAT3 pathway is
involved in OS cell proliferation, a dose-response analysis of
FLLL32, a specific JAK2/STAT3 inhibitor on a human 143.98.2 OS
cells, was conducted. After 4 days of treatment, FLLL32 decreased
cell survival in a dose-dependent manner. The average
IC50 was ~500 nM (Fig.
1A). The effects of another STAT3 inhibitor, NSC74859, on the
same human 143.98.2 OS cell line were then observed. NSC74859
showed similar effects as FLLL32, decreasing human OS cell survival
(Fig. 1B). These results suggest
that JAK2/STAT3 mediated STAT3 signaling contributes to
osteosarcaoma cell proliferation in vitro. JAK2/STAT3
inhibition prevents OS cell growth.
Pharmacological inhibition of JAK2/STAT3
delays OS xeno- graft growth in vivo
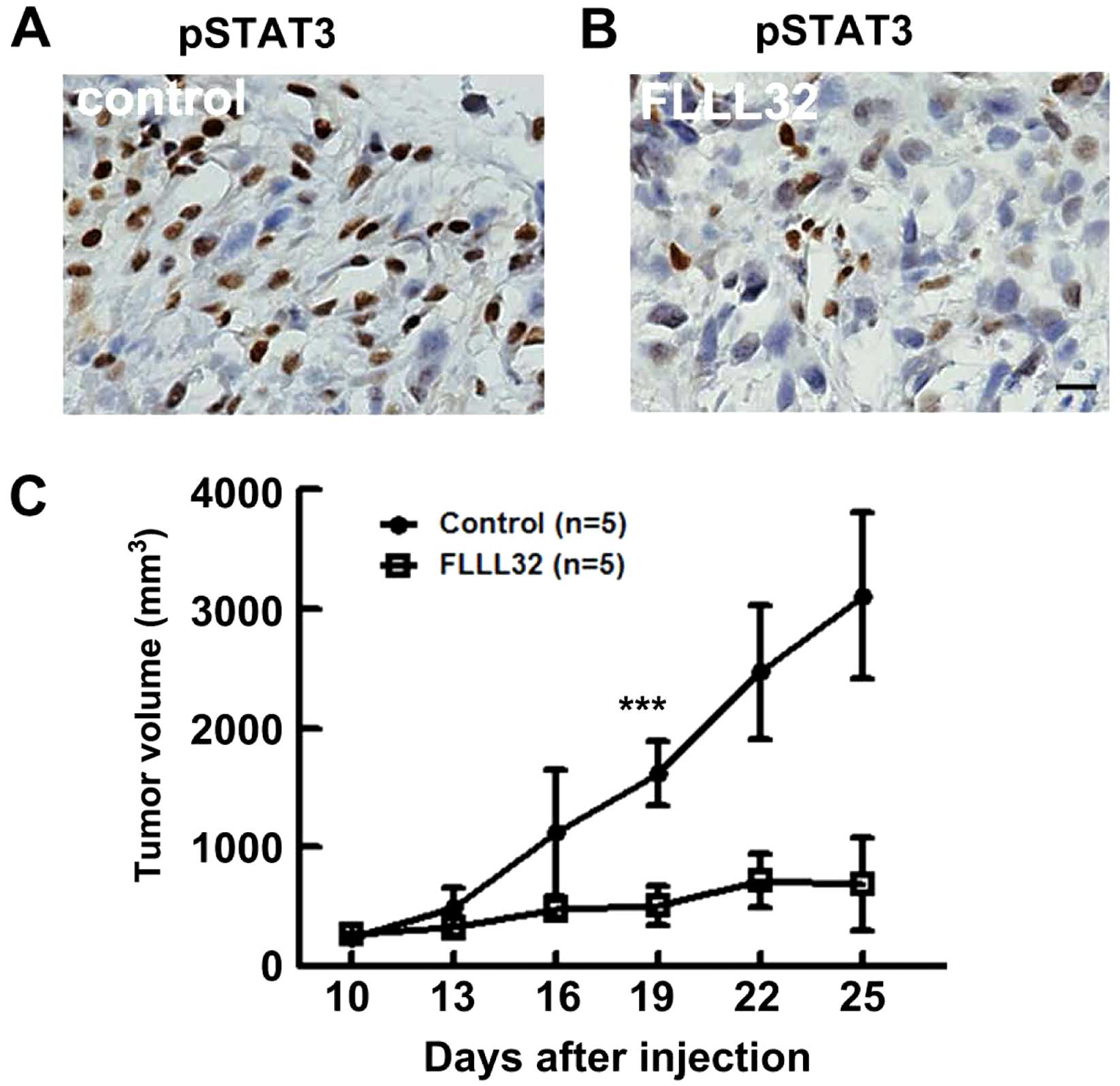
It was then analyzed whether blocking the JAK2/STAT3
pathway may affect growth in a human 143.98.2 OS xenograft model.
Nude mice with average OS tumors ~150 mm3 (n=5) were
treated for 10 days after cell transplantation with FLLL32 (200
mg/kg) daily via intraperitoneal injection. All mice survived the
treatment period without significant weight loss. Following
sacrifice, immunostaining was performed to determine pSTAT3 levels
at the end of the experiment with or without FLLL32 treatment. The
PSTAT3 level was significantly lower in the treatment group
compared with that of vehicle controls, indicating that the drug
was effective at this time point (Fig.
2A and B). Tumor volume measurements demonstrated that FLLL32
decreased OS growth significantly compared with vehicle controls
(P<0.001) (Fig. 2C).
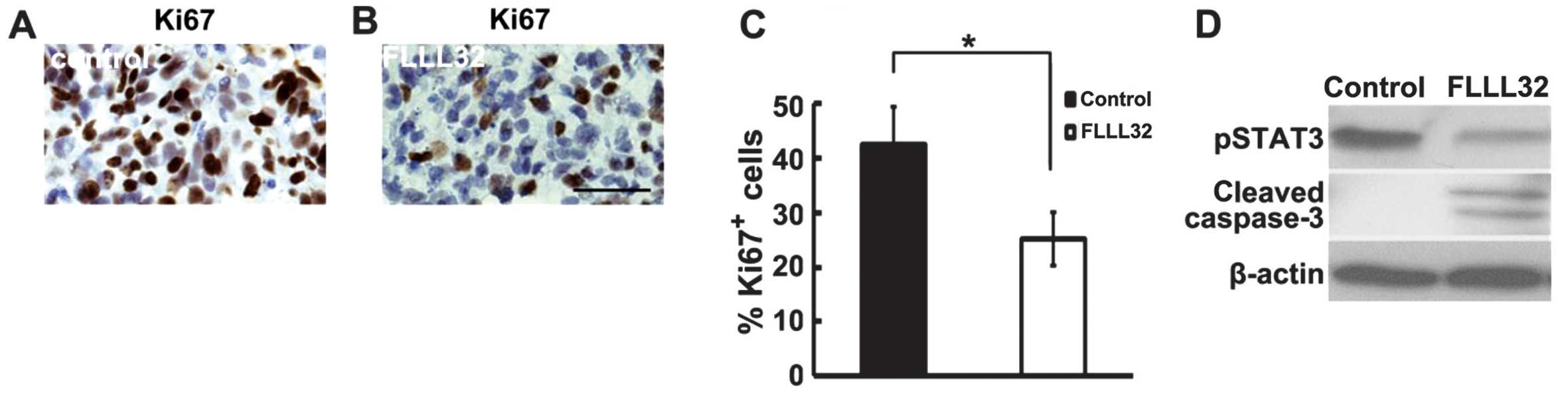
Cell proliferation and cell death were then analyzed
in paraffin sections of excised OSs by Ki67 staining and western
blot analysis, respectively. There was a significant difference in
the percentage of Ki67+ proliferating cells in
FLLL32-treated OS (n=5) compared with vehicle controls (n=5,
P<0.05) (Fig. 3A–C). Western
blot analysis demonstrated decreased pSTAT3 and increased cleaved
caspase 3 in FLLL32-treated OS xenografts as compared with vehicle
control (Fig. 3D). The results
suggest that FLLL32 delayed OS xenograft growth by inhibiting cell
proliferation and inducing cell apoptosis.
STAT3 deficiency delayed OS formation in
vivo
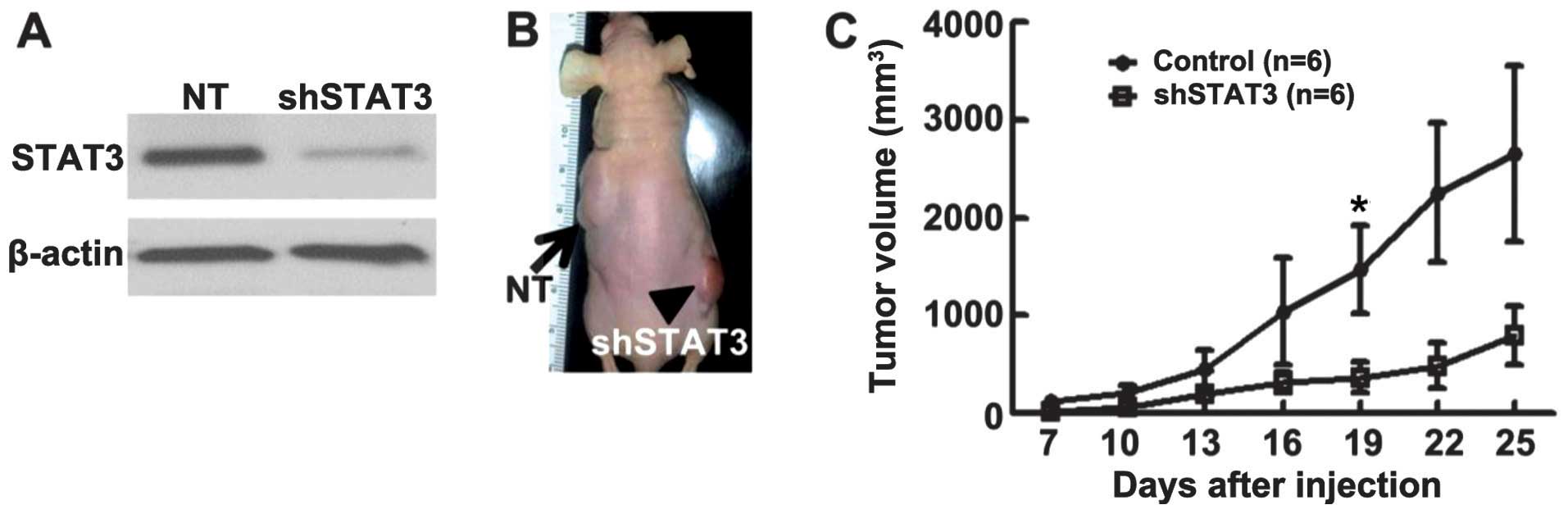
With any chemical inhibitor, there is the potential
for lack of specificity. Therefore, to confirm the role of STAT3 in
OS formation, 143.98.2 OS cells were infected with a lentivirus
encoding shRNA targeting STAT3. A non-targeting shRNA with the same
backbone served as a control. Western blot analysis confirmed that
the STAT3 level was decreased following infection of OS cells with
this vector (Fig. 4A). MTS assay
of shSTAT3 expressing cells revealed decreased numbers of viable
cells, but in contrast to cells exposed to JAK/STAT3 inhibitors,
cleaved caspase-3 was not detectable, suggesting that these shSTAT3
infected cells were not dying by apoptosis (data not shown).
sh-non-target control 143.98.2 OS cells were injected
subcutaneously into the left side of nu/nu (nude) mice, and shSTAT3
transduced 143.98.2 OS cells to the right side of nu/nu mice (n=6
for each group). Tumor volumes were measured every 3 days starting
at 7 days after transplantation. Tumor growth was detected on both
sides of the animals (Fig. 4B,
control: black arrow; shSTAT3: black arrowhead). However, the left
side, which received the non-target shRNA-expressing OS cells,
exhibited significant tumor growth compared with the right side,
which received the same number of shSTAT3-expressing OS cells
(Fig. 4C). Mice were sacrificed at
day 25, when volumes of control non-target tumors reached allowable
limits. These results confirm a key role for STAT3 in OS tumor
formation in xenografts.
Discussion
The present study demonstrated that the JAK2/STAT3
inhibitor, FLLL32, inhibited human OS cell growth in vitro
as well as delayed human OS xenograft growth in vivo.
Genetic knockdown of STAT3 by shSTAT3 significantly delayed OS
xenograft tumor formation in vivo. These data support a
vital role of the JAK2/STAT3 pathway in OS formation.
The data indicate that STAT3 activation
(phosphorylation) contributes to the survival and proliferation of
OS cells, and is crucial in the development and progression of OS
by promoting cell proliferation and protecting against apoptosis,
providing a potential promising molecular target for gene therapy
in human OSs. Similar results were detected using a STAT3 inhibitor
S31-201 (19). Current OS
chemotherapies that prolong life do not specifically target the OS
initiating cells (OS stem-like cells), such as CD271+ OS
cells. These cells possess the self-renewal, differentiation and
proliferation properties of stem cells. They also overexpress
Nanog, Oct3/4 and STAT3 and are often resistant to traditional
radiation chemotherapies (20).
The results of the present study indicated that STAT3 may be
important to the CD271+ tumor initiating cells in OS,
and blockade of the STAT3 pathway may provide a promising target
for removing these OS initiating cells that potentially contribute
to OS formation.
The partial delay in tumor growth following
treatment with FLLL32 suggests that drug combination may synergize
the therapeutic effects. The blockade of STAT3 protein signaling
can be achieved by various means, including dominant-negative
mutants, antisense methods, inhibition of upstream signaling,
phosphotyrosyl peptides, the double-stranded DNA decoy method and
RNA interference (21–24). Testing different combinations of
these treatments is required to determine an effective therapeutic
strategy.
