Introduction
Lung cancer is currently the leading cause of
cancer-associated mortality worldwide, being responsible for an
estimated 87,750 mortalities in males and 72,590 in females in 2012
(1). The majority of patients have
advanced disease (stage III/IV) at initial diagnosis, which
contributes to the poor prognosis of these patients. It is reported
that the 5-year overall survival rate is <5% among patients with
advanced-stage disease (2).
Chemotherapy, including cisplatin (CDDP), carboplatin and
oxaliplatin, remains a crucial treatment for advanced patients
(3). Platinum drugs cross-link at
the nucleophilic centers on the DNA of tumor cells, forming intra-
(on the same strand) and/or inter-strand (between two opposite
strands) adducts between guanines or guanine and adenine, which
inhibit DNA replication and trigger cell cycle arrest and apoptosis
(4,5). However, the major limitation in the
clinical applications of platinum compounds is the development of
tumor drug resistance (6–8). Therefore, it is essential to develop
more effective treatment strategies to overcome this drug
resistance.
Integrin-linked kinase (ILK), a highly conserved, 59
kDa serine/threonine kinase, has been implicated in the regulation
of various biological processes that are crucial to the progression
of malignant disease (9). ILK
expression and activity have been revealed to be increased in
association with tumor grade, T status, lymph node metastasis and
survival in lung cancer patients (10–12).
ILK promotes lung cancer cell migration and invasion via
upregulation of matrix metal-loproteinase-9 (MMP-9) (13) and epithelial-mesenchymal
transition-associated genes, including vimentin, fibronectin, Snail
and Slug (14). Further studies
indicate that silencing ILK through targeting small interfering RNA
(siRNA) inhibits cell proliferation and growth, induces cell cycle
arrest and the apoptosis of bladder (15) and pancreatic cancer cells (16), suggesting the inhibition of ILK may
be a novel approach for treating lung cancer. Notably, Song et
al (17) previously
demonstrated that downregulation of ILK by siRNA arrests the growth
and increases the CDDP sensitivity and apoptotic rate of human
gastric cell line cells that are resistant to SGC7901/CDDP. Thus,
it is hypothesized that there may be a synergistic interaction
between downregulation of ILK and CDDP administration for treating
lung cancer by creating cytotoxic DNA lesions and affecting
apoptosis in lung cancer A549 cells. To the best of our knowledge,
the present study is the first to examine this mechanism.
Materials and methods
Cell culture
The human lung adenocarcinoma cell line A549 and
human embryo kidney (HEK) 293T cells (American Type Culture
Collection, Manassas, VA, USA) were maintained in Dulbecco’s
modified Eagle’s medium (Invitrogen Life Technologies, Carlsbad,
CA, USA) containing 10% fetal bovine serum (Invitrogen Life
Technologies) and cultured in a humidified atmosphere of 5%
CO2 at 37°C.
Construction of lentiviral vectors
expressing siRNA targeting ILK and transfection
The oligonucleotides encoding a negative control
(NC) siRNA with no homology to the human genome (5′-AAT GTA CTG CGC
GTG GAG A-3′) and ILK siRNA (5′-CCT TCA ACT TTG TGC TCA T-3′) were
designed and synthesized by Shanghai Jikai Gene Chemical Co., Ltd,
(Shanghai, China) and cloned into the Age I/EcoRI
linearized pGCSIL-GFP viral vector (GeneChem, Shanghai, China) to
generate the lentiviral vectors. Expression of the lentiviral shRNA
was confirmed by DNA sequencing. Plasmids along with 20 μg
of pGCSIL-shILK or -shNC lentiviral vectors, 15 μg of
packaging vectors pHelper 1.0 and 10 μg of packaging vectors
pHelper 2.0 were mixed with 200 μl of Opti-MEM and 15
μl of Lipofectamine 2000 and then transfected into HEK293T
cells. After 48 h transfection, lentiviruses were harvested in
serum-free medium, filtered and concentrated in primed Centricon
Plus-20 filter devices (Millipore, Billerica, MA, USA).
Subsequently, A549 cells were infected with ILK-shRNA lentivirus or
control lentivirus at a multiplicity of infection of 20. The number
of green fluorescent protein (GFP)-positive cells was determined
microscopically (MicroPublisher 3.3RTV; Olympus, Tokyo, Japan)
three days post-transduction.
RNA extraction and reverse transcription
quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells with TRