Introduction
Asthma is a chronic inflammatory disorder of the
airways, and is characterized by reversible bronchoconstriction,
airway hyper-responsiveness and airway remodeling (1). Various cells that migrate from the
bloodstream to the bronchial tree are sources of local
inflammation. Numerous eosinophils, T lymphocytes and
polymorphonuclear cells (PMNs) infiltrate peribronchial tissues in
asthmatics, introducing into the lungs an increased capacity to
generate proinflammatory mediators, cytokines and chemokines,
including leukotriene (LT) B4, cysteinyl leukotrienes (CysLTs),
T-helper lymphocyte 2 (Th2) cytokines and interleukins (ILs)
(2,3). IL-1β has a pivotal role in the
progression of allergic airway inflammation in asthma (3,4).
Leukotrienes (LTs) are lipids synthesized from arachidonic acids by
mast cells, eosinophils and alveolar macrophages in the lungs, and
have been considered to have an important role in the pathogenesis
of asthma (2). LTB4 acts as a
chemoattractant pro-adhesive agent and secretagogue for PMNs,
eosinophils and T lymphocytes (4,5).
CysLTs cause bronchoconstriction, which contributes to the
responses to an inhaled allergen challenge (6). In contrast to proinflammatory LTs,
lipoxins, a separate class of eicosanoids that are distinct in
structure and function from CysLTs and LTB4, act as potent
anti-inflammatory agents (7).
Lipoxin A4 (LXA4) and its analogs inhibit LTB4-induced PMN and
eosinophil chemotaxis (8). A
previous study by our group also demonstrated that LXA4 suppressed
the production of LTB4 in PMNs (9). In addition, LXA4 and its analogs
blocked CysLTs-triggered airway obstruction and hypersecretion of
CysLTs in bronchoalveolar lavage fluids (BALF) obtained from
asthmatic mice (10,11). Furthermore, LXA4 analogs inhibited
airway hyper-responsiveness and pulmonary inflammation in murine
models of asthma, as shown by decreased leukocytes and mediators in
BALF, including IL-5, IL-13, eotaxin, prostanoids and CysLTs
(11). Moreover, transgenic
overexpression of LXA4 receptor in leukocytes obtained from
asthmatic mice led to a significant inhibition of pulmonary
inflammation and eosinophil infiltration in the lungs (11). In addition, LXA4 repressed the
levels of IL-8 released from peripheral blood mononuclear cells
obtained from patients with asthma (12). To date, the signaling pathways
involved in LXA4 action in asthmatic models remain to be fully
elucidated. A previous study by our group showed that LXA4
abrogated the synthesis of IL-1β, IL-6 and IL-8 induced by
lipopolysaccharide (LPS) via downregulation of nuclear factor
(NF)-κB pathway-dependent signal transduction in pulmonary
microvascular endothelial cells (13). LPS activates NF-κB through
toll-like receptor (TLR) 4 and mycloid differentiation factor 88
(MyD88) in endothelial cells (14). In this regard, increasing evidence
suggested that toll-like receptors (TLRs) may be potential
mediators or modulators of inflammation within the lungs (15–17).
TLRs are the best characterized class of pattern recognition
receptors of the innate immune system, and trigger anti-microbial
host defense responses (18).
Various cells, including leukocytes and airway epithelial cells,
express TLRs (16). Signaling
through the TLRs leads to transcription and translation of a
variety of cytokines and mediators (16). However, previous studies
investigating the role of TLRs in asthma have been inconclusive.
For example, Redecke et al (17) demonstrated that activation of TLR2
induced a Th2 immune response and promoted experimental asthma.
Conversely, Velasco et al (19) reported that TLR4 and TLR2 agonists
decreased allergic inflammation. Therefore, the present study was
designed to examine the changes in the TLR2/MyD88/NF-κB signaling
pathway in asthmatic mice, and also to investigated whether the
TLR2/MyD88/NF-κB signaling pathway is involved in the inhibitory
effects of LXA4 on pulmonary inflammation in asthmatic mice, and to
determine whether IL-1β modulates the changes in the
TLR2/MyD88/NF-κB signaling pathway in asthmatic mice.
LXA4 action is mediated by the LXA4 receptor (ALX)
expressed on the membrane of various cell types, including airway
epithelial cells and leukocytes, and ALX can be upregulated by
specific inflammatory mediators (7). Allergen sensitization and challenge
with ovalbumin (OVA) increases ALX expression in infiltrating
leukocytes and airway epithelial cells in the lungs of asthmatic
mice (11). Following stimulation
by mediators, LXA4 is rapidly generated at sites of inflammation,
acts locally and is then rapidly inactivated by metabolic enzymes
(7). Thus, the use of LXA4 may not
be suitable for in vivo experiments. Instead, stable analogs
of LXA4 and LXA4 receptor agonist, including BML-111 and CGEN-855A,
were used for in vivo experiments (10,11,20–22).
Accordingly, the present study used BML-111, a potent ALX agonist
with an inhibitory activity on LTB4-induced PMN chemotaxis similar
to that of LXA4 (21), was used in
the in vivo experiment.
Materials and methods
Animals
Male BALB/c mice weighing 19–21 g were obtained from
the Laboratory Animal Center of Nanjing First Hospital (Nanjing,
China), and quarantined for one week prior to the experiment and
bled to establish that they were virus free. The mice were housed
in the animal facility that was maintained at 22–24°C with a 12-h
dark/light cycle, and fed with commercial pelleted mouse food and
water ad libitum under specific pathogen-free conditions.
The present study was performed in strict accordance with the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. The protocol was
approved by the Committee on the Ethics of Animal Experiments of
Nanjing First Hospital affiliated to Nanjing Medical University
(permit number, 2013-6135). All surgical procedures were performed
under sodium pentobarbital (Sigma-Aldrich, St. Louis, MO, USA)
anesthesia, and all efforts were made to minimize suffering.
Induction of asthmatic models
The mice were randomly divided into six groups,
i.e., normal controls (NC), asthmatic mice (AM), BML-111-treated
asthmatic mice (BAM), vehicle (0.1 ml of ethanol) of
BML-111-treated asthmatic mice (VAM), anti-IL-1β antibody-treated
asthmatic mice (AAM) and rabbit immunoglobulin (Ig)G-treated
asthmatic mice (RAM). Each group consisted of 10 mice, and 5 mice
were used for BALF collection, another 5 mice were used for blood
collection and pathologic studies. For induction of asthmatic
models, BALB/c mice were sensitized with 10 μg of OVA
(Sigma-Aldrich) emulsified in 20 mg aluminum hydroxide
(Sigma-Aldrich) as adjuvant in a total volume of 0.1 ml of normal
saline by intraperitoneal injection at days 0, 7 and 14.
Subsequently, the OVA challenges were given once a day at days 25,
27, 29, 31 and 33 with 1% aerosolized OVA in saline for 30 min
using an atomizer (PARI BOY N085; PARI GmbH, Starnberg, Germany).
The mice were sacrificed 24 h after the final OVA challenge by
cervical dislocation. The NC mice were sensitized and challenged by
using the same protocol but using saline instead of OVA. The mice
from the AM, VAM and RAM groups but not those from the NC, BAM or
AAM groups, had the asthmatic symptoms, including dysphoria, short
breath, tachypnea and apathism from days 31–33.
BML-111 treatment of asthmatic mice
BML-111 (5S,6R, 7-trihydroxyheptanoic
acid methyl ester; Cayman Chemicals, Ann Arbor, MI, USA) was
diluted in ethanol (Sigma-Aldrich) at a concentration of 200
μg/ml. Following the final OVA sensitization, the BAM mouse
was intraperitoneally injected with 0.1 ml BML-111 at a dose of 1
μg/g body weight (or 0.1 ml ethanol for the VAM mouse) 30
min prior to the OVA challenges at days 25, 27 and 29. Twenty-four
hours following the final OVA challenge, the mouse was anesthetized
by an intraperitoneal injection of 0.6 mg of sodium pentobarbital.
Subsequently, the blood and BALF were collected and stored at −20°C
until use. The lungs were removed and excised. The low lobes of the
left lungs were processed for western blot analysis and
electrophoretic mobility shift assay (EMSA), while the low lobes of
the right lungs were fixed with 4% paraformaldehyde for 24 h and
then stained for pathological examination.
Anti-IL-1β antibody treatments of
asthmatic mice
Rabbit anti-mouse IL-1β antibody (Thermo Fisher
Scientific, Waltham, MA, USA) was diluted in sterile water. After
the final OVA sensitization, mice in the AAM group were
subcutaneously injected with 1 μg/g body weight of
anti-IL-1β antibody in a total volume of 0.1 ml sterile water 30
min prior to the OVA challenges at days 25, 27, 29, 31 and 33.
Twenty-four hours after the final OVA challenge, the mice were
sacrificed and the samples were collected in the same way as
mentioned above. Mice in the RAM group were treated by using the
same protocol but using rabbit immunoglobulin (Ig)G (1 μg/g,
Santa Cruz Bio Biotechnology, Inc., Dallas, TX, USA) instead of
anti-IL-1β antibody.
ELISA of IL-1β, IL-4, IL-8 and
interferon-γ (IFN-γ)
Serum levels of IL-1β, IL-4, IL-8 and IFN-γ were
measured using ELISA kits (USCN Life Science, Wuhan, China)
following the manufacturer’s instructions. The limits of detection
for IL-1β, IL-4, IL-8 and IFN-γ by the ELISA were 1 pg/ml.
Leukocyte counts and ELISA of BALF
Bilateral BALF was obtained by four injections of 1
ml saline through a tracheal cannula into the lungs and four
repeats of bronchial lavage. The BALF, obtained from each mouse at
an average of 3.5 ml, was centrifuged and resuspended as two
aliquots of 1 ml of phosphate-buffered saline (PBS; Sigma-Aldrich)
plus 0.6 mM EDTA (Sigma-Aldrich). Subsequently, the cells in BALF
were enumerated and identified following Wright-Giemsa
(Sigma-Aldrich) staining with a hemocytometer, and the leukocytes
were differentiated into eosinophils, neutrophils and lymphocytes
according to standard cellular morphology and staining
characteristics. The leukocytes were counted by two independent
investigators in a single-blinded study, which analyzed at least
200 leukocytes for each mouse from four different random locations
using a microscope (CX22; Olympus Corporation, Tokyo, Japan). The
levels of IL-1β, IL-4, IL-8 and OVA-specific IgE in BALF were
determined using ELISA kits (for ILs, USCN Life Science, Wuhan,
China; for OVA-IgE, Shibayagi, Gunma, Japan) according to the
manufacturer’s instructions.