Overall, the present data demonstrated that STAT3
appears to govern the propagation of OS initiating cells. Current
cancer therapies that prolong life do not specifically target
cancer-initiating cells, and these cells are often resistant to
traditional radiation and chemotherapies. Targeted inhibition of
STAT3 in these OS initiating cells provides a novel approach for
the treatment of OS.
References
|
1
|
Messerschmitt PJ, Garcia RM, Abdul-Karim
FW, Greenfield EM and Getty PJ: Osteosarcoma. J Am Acad Orthop
Surg. 17:515–527. 2009.PubMed/NCBI
|
|
2
|
Jaffe N: Osteosarcoma: review of the past,
impact on the future. The American experience. Cancer Treat Res.
152:239–262. 2009.
|
|
3
|
Federman N, Bernthal N, Eilber FC and Tap
WD: The multidisciplinary management of osteosarcoma. Curr Treat
Options Oncol. 10:82–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janeway KA and Grier HE: Sequelae of
osteosarcoma medical therapy: a review of rare acute toxicities and
late effects. Lancet Oncol. 11:670–678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scotlandi K, Picci P and Kovar H: Targeted
therapies in bone sarcomas. Curr Cancer Drug Targets. 9:843–853.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O’Day K and Gorlick R: Novel therapeutic
agents for osteosarcoma. Expert Rev Anticancer Ther. 9:511–523.
2009. View
Article : Google Scholar
|
|
7
|
Whelan J, Patterson D, Perisoglou M, et
al: The role of interferons in the treatment of osteosarcoma.
Pediatr Blood Cancer. 54:350–354. 2010. View Article : Google Scholar
|
|
8
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murase M, Kano M, Tsukahara T, et al: Side
population cells have the characteristics of cancer stem-like
cells/cancer-initiating cells in bone sarcomas. Br J Cancer.
101:1425–1432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeda K, Noguchi K, Shi W, et al:
Targeted disruption of the mouse Stat3 gene leads to early
embryonic lethality. Proc Natl Acad Sci USA. 94:3801–3804. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
et al: Stat3 as an oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang YC, Zheng LH, Ma BA, et al: Clinical
value of signal transducers and activators of transcription 3
(STAT3) gene expression in human osteosarcoma. Acta Histochem.
113:402–408. 2011. View Article : Google Scholar
|
|
13
|
Fossey SL, Liao AT, McCleese JK, et al:
Characterization of STAT3 activation and expression in canine and
human osteosarcoma. BMC Cancer. 9:812009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gibbs CP, Kukekov VG, Reith JD, et al:
Stem-like cells in bone sarcomas: implications for tumorigenesis.
Neoplasia. 7:967–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang YC, Zheng LH, Ma BA, et al: Clinical
value of signal transducers and activators of transcription 3
(STAT3) gene expression in human osteosarcoma. Acta Histochem.
113:402–408. 2011. View Article : Google Scholar
|
|
16
|
Ryu K, Choy E, Yang C, et al: Activation
of signal transducer and activator of transcription 3 (Stat3)
pathway in osteosarcoma cells and overexpression of
phosphorylated-Stat3 correlates with poor prognosis. J Orthop Res.
28:971–978. 2010.PubMed/NCBI
|
|
17
|
Lin L, Hutzen B, Zuo M, et al: Novel STAT3
phosphorylation inhibitors exhibit potent growth-suppressive
activity in pancreatic and breast cancer cells. Cancer Res.
70:2445–2454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller SJ, Rangwala F, Williams J, et al:
Large-scale molecular comparison of human schwann cells to
malignant peripheral nerve sheath tumor cell lines and tissues.
Cancer Res. 66:2584–2591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Goldstein D, Crowe PJ and Yang JL:
Impact of STAT3 inhibition on survival of osteosarcoma cell lines.
Anticancer Res. 34:6537–6545. 2014.PubMed/NCBI
|
|
20
|
Tian J, Li X, Si M, Liu T and Li J:
CD271+ osteosarcoma cells display stem-like properties.
PLoS One. 9:e985492014. View Article : Google Scholar
|
|
21
|
Gartel AL and Kandel ES: RNA interference
in cancer. Biomol Eng. 23:17–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hokaiwado N, Takeshita F, Banas A and
Ochiya T: RNAi-based drug discovery and its application to
therapeutics. IDrugs. 11:274–278. 2008.PubMed/NCBI
|
|
23
|
Krishnamachary B, Glunde K, Wildes F, et
al: Noninvasive detection of lentiviral-mediated choline kinase
targeting in a human breast cancer xenograft. Cancer Res.
69:3464–3471. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Piao YF, Jiang Z, et al: Silencing
of signal transducer and activator of transcription 3 expression by
RNA interference suppresses growth of human hepatocellular
carcinoma in tumor-bearing nude mice. World J Gastroenterol.
15:2602–2608. 2009. View Article : Google Scholar : PubMed/NCBI
|