Lung tissue pathological studies
The lung sections were stained with hematoxylin and
eosin (Sigma-Aldrich) and observed using light microscopy.
Assessment of grades of bronchial and peribronchial inflammation
was based on a six-scale score as previously described (23). Expressions of TLR2 and
phosphorylated NF-κB p65 were determined using immunohistochemical
staining. Briefly, the sections of lungs were deparaffinized and
treated with hydrogen peroxide to block endogenous peroxidase
activity. Sections were incubated with either rabbit anti-mouse
polyclonal TLR2 antibody (cat. no. sc-10739) or rabbit anti-mouse
polyclonal phosphorylated NF-κB p65 (Ser 276) antibody (cat. no.
sc-109; Santa Cruz Biotechnology, Inc.) at 1:100 dilution.
Subsequently, biotinylated goat anti-rabbit IgG antibody (Vector
Labs, Burlingame, CA, USA) was applied. The sections were exposed
to avidin-biotinylated horseradish peroxidase (HRP) and
diaminobenzidine tetrahydrochloride (Vector Labs). Hematoxylin
staining was used for counterstaining. The mean ratios of TLR2- and
NF-κB-positive cellular areas (deep brown) in five fields of view
per section from each mouse were assessed by a JD-801
computer-aided image analyzer (Jeda Co., Nanjing, China) under high
power magnification (x200; Olympus CX22).
Western blot analysis of TLR2 and
MyD88
The lung tissues were homogenized in lysis buffer
with protease inhibitors. Proteins in the lysates were extracted
using Protein Extraction kits (Active Motif, Carlsbad, CA, USA)
according to the manufacturer’s instructions. After total protein
was measured using the Bradford method, 10 μl lysate was
purified using 10% SDS-PAGE for 4 h prior to blotting onto
polyvinylidene difluoride membranes (Amersham, Arlington, IL, USA).
Non-specific sites on the membranes were blocked with 5% non-fat
milk (Sigma-Aldrich). The blots were incubated with primary rabbit
anti-mouse polyclonal antibodies against TLR2 (cat. no. sc-10739),
MyD88 (cat. no. sc-11356) or β-actin (cat. no. sc-130656; Santa
Cruz Biotechnology, Inc.) at 1:2,000 dilution overnight followed by
incubation for 2 h with an HRP-conjugated goat anti-rabbit IgG
antibody (Jackson, West Grove, PA, USA) at 1:5,000 dilution.
Following washing, the membranes were incubated with an enhanced
chemiluminance reagent system (Amersham) and then exposed to Kodak
Biomax films (Eastman Kodak, Rochester, NY, USA). Semiquantitative
analysis was performed using UVP-gel densitometry (UVP Co., San
Gabriel, CA, USA). Arbitrary unit =
(ATLR2/Aβ-actin) ×100%, or
(AMyD88/Aβ-actin) ×100%.
EMSA of NF-κB activation
Nuclear proteins were extracted using a Nuclear
Protein Extraction kit (Active Motif). EMSA was performed using a
Gel Shift Assay kit (Promega, Madison, WI, USA) following the
manufacturer’s instructions. The nuclear extracts containing 30
μg of total protein were pre-incubated with gel shift
binding buffer for 10 min, followed by the addition of
γ-(32P)-labeled double-stranded oligonucleotide probes
of NF-κB (Santa Cruz Biotechnology, Inc.) and further incubated for
20 min in a binding reaction mixture. The oligonucleotide pairs for
NF-κB were 5′-AGTTGAGGGGACTTTCCCAGGC-3′ and
5′-GCCTGGGAAAGTCCCCTCAACT-3′. Formed nuclear protein-DNA complexes
were dissolved in 4% non-denaturing polyacrylamide gels.
Electrophoresis was performed under 90 V for 2 h. The gels were
dried and exposed to Kodak X-ray films (Eastman Kodak) at −70°C for
36 h. Semiquantitative analysis was performed using UVP-gel
densitometry. Arbitrary unit = (ANC or AAM or
ABAM or AVAM or AAAM/Aunlabeled
probes) ×100%. To assess the specificity of the reaction,
competition assays were performed with 100-fold excess of unlabeled
consensus oligonucleotide pairs of NF-κB, which were added to the
binding reaction mixture 10 min prior to the addition of the
labeled probes.
ELISA of IL-4, IL-6 and IL-8 released
from leukocytes
A whole blood sample was obtained from an NC mouse
and drawn into 5-ml tubes containing heparin (Sigma-Aldrich).
Leukocytes were separated from the blood as previously described
(9). Following washing with PBS,
the leukocytes were resuspended and adjusted to a density of
2×106 cells/ml in RPMI-1640 supplemented with 0.5% FCS
and antibiotic-anti-mycotic solution (Gibco-BRL, Invitrogen Life
Technologies, Inc., Carlsbad, CA, USA). The cells were then seeded
into a 24-well plate in 1 ml medium and cultured at 37°C in a 5%
CO2 incubator. Subsequently, the cells were stimulated
with IL-1β (10 ng/ml; Sigma-Aldrich) with or without pretreatment
with BML-111 (1 mM) for 30 min, N-t-Boc-Phe-Leu-Phe-Leu-Phe
(BOC1, an antagonist of G protein-coupled LXA4 receptor; 100
μM; Calbiochem, San Diego, CA, USA) for 30 min, goat
anti-mouse TLR2-neutralizing polyclonal antibody (TLR2Ab; 1
μg/ml; Santa Cruz Biotechnology, Inc.) for 1 h, goat IgG (1
μg/ml; Santa Cruz Biotechnology, Inc.) for 1 h, ST2825 (a
MyD88 dimerization inhibitor; 20 μM; MedChem Express,
Princeton, NJ, USA) for 1 h, and thiophene-3-car-boxamide 1
(TPCA-1; an inhibitor of IκB kinase β; 10 μM; Sigma-Aldrich)
for 1 h. Following co-incubation for 24 h, the cellular
supernatants were collected for measurement of IL-4, IL-6 and IL-8
proteins using ELISA kits (USCN Life Science) following the
manufacturer’s instructions. The cell viability was measured by
trypan blue exclusion assay (Sigma-Aldrich) in pilot experiments
and the percentage of viable cells was >95% following exposure
to the abovementioned mediators.
Western blot analysis of TLR2 and MyD88
in leukocytes
The leukocytes were treated with the mediators in
the same way as mentioned above. Following co-incubation for 30
min, the cells were collected, total protein in the lysates was
extracted, and the expression of TLR2, MyD88 and β-actin were
assessed with using western blot analysis in a similar way to the
abovementioned procedure.
Determination of NF-κB activation in
leukocytes
The leukocytes were treated with the mediators in
the same way as mentioned above. Following co-incubation for 1 h,
the cytosolic or nuclear extracts were obtained using NE-PER
Nuclear and Cytoplasmic Extraction reagents (Fisher Thermo
Scientific) in the presence of protease inhibitors and phosphatase
inhibitor (Roche Diagnostics, Indianapolis, IN, USA). Denatured
proteins were purified using 10% SDS-PAGE (Amersham). Following
electrophoresis, separated proteins were transferred to
polyvinylidene difluoride membranes (Amersham). Nonspecific sites
were blocked with 5% nonfat milk for 2 h. Subsequently, the
cytosolic protein blots were incubated with rabbit anti-mouse NF-κB
p65 or IκBα or anti-GAPDH antibodies (Cell Signaling Technology,
Beverly, MA, USA), and the nuclear protein blots were incubated
with rabbit anti-mouse phosphorylated NF-κB p65 (Ser 276) antibody
(Santa Cruz Biotechnology, Inc.) or rabbit anti-mouse histone 3
antibody (Cell Signaling Technology) overnight at 4°C. The blots
were then incubated for 2 h with an HRP-conjugated goat anti-rabbit
IgG antibody (Jackson, West Grove, PA, USA) at 1:5,000 dilution.
Following washing, the membranes were incubated with an enhanced
chemilu-minance reagent system (Amersham) and then exposed to Kodak
Biomax films (Eastman Kodak). Semiquantitative analysis was
performed by using UVP-gel densitometry (UVP, Co.). Arbitrary unit
= (AIκBα/AGAPDH) ×100%, or (Acytosolic
p65/AGAPDH) ×100%, or (Anuclear
p65/Ahistone 3) ×100%.
Statistical Analysis
Values are expressed as the mean ± standard
deviation (SD). Experimental values are analyzed using one-way
analysis of variance followed by F and q tests. The degree of
peribronchial inflammation was analyzed using the χ2
test. All analyses were performed using the Statistical Package for
Social Sciences version 16.0 (SPSS, Inc., Chicago, IL, USA).
Differences between values were considered to be statistically
significant when P<0.05.
Results
Serum IL-1β, IL-4, IL-8 and IFN-γ
levels
As shown in Fig. 1,
OVA immunization upregulated the serum levels of IL-1β, IL-4 and
IL-8, downregulated the serum levels of IFN-γ, and therefore
increased serum IL-4/IFN-γ ratios in mice. However, treatment of
asthmatic mice with BML-111 partially but significantly abrogated
the abovementioned changes induced by OVA. Treatment with
anti-IL-1β antibody significantly reduced the serum levels of
IL-1β, IL-4 and IL-8, and augmented the serum levels of IFN-γ
reduced by OVA immunization.
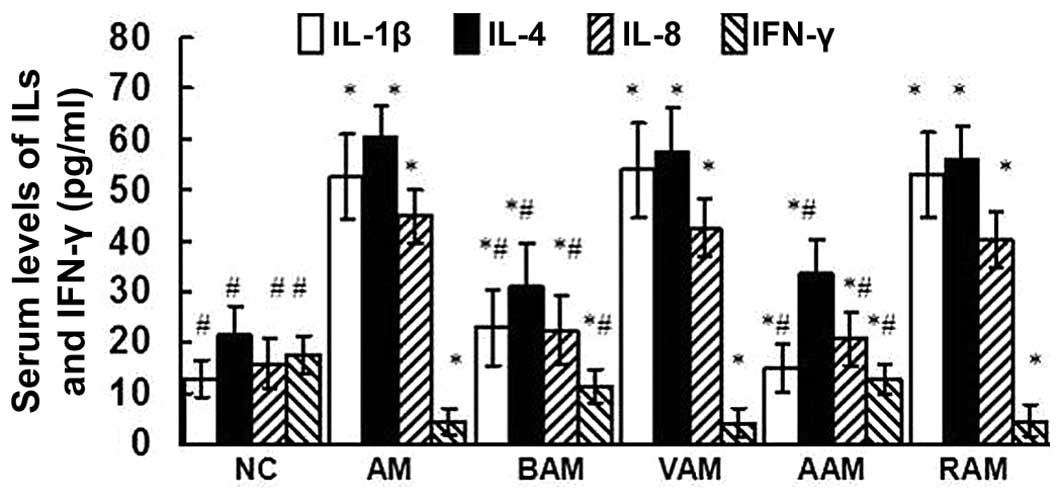 | Figure 1Serum levels of IL-1β, IL-4, IL-8 and
IFN-γ assessed using ELISA. Values are expressed as mean ± standard
deviation of five mice in each group. *P<0.05, as
compared with the levels of the same cytokine in serum obtained
from NC mice. #P<0.05, as compared with the levels of
the same cytokine in serum obtained from AM mice. NC, normal
control; AM, ovalbumin immunization-induced asthmatic mice; BAM,
BML-111-treated AM; VAM, vehicle of BML-111-treated AM; AAM,
anti-IL-1β antibody-treated AM; RAM, rabbit immunoglobulin
G-treated AM; IL, interleukin; IFN, interferon. |
Leukocyte counts, ILs and OVA-IgE in
BALF
As indicated in Fig.
2A, the number of total leukocytes, eosinophils, neutrophils
and lymphocytes increased in the BALF obtained from the AM mice
compared to those in normal controls. However, OVA immunization did
not affect the number of monocytes/macrophages compared with those
in normal controls (data not shown). Treatment with BML-111
partially but significantly suppressed the increments in the number
of total leukocytes, eosinophils, neutrophils and lymphocytes in
BALF induced by OVA immunization. Similarly, treatment with
anti-IL-1β antibody also reduced the number of total leukocytes,
eosinophils, neutrophils and lymphocytes in BALF stimulated by OVA
immunization. In addition, BML-111 treatment decreased the levels
of IL-1β, IL-4, IL-8 and OVA-IgE in BALF induced by OVA (Fig. 2B). Interestingly, treatment with
anti-IL-1β antibody also restrained the levels of IL-1β, IL-4, IL-8
and OVA-IgE in BALF induced by OVA (Fig. 2B).
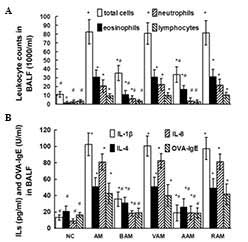 | Figure 2(A) Leukocyte counts and (B) levels
of IL-1β, IL-4, IL-8 and OVA-IgE assessed using ELISA in BALF.
Values are expressed as the mean ± standard deviation of five mice
in each group. *P<0.05, as compared with the number
of the same cells in A or levels of the same cytokine in BALF
obtained from NC mice in B. #P<0.05, as compared with
the number of the same differential cells in A or the levels of the
same cytokine in BALF obtained from AM mice in B. NC, normal
control; AM, ovalbumin immunization-induced asthmatic mice; BAM,
BML-111-treated AM; VAM, vehicle of BML-111-treated AM; AAM,
anti-IL-1β antibody-treated AM; RAM, rabbit IgG-treated AM; BALF,
bronchoalveolar lavage fluid; Ig, immunoglobulin. |
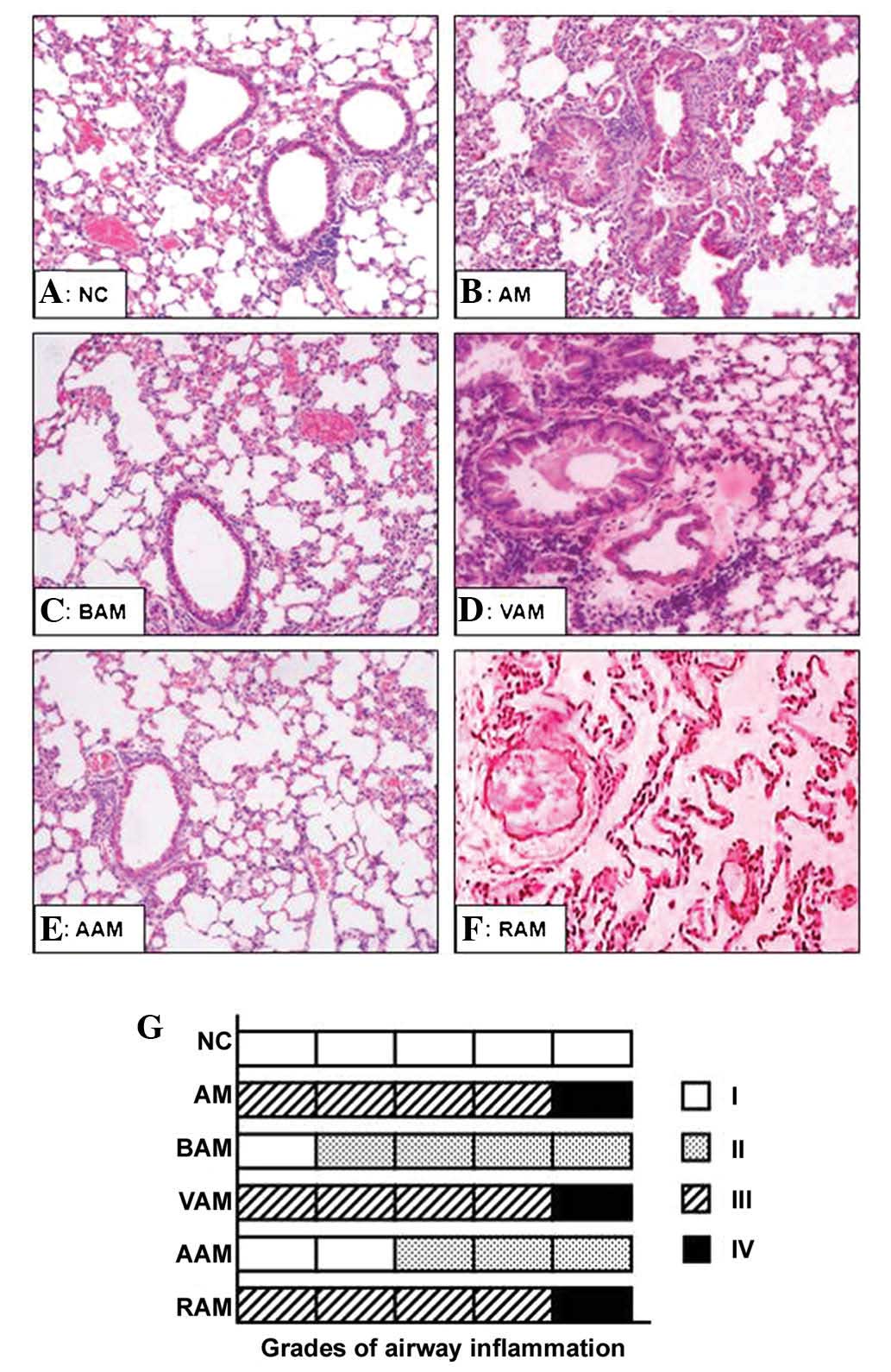
Lung tissue pathological studies
As shown in Fig. 3,
the lung sections obtained from mice in the AM, VAM and RAM groups
exhibited an obvious peribronchial and perivascular inflammatory
cell infiltration, as well as a marked cellular swelling, hydropic
degeneration, hyperplasia and necrosis in the airway epithelial
cells, and an excessive mucus in the lumen of bronchi with
thickened bronchial mucosa and smooth muscle as compared with that
in normal controls (Fig. 3B, D and
F). Treatment with BML-111- and IL-1β antibodies abolished the
inflammatory cell infiltration, airway epithelial cell swelling and
hydropic degeneration, as well as bronchial mucus hypersecretion
induced by OVA (Fig. 3C and E).
OVA immunization induced significant airway inflammation, as shown
by higher grades of the bronchial and peribronchial inflammation
scale (Fig. 3G). However,
treatment with BML-111 and anti-IL-1β antibodies significantly
inhibited OVA-induced airway inflammation (Fig. 3G; χ2=28.14;
P<0.001).
Expression of TLR2 and MyD88
As shown in Figs.
4, 5 and 6, the expression of TLR2 and MyD88
assessed using western blot analysis and immunohistochemistry in
the lungs signifi-cantly increased in the mice with AM compared
with those in the normal controls. However, these elevated
expression levels of TLR2 and MyD88 in the lungs induced by OVA
were partially but significantly suppressed by BML-111 treatment
(P<0.05). In a similar manner, treatment with anti-IL-1β
antibody also inhibited the expression of TLR2 and MyD88 induced by
OVA.
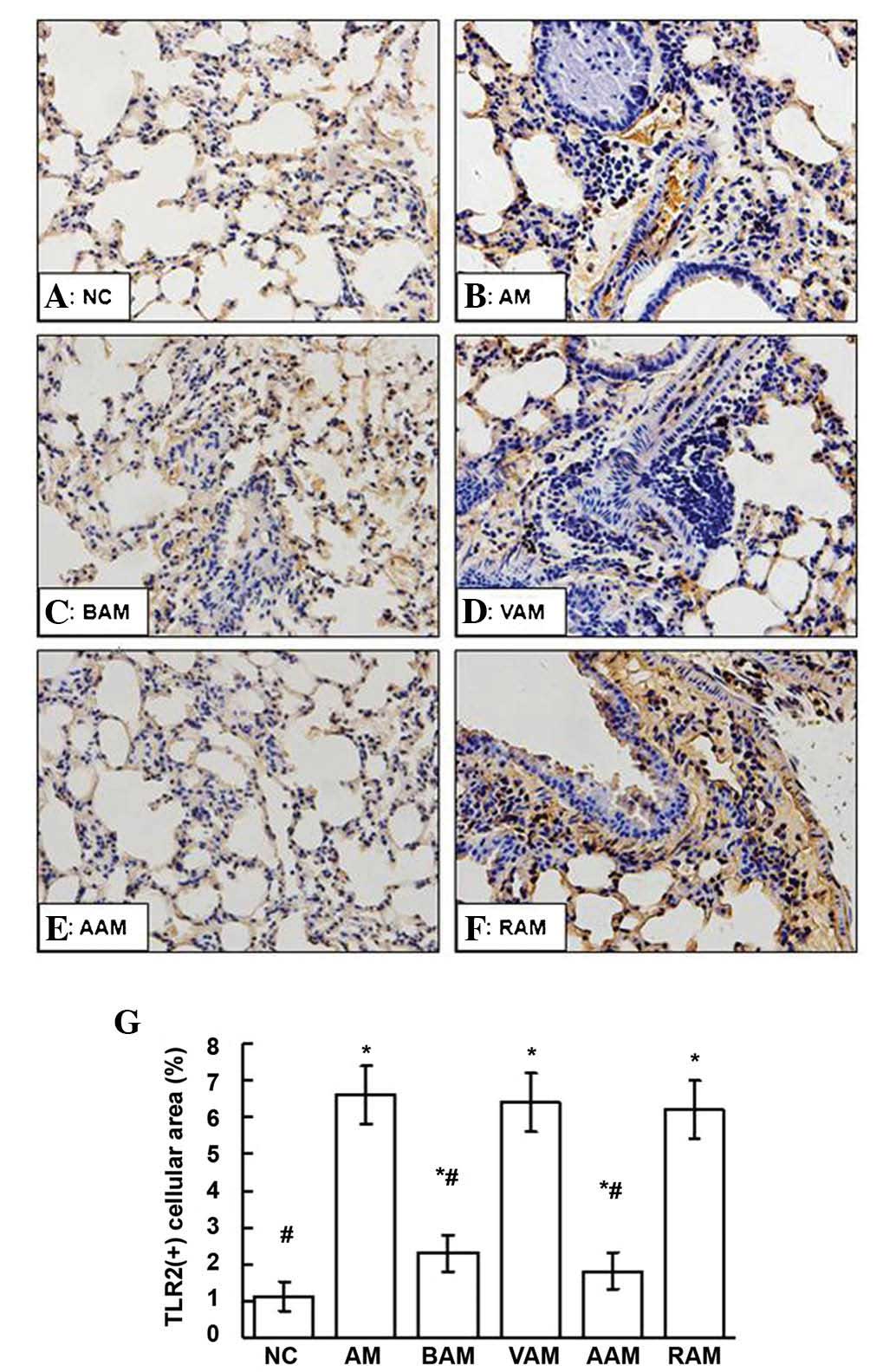 | Figure 4(A–F) Expression of TLR2 in lung
tissue from the mice in each group was assessed using
immunohistochemistry (deep brown cellular area indicates positive
staining; magnification, x200). (G) Quantification of TLR2-positive
staining in five fields of view per section from each mouse was
assessed by JD-801 computer-aided image analyzer under high-power
magnification (x200). Values are expressed as the mean ± standard
deviation of five mice in each group. *P<0.05, as
compared with the NC group. #P<0.05, as compared with
the AM group. NC, normal control; AM, ovalbumin
immunization-induced asthmatic mice; BAM, BML-111-treated AM; VAM,
vehicle of BML-111-treated AM; AAM, anti-IL-1β antibody-treated AM;
RAM, rabbit IgG-treated AM; TLR, of toll-like receptor. |
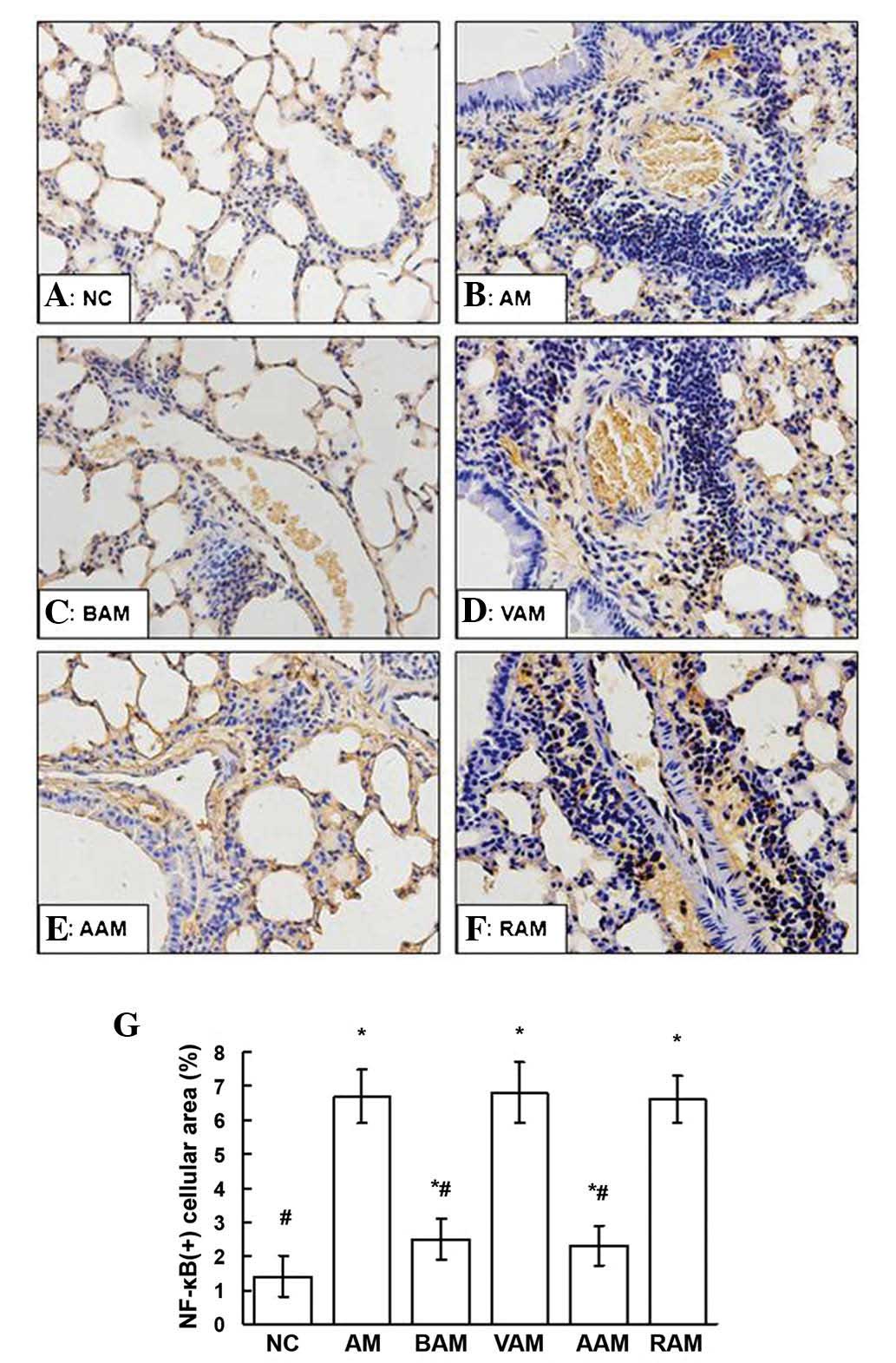 | Figure 5(A–F) Expression of NF-κB p65 in lung
tissue assessed using immunohistochemistry in the mice in each
group (deep brown cellular area indicates positive staining;
magnification, x200). (G) The mean ratio of the NF-κB-positive
cellular area in five fields of view per section of each mouse was
assessed by JD-801 computer-aided image analyzer under high-power
magnification (x200). Values are expressed as the mean ± standard
deviation of five mice in each group. *P<0.05, as
compared with the NC group. #P<0.05, as compared with
the AM group. NC, normal controls; AM, ovalbumin
immunization-induced asthmatic mice; BAM, BML-111-treated AM; VAM,
vehicle of BML-111-treated AM; AAM, anti-IL-1β antibody-treated AM;
RAM, rabbit immunoglob-ulin G-treated AM; NF-κB, nuclear
factor-κB. |
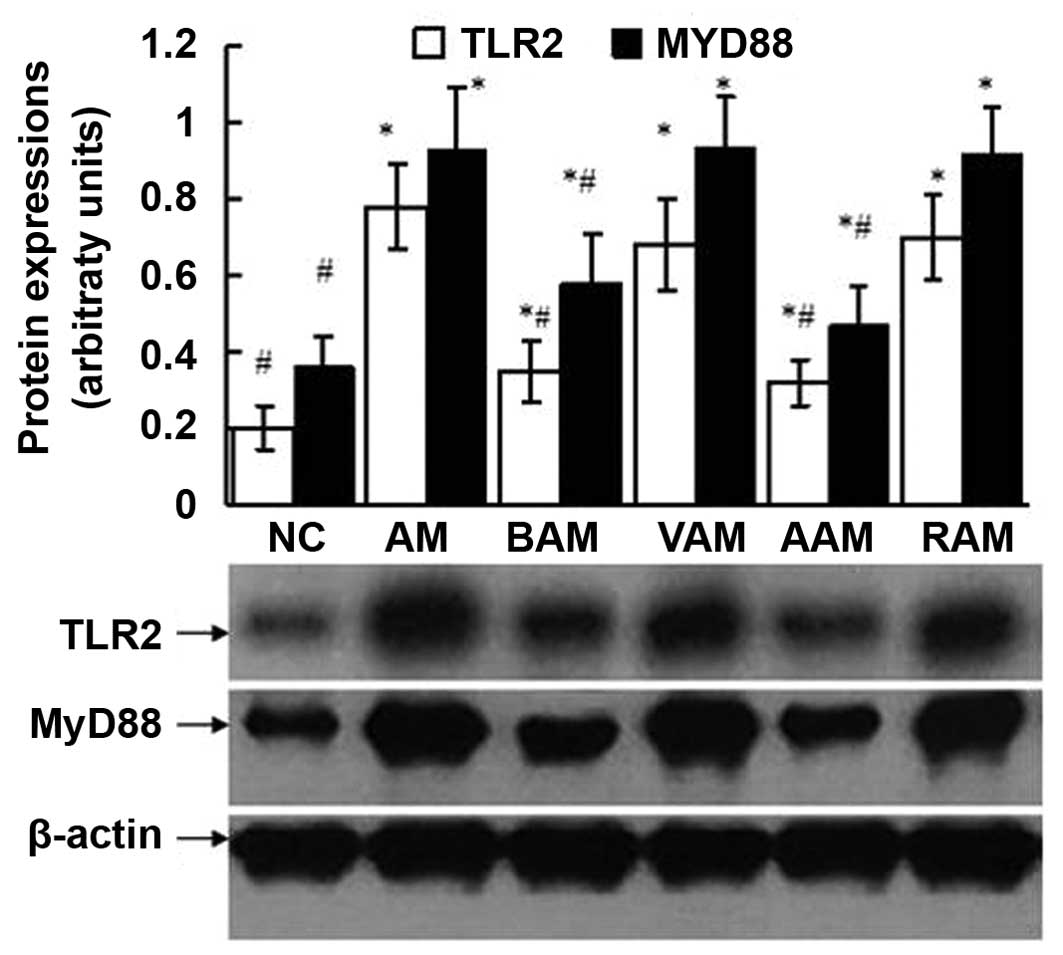 | Figure 6Expressions of TLR2 and MyD88
assessed using western blot analysis in lung tissue obtained from
the mice in each group. The western blot shown is a representative
of five independent experiments, and β-actin protein served as a
loading control. Semiquantitative analysis was performed by using
UVP-gel densitometry. Arbitrary unit =
(ATLR2/Aβ-actin) × 100%, or
(AMyD88/Aβ-actin) ×100%. Values are expressed
as mean ± standard deviation of five mice in each group.
*P<0.05, as compared with the arbitrary units of the
same protein in the NC group. #P<0.05, as compared
with the arbitrary units of the same protein in the AM group. NC,
normal control; AM, ovalbumin immunization-induced asthmatic mice;
BAM, BML-111-treated AM; VAM, vehicle of BML-111-treated AM; AAM,
anti-IL-1β antibody-treated AM; RAM, rabbit immunoglobulin
G-treated AM; MyD88, mycloid differentiation factor 88; TLR,
toll-like receptor. |
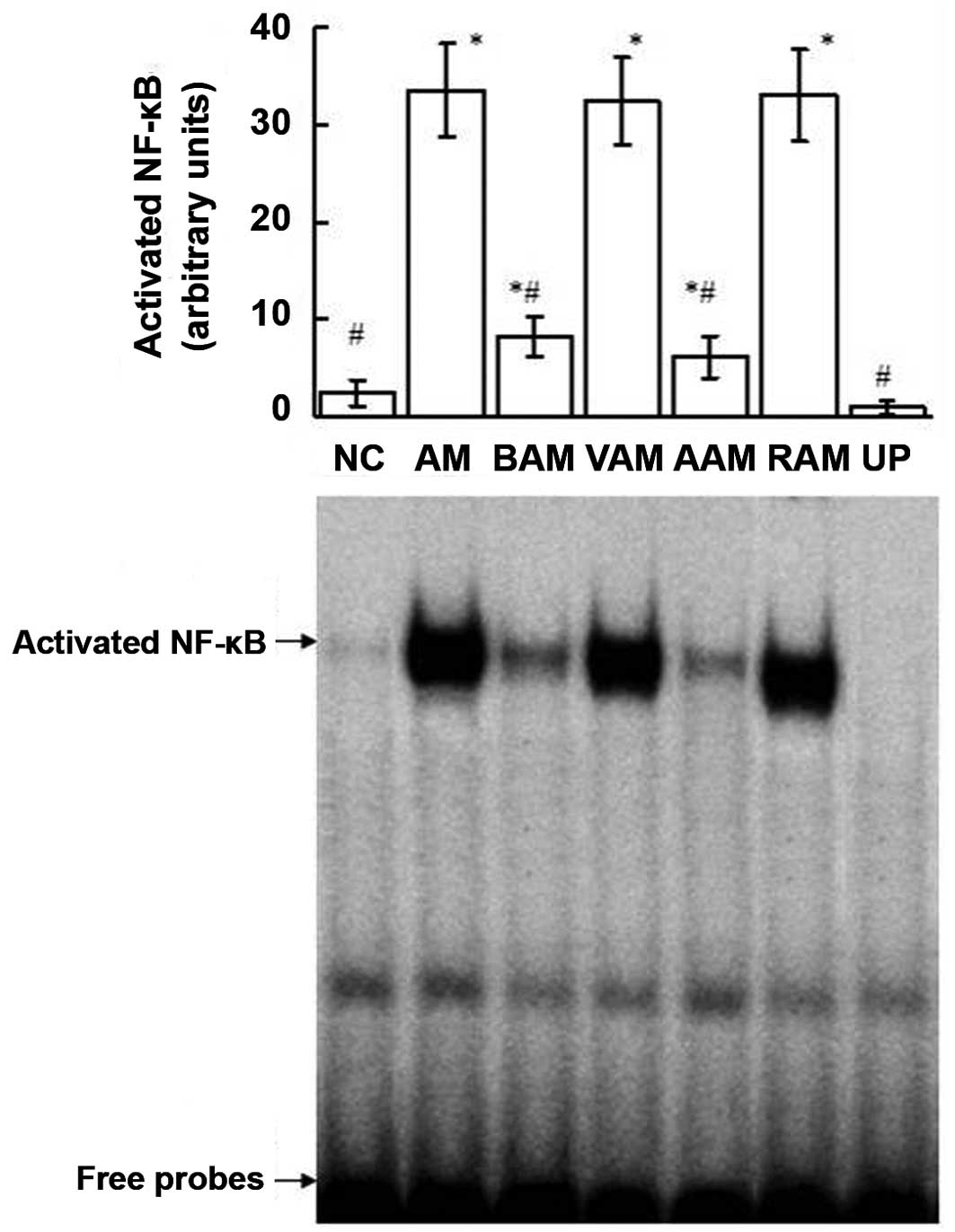
Activity of NF-κB
As indicated in Fig.
7, OVA immunization upregulated the DNA-binding activities of
NF-κB in the lungs. However, treatment with BML-111 significantly
inhibited the activity of NF-κB induced by OVA. Additionally,
treatment with anti-IL-1β antibody also suppressed the OVA-induced
activity of NF-κB. The competition assay performed with unlabeled
probes demonstrated the specificity of the DNA-binding activities
of NF-κB detected using EMSA.
Effects of BOC1, TLR2Ab, ST2825 and
TPCA-1 on ILs released from leukocytes
As presented in Fig.
8, the levels of IL-4, IL-6 and IL-8 released from leukocytes
increased following exposure of the cells to IL-1β. However, these
enhanced IL levels were significantly diminished by BML-111
pre-treatment. These inhibitory effects of BML-111 were mediated by
LXA4 receptor, as pre-treatment with BOC1 abolished the inhibitory
effects of BML-111 on the IL hypersecretion induced by IL-1β. The
enhanced levels of the ILs induced by IL-1β were also blocked by
pre-treatment of the cells with TLR2Ab, ST2825 and TPCA-1.
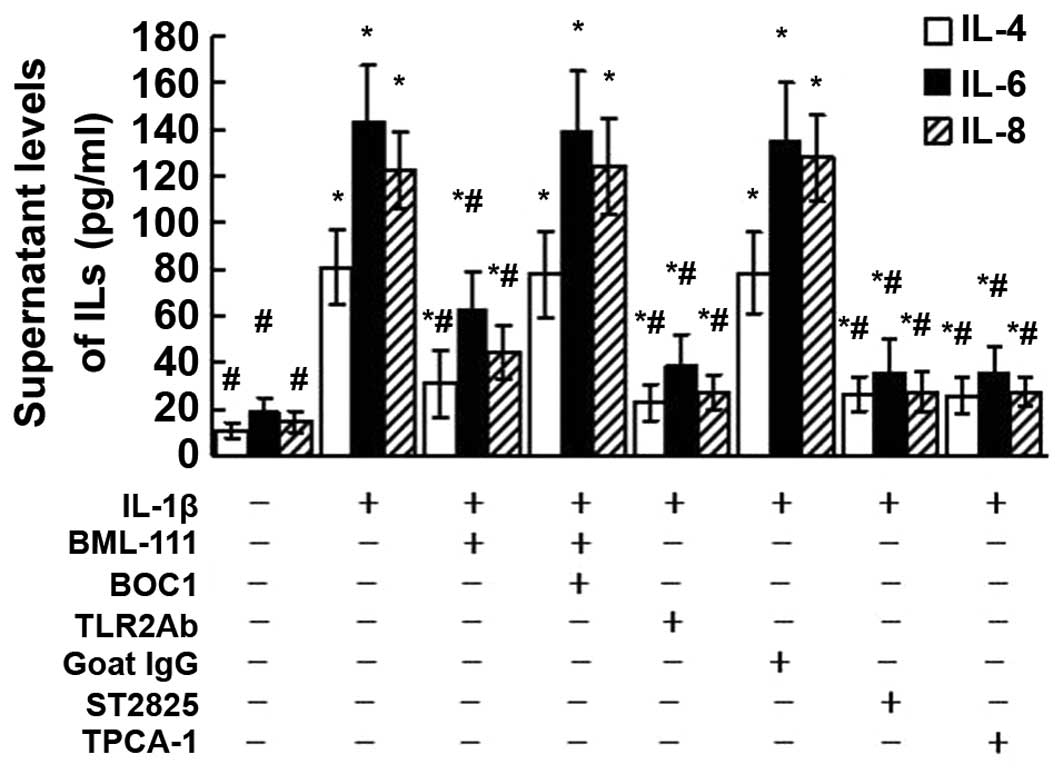 | Figure 8Supernatant levels of IL-4, IL-6 and
IL-8 released from leukocytes exposed to IL-1β were assessed using
ELISA. The cultured leukocytes were obtained from a mouse from the
normal control group and stimulated with IL-1β (10 ng/ml) for 24 h
with or without pre-treatment of BML-111 (1 mM) for 30 min,
antagonist of G protein-coupled LXA4 receptor BOC1 (100 μM)
for 30 min, TLR2Ab (1 μg/ml) for 1 h, goat IgG (1
μg/ml) for 1 h, MyD88 dimerization inhibitor ST2825 (20
μM) for 1 h and TPCA-1 (10 μM) for 1 h. Values are
expressed as the mean ± standard deviation of five independent
experiments. *P<0.05, as compared to the levels of
same cytokine released from the cells without treatment.
#P<0.05, as compared to the levels of same cytokine
released from the cells treated with IL-1β alone. IL, interleukin;
IgG, immunoglobulin G; TLR, toll-like receptor; TPCA-1,10,
thiophene-3-carboxamide 1; TLR2Ab, toll-like receptor
2-neutralizing antibody; BOC1,
N-t-Boc-Phe-Leu-Phe-Leu-Phe. |
Effects of BOC1, TLR2Ab and ST2825 on the
expression of TLR2 and MyD88 in leukocytes
As shown in Fig. 9,
treatment of the leukocytes with IL-1β augmented the expression of
TLR2 and MyD88. However, these increments were alleviated by
BML-111 pre-treatment. These inhibitory effects of BML-111 were
mediated by the LXA4 receptor, as pretreatment with BOC1 abrogated
the inhibitory effects of BML-111 on the expression of TLR2 and
MyD88 induced by IL-1β. The IL-1β-triggered upregulation of TLR2
and MyD88 were repressed by TLR2Ab but not by ST2825. Since ST2825
is a dimerization inhibitor of MyD88 and inhibits the interaction
of Flag-MyD88 and Myc-MyD88 proteins according to a previous
co-immunoprecipitation assay (24), the total protein expression of
MyD88 may be not affected by ST2825.
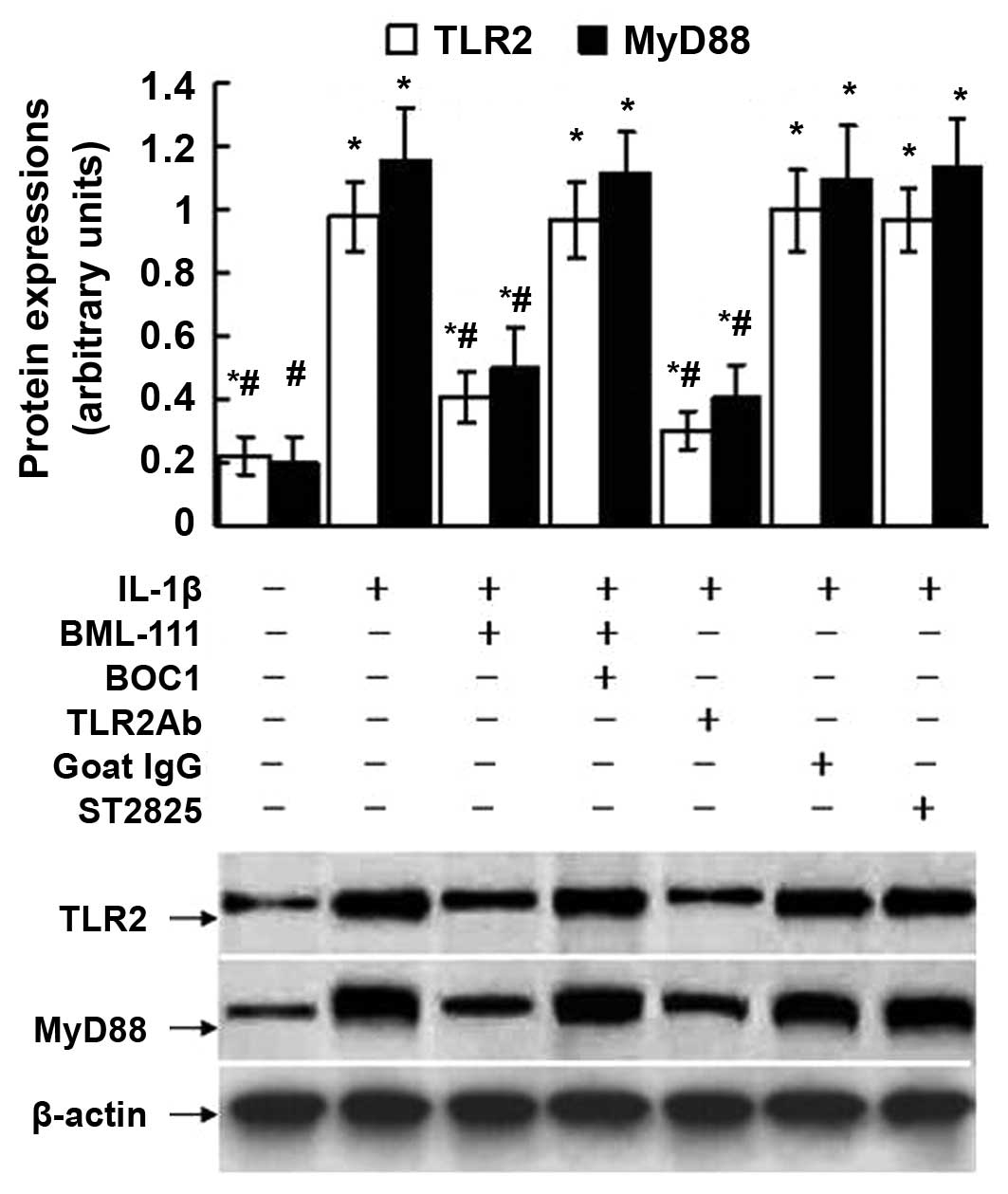 | Figure 9Expression of TLR2 and MyD88 assessed
using western blot analysis of leukocytes exposed to IL-1β. The
cultured leukocytes were obtained from a normal control mouse and
stimulated with IL-1β (10 ng/ml) for 30 min with or without
pre-treatment of BML-111 (1 mM) for 30 min, antagonist of G
protein-coupled LXA4 receptor BOC1 (100 μM) for 30 min,
TLR2Ab (1 μg/ml) for 1 h, goat IgG (1 μg/ml) for 1 h
and MyD88 dimerization inhibitor ST2825 (20 μM) for 1 h. The
western blot is representative of five independent experiments, and
the lower panel shows β-actin protein, which served as a loading
control. Semiquantitative analysis was performed by using UVP-gel
densitometry. Arbitrary unit =
(ATLR2/Aβ-actin) ×100%, or
(AMyD88/Aβ-actin) ×100%. Values are expressed
as the mean ± standard deviation of five independent experiments.
*P<0.05, as compared to the arbitrary units of the
same protein in the cells without treatment. #P<0.05,
as compared to the arbitrary units of the same protein in the cells
treated with IL-1β alone. IL, interleukin; IgG, immunoglobulin;
TLR, toll-like receptor; TLR2Ab, toll-like receptor 2-neutralizing
antibody; BOC1, N-t-Boc-Phe-Leu-Phe-Leu-Phe. |
Effects of BOC1, TLR2Ab, ST2825 and
TPCA-1 on NF-κB activation in leukocytes
As indicated in Fig.
10, treatment of the leukocytes with IL-1β induced the
degradation of IκBα and nuclear translocation of p65, as the levels
of phosphorylated p65 subunit in the nuclear extract increased and
the expression of p65 in the cytoplasmic extract decreased.
However, BML-111 pre-treatment blocked the degradation of IκBα, as
well as nuclear translocation of p65 induced by IL-1β. These
inhibitory effects of BML-111 were mediated by the LXA4 receptor,
as pretreatment with BOC1 abolished the inhibitory effects of
BML-111 on the degradation of IκBα and nuclear translocation of p65
induced by IL-1β. The degradation of IκBα and nuclear translocation
of p65 induced by IL-1β were also dramatically blocked by
pretreatment of the cells with TLR2Ab, ST2825 and TPCA-1. LXA4 and
its analog inhibited degradation, but not phosphorylation of IκBα
in epithelial cells and keratinocytes (25,26),
and in analogy with this, the present study detected degradation of
IκBα but not phosphorylation of IκBα.
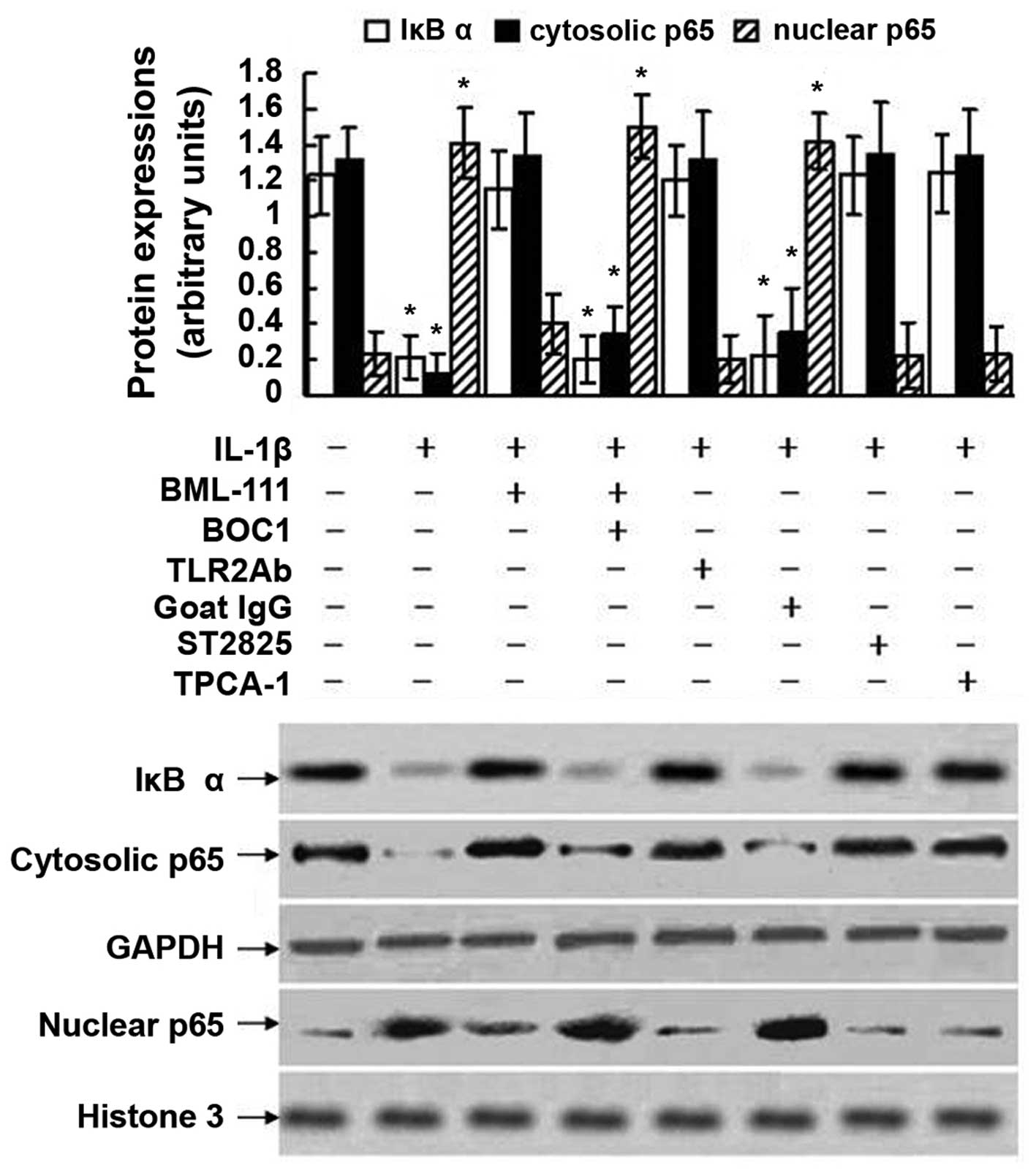 | Figure 10Activation of NF-κB assessed using
western blot analysis in leukocytes exposed to IL-1β. The cultured
leukocytes were obtained from a normal control mouse and stimulated
with IL-1β (10 ng/ml) for 1 h with or without pre-treatment of
BML-111 (1 mM) for 30 min, BOC1 (100 μM) for 30 min, TLR2Ab
(1 μg/ml) for 1 h, goat IgG (1 μg/ml) for 1 h, ST2825
(20 μM) for 1 h or TPCA-1 (10 μM) for 1 h. The
western blot is representative of five independent experiments.
GAPDH served as a loading control of cytosolic proteins. Histone 3
protein served as a loading control of nuclear proteins.
Semiquantitative analysis was performed using UVP-gel densitometry.
Arbitrary unit = (AIκBα/AGAPDH) ×100%, or
(Acytosolic p65/AGAPDH) ×100%, or
(Anuclear p65/Ahistone 3) ×100%. Values are
expressed as the mean ± standard deviation of five independent
experiments. *P<0.05, as compared to the arbitrary
units of the same protein in the cells without treatment. IL,
interleukin; IgG, immunoglobulin G; TPCA-1,10,
thiophene-3-carbox-amide 1; TLR2Ab, toll-like receptor
2-neutralizing antibody; BOC1, N-t-Boc-Phe-Leu-Phe-Leu-Phe;
NF-κB, nuclear factor-κB; IκBα, inhibitor of kappa B alpha. |
Discussion
TLRs are important recognition receptors in the host
innate defense against invading pathogens. TLR2 is particularly
involved in signal transduction of cellular responses to
lipoproteins/lipopeptides, Gram-positive bacteria and mycobacterial
wall constituents (16). The IL-1
receptor (IL-1R) shares sequence homology with the cytosolic domain
of the TLRs. The homologous cytoplasmic domain of IL-1R and TLRs is
referred to as the Toll/IL-1R (TIR) homology domain (27,28).
TLRs and IL-1R recruit the intracellular protein MyD88 via
respective TIR domain interactions. These interactions result in
the recruitment of IL-1R-associated kinase 1 (IRAK-1) to the
receptor complex, where it interacts with tumor necrosis factor
receptor-associated factor 6 (TRAF6), resulting in the downstream
activation of NF-κB, which triggers the induction of a variety of
effector genes, including genes of cytokines/mediators and
ultimately results in upregulation of co-stimulatory molecules,
secretion of cytokines, as well as enhanced uptake and presentation
of antigen (16,27,28).
The present study provided evidence that OVA-induced airway
inflammation was mediated by upregulation of the TLR2/MyD88/NF-κB
pathway. First, OVA immunization upregulated the protein expression
of TLR2 and MyD88, and activated NF-κB in the lungs. Moreover,
OVA-induced upregulation of the TLR2/MyD88/NF-κB pathway was
further demonstrated by immunohistochemical staining in the lungs.
Finally, consistent with the downregu-lation of TLR2/MyD88/NF-κB
pathway proteins following treatment with BML-111- and anti-IL-1β
antibodies, the OVA-induced airway inflammation was restrained, as
shown by the reduced serum levels of IL-1β, IL-4 and IL-8, as well
as BALF levels of IL-1β, IL-4, IL-8, OVA-IgE, and leukocyte counts;
furthermore, peribronchial inflammation of lungs induced by OVA was
ameliorated by BML-111- and anti-IL-1β antibodies. The findings of
the present study are supported by those of other studies (17,29–32).
For example, Lee et al (29) reported that TLR2 and TLR4
expression in lungs obtained from OVA-immunized mice was activated.
Furthermore, in fatal asthma, elevated expression of TLR2, -3 and
-4 in airways may have contributed to the acute inflammation
causing asthma mortalities (29).
Expression of TLR2 and TLR4 as well as that of the pro-inflammatory
cytokines IL-8 and IL-1 increased in subjects with neutrophilic
asthma as compared with that in other asthma subtypes and controls
(31). In addition, activation of
TLR2 induced a T helper type 2 immune response and promoted
experimental asthma (17).
Recently, it was demonstrated that organic dust extract-induced
airway hyperresponsiveness, neutrophil influx and
cytokine/chemokine production were nearly absent in MyD88 knockout
mice, suggesting that acute organic dust-induced airway
inflammatory response is highly dependent on MyD88 signaling, and
is dictated by important contributions from upstream TLRs (32). In the present study, the ligands
which activated the TLR2/MyD88/NF-κB pathway remain elusive;
however, based on the in vivo and in vitro
experiments, the most likely mediator was IL-1β.
As mentioned above, Toll/IL-1R is involved in the
activation of several TLRs and the MyD88/NF-κB pathway. Therefore,
the present study also explored the role of IL-1 in the
pathogenesis of airway inflammation in OVA-immunized mice. The
results of the present study demonstrated that IL-1β has a critical
role in OVA-induced airway inflammation. First, the levels of IL-1β
increased in the serum and BALF obtained from OVA-immunized mice.
Treatment with anti-IL-1β antibody significantly suppressed the
number of total leukocytes, eosinophils, neutrophils and
lymphocytes in BALF stimulated by OVA; furthermore, it decreased
the serum levels of IL-1β, IL-4 and IL-8, as well as the BALF
levels of IL-1β, IL-4, IL-8 and OVA-IgE stimulated by OVA. Finally,
treatment with anti-IL-1β antibody also blocked inflammatory cell
infiltration and bronchial mucus hypersecretion induced by OVA. The
results of the present study were supported by those of previous
studies (33–37). For example, IL-1 is required for
allergen-specific Th2 cell activation, synthesis of IgE and
proinflammatory cytokines, and development of the airway
hypersensitivity response (34–36).
A recent study showed that inhalation of aerosolized anti-IL-1β
antibody repressed the pathological responses in the pulmonary
tissues of guinea pigs with asthma, and this inhibitory activity
may be attributed to the decreased number of eosinophils and
neutrophils and the reduced levels of inflammatory cytokines and
IgE in the peripheral blood and BALF (37). The present study indicated that the
inhibitory effects of anti-IL-1β antibody on OVA-induced airway
inflammation may be mediated by the inhibitory role of anti-IL-1β
antibody on the TLR2/MyD88/NF-κB pathway. First, treatment with
anti-IL-1β antibody downregulated the expression of TLR2 and MyD88,
and decreased the expression and activity of NF-κB in lungs induced
by OVA. Secondly, IL-1β augmented the expression of TLR2 and MyD88
as well as the activity of NF-κB in leukocytes. Moreover,
activation of NF-κB as well as enhanced levels of IL-4, IL-6 and
IL-8 released from leukocytes exposed to IL-1β were blocked by
TLR2Ab and ST2825. The results of the present study were supported
by those of previous studies (24,38).
For example, a previous study demonstrated that IL-1β stimulation
significantly increased mRNA and protein expression of TLR2 and
TLR4 in articular chondrocytes (38). Moreover, the MyD88 inhibitor ST2825
blocked IL-1β-induced synthesis of IL-6 in vivo as well as
activation of NF-κB in treated mice, indicating that TLRs/MyD88
mediated IL-1β-induced, NF-κB-dependent production of IL-6
(24).
Signaling pathways involved in LXA4 activity in
asthma remain elusive, although it was demonstrated that an LXA4
analog blocked airway hyper-responsiveness and pulmonary
inflammation in a murine model of asthma (11). The present study showed that LXA4
receptor activation by BML-111 inhibited the production of IL-1β,
IL-4 and IL-8 in serum and BALF, restored the production of IFN-γ
in serum obtained from OVA-immunized mice, and suppressed airway
inflammation. These effects of BML-111 may be attributed to the
BML-111-induced downregulation of the TLR2/MyD88/NF-κB pathway.
First, BML-111 treatment downregulated the expression of TLR2 and
MyD88, as well as the expression and activity of NF-κB in the lungs
induced by OVA. Consistent with BML-111-induced downregulation of
the TLR2/MyD88/NF-κB pathway, the OVA-induced airway inflammation
was restrained, as shown by the reduced serum levels of IL-1β, IL-4
and IL-8, as well as BALF levels of IL-1β, IL-4, IL-8, OVA-IgE.
Furthermore, leukocyte counts were reduced and peribronchial
inflammation was ameliorated in lungs induced by OVA. Finally, the
elevated levels of IL-4, IL-6 and IL-8, expression of TLR2 and
MyD88, and activation of NF-κB in leukocytes were all diminished by
BML-111. In the present study, BML-111-induced downregulation of
the TLR2/MyD88/NF-κB pathway in OVA-immunized mice may be
attributed to the following reasons: First, BML-111 itself
downregulated the TLR2/MyD88/NF-κB pathway; second, BML-111 reduced
the levels of IL-1β in the serum and BLAF, and therefore inhibited
the IL-1β-induced upregulation of the TLR2/MyD88/NF-κB pathway; and
third, BML-111 decreased the pulmonary infiltration of leukocytes
and this decrement in the number of total cells in the lungs may
have contributed to the reduced protein expression of the
TLR2/MyD88/NF-κB pathway in the lung tissue.
In conclusion, the present study identified that
first, OVA-induced airway inflammation is mediated by upregulation
of the TLR2/MyD88/NF-κB pathway; second, IL-1β has a pivotal role
in the airway inflammation and upregulation of the TLR2/MyD88/NF-κB
pathway induced by OVA; and third, LXA4 receptor activation by
BML-111 restrains the OVA-induced airway inflammation via
downregulation of the TLR2/MyD88/NF-κB pathway. Coupled with a
previous demonstration of the efficacy of LXA4 and LXA4 analog in
the treatment of asthmatic mice and humans (11,39),
the present study provided a theoretical basis and supported a
therapeutic value for clinical use of LXA4 analogs in the treatment
of asthma. The efficacy of anti-IL-1β antibody was previously
proven in the treatment of asthmatic guinea pigs (37), and the present study also supports
a therapeutic value for anti-IL-1β antibody in the treatment of
refractory asthma.
Acknowledgments
This study was funded by the Priority Academic
Program Development (PAPD) of Jiangsu Higher Education Institutions
(grant no. JX10231801).
References
|
1
|
WHO/NHLBI Workshop report: Global strategy
for asthma management and prevention. National Institutes of
Health/National Heart. Lung and Blood Institue. (NIH Publication
02-3659). Bethesda: 2005
|
|
2
|
Leff AR: Role of leukotrienes in bronchial
hyperresponsiveness and cellular responses in airways. Am J Respir
Crit Care Med. 161:S125–S132. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Broide DH, Lotz M, Cuomo AJ, Coburn DA,
Federman EC and Wasserman SI: Cytokines in symptomatic asthma
airways. J Allergy Clin Immunol. 89:958–967. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krause K, Metz M, Makris M, Zuberbier T
and Maurer M: The role of interleukin-1 in allergy-related
disorders. Curr Opin Allergy Clin Immunol. 12:477–484. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luster AD and Tager AM: T-cell trafficking
in asthma: lipid mediators grease the way. Nat Rev Immunol.
4:711–724. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
O’Byrne PM: Leukotriene
bronchoconstriction induced by allergen and exercise. Am J Respir
Crit Care Med. 161:S68–S72. 2000. View Article : Google Scholar
|
|
7
|
Serhan CN: Lipoxins and aspirin-triggered
15-epi-lipoxin biosynthesis: an update and role in
anti-inflammation and pro-resolution. Prostaglandins Other Lipid
Mediat. 68:433–455. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee TH, Lympany P, Crea AK and Spur BW:
Inhibition of leukotriene B4-induced neutrophil migration by
lipoxin A4: structure-function relationships. Biochem Biophys Res
Commun. 180:1416–1421. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu SH, Liao PY, Yin PL, Zhang YM and Dong
L: Elevated expressions of 15-lipoxygenase and lipoxin A4 in
children with acute poststreptococcal glomerulonephritis. Am J
Pathol. 174:115–122. 2009. View Article : Google Scholar :
|
|
10
|
Levy BD, Lukacs NW, Berlin AA, Schmidt B,
Guilford WJ, Serhan CN and Parkinson JF: Lipoxin A4 stable analogs
reduce allergic airway responses via mechanisms distinct from
CysLT1 receptor antagonism. FASEB J. 21:3877–3884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levy BD, De Sanctis GT, Devchand PR, Kim
E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM and Serhan CN:
Multi-pronged inhibition of airway hyper-responsiveness and
inflammation by lipoxin A4. Nat Med. 8:1018–1023. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bonnans C, Vachier I, Chavis C, Godard P,
Bousquet J and Chanez P: Lipoxins are potential endogenous
antiinflammatory mediators in asthma. Am J Respir Crit Care Med.
165:1531–1535. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu SH, Liao PY, Dong L and Chen ZQ: Signal
pathway involved in inhibition by lipoxin A(4) of production of
interleukins induced in endothelial cells by lipopolysaccharide.
Inflamm Res. 57:430–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Faure E, Equils O, Sieling PA, Thomas L,
Zhang FX, Kirschning CJ, Polentarutti N, Muzio M and Arditi M:
Bacterial lipopolysaccharide activates NF-kappaB through toll-like
receptor 4 (TLR4) in cultured human dermal endothelial cells.
Differential expression of TLR-4 and TLR-2 in endothelial cells. J
Biol Chem. 275:11058–11063. 2000. View Article : Google Scholar
|
|
15
|
Hoth JJ, Wells JD, Brownlee NA, Hiltbold
EM, Meredith JW, McCall CE and Yoza BK: Toll-like receptor
4-dependent responses to lung injury in a murine model of pulmonary
contusion. Shock. 31:376–381. 2009. View Article : Google Scholar
|
|
16
|
Sabroe I, Parker LC, Wilson AG, Whyte MK
and Dower SK: Toll-like receptors: their role in allergy and
non-allergic inflammatory disease. Clin Exp Allergy. 32:984–989.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Redecke V, Häcker H, Datta SK, Fermin A,
Pitha PM, Broide DH and Raz E: Cutting edge: activation of
Toll-like receptor 2 induces a Th2 immune response and promotes
experimental asthma. J Immunol. 172:2739–2743. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eisenbarth SC and Flavell RA: Innate
instruction of adaptive immunity revisited: the inflammasome. EMBO
Mol Med. 1:92–98. 2009. View Article : Google Scholar
|
|
19
|
Velasco G, Campo M, Manrique OJ, Bellou A,
He H, Arestides RS, Schaub B, Perkins DL and Finn PW: Toll-like
receptor 4 or 2 agonists decrease allergic inflammation. Am J
Respir Cell Mol Biol. 32:218–224. 2005. View Article : Google Scholar
|
|
20
|
Wu SH, Wu XH, Liao PY and Dong L: Signal
transduction involved in protective effects of 15(R/S)-methyl-
lipoxin A(4) on mesangioproliferative nephritis in rats.
Prostaglandins Leukot Essent Fatty Acids. 76:173–180. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Zhang X, Wu P, Li H, Jin S, Zhou
X, Li Y, Ye D, Chen B and Wan J: BML-111, a lipoxin receptor
agonist, modulates the immune response and reduces the severity of
collagen-induced arthritis. Inflamm Res. 57:157–162. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hecht I, Rong J, Sampaio AL, Hermesh C,
Rutledge C, Shemesh R, Toporik A, Beiman M, Dassa L, et al: A novel
peptide agonist of formyl-peptide receptor-like 1 (ALX) displays
anti-inflammatory and cardioprotective effects. J Pharmacol Exp
Ther. 328:426–434. 2009. View Article : Google Scholar
|
|
23
|
Cho JY, Miller M, Baek KJ, Castaneda D,
Nayar J, Roman M, Raz E and Broide DH: Immunostimulatory DNA
sequences inhibit respiratory syncytial viral load, airway
inflammation, and mucus secretion. J Allergy Clin Immunol.
108:697–702. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Loiarro M, Capolunghi F, Fantò N, Gallo G,
Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, et al:
Pivotal advance: inhibition of MyD88 dimerization and recruitment
of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc
Biol. 82:801–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gewirtz AT, Collier-Hyams LS, Young AN,
Kucharzik T, Guilford WJ, Parkinson JF, Williams IR, Neish AS and
Madara JL: Lipoxin a4 analogs attenuate induction of intestinal
epithelial proinflammatory gene expression and reduce the severity
of dextran sodium sulfate-induced colitis. J Immunol.
168:5260–5267. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu SH, Liu B, Dong L and Wu HJ: NF-kappaB
is involved in inhibition of lipoxin A4 on dermal inflammation and
hyperplasia induced by mezerein. Exp Dermatol. 19:e286–e288. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kaisho T and Akira S: Toll-like receptor
function and signaling. J Allergy Clin Immunol. 117:979–987. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X and Qin JJ: Modulation of
Toll-interleukin 1 receptor mediated signaling. J Mol Med (Berl).
83:258–266. 2005. View Article : Google Scholar
|
|
29
|
Lee CC, Lai YT, Chang HT, Liao JW, Shyu
WC, Li CY and Wang CN: Inhibition of high-mobility group box 1 in
lung reduced airway inflammation and remodeling in a mouse model of
chronic asthma. Biochem Pharmacol. 86:940–949. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferreira DS, Annoni R, Silva LF, Buttignol
M, Santos AB, Medeiros MC, Andrade LN, Yick CY, Sterk PJ, et al:
Toll-like receptors 2, 3 and 4 and thymic stromal lymphopoietin
expression in fatal asthma. Clin Exp Allergy. 42:1459–1471. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Simpson JL, Grissell TV, Douwes J, Scott
RJ, Boyle MJ and Gibson PG: Innate immune activation in
neutrophilic asthma and bronchiectasis. Thorax. 62:211–218. 2007.
View Article : Google Scholar
|
|
32
|
Bauer C, Kielian T, Wyatt TA, Romberger
DJ, West WW, Gleason AM and Poole JA: Myeloid differentiation
factor 88-dependent signaling is critical for acute organic
dust-induced airway inflammation in mice. Am J Respir Cell Mol
Biol. 48:781–789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krause K, Metz M, Makris M, Zuberbier T
and Maurer M: The role of interleukin-1 in allergy-related
disorders. Curr Opin Allergy Clin Immunol. 12:477–484. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakae S, Komiyama Y, Yokoyama H, Nambu A,
Umeda M, Iwase M, Homma I, Sudo K, Horai R, et al: IL-1 is required
for allergen-specific Th2 cell activation and the development of
airway hypersensitivity response. Int Immunol. 15:483–490. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sim TC, Hilsmeier KA, Reece LM, Grant JA
and Alam R: Interleukin-1 receptor antagonist protein inhibits the
synthesis of IgE and proinflammatory cytokines by
allergen-stimulated mononuclear cells. Am J Respir Cell Mol Biol.
11:473–479. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nagarkar DR, Poposki JA, Comeau MR,
Biyasheva A, Avila PC, Schleimer RP and Kato A: Airway epithelial
cells activate TH2 cytokine production in mast cells through IL-1
and thymic stromal lymphopoietin. Allergy Clin Immunol.
130:225–232. 2012. View Article : Google Scholar
|
|
37
|
Wei-xu H, Qin X, Zhu W, Yuan-yi C, Li-feng
Z, et al: Therapeutic potential of anti-IL-1β IgY in guinea pigs
with allergic asthma induced by ovalbumin. Mol Immunol. 58:139–149.
2014. View Article : Google Scholar
|
|
38
|
Campo GM, Avenoso A, D’Ascola A, Scuruchi
M, Prestipino V, Nastasi G, Calatroni A and Campo S: Adenosine A2A
receptor activation and hyaluronan fragment inhibition reduce
inflammation in mouse articular chondrocytes stimulated with
interleukin-1β. FEBS J. 279:2120–2133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christie PE, Spur BW and Lee TH: The
effects of lipoxin A4 on airway responses in asthmatic subjects. Am
Rev Respir Dis. 145:1281–1284. 1992. View Article : Google Scholar : PubMed/NCBI
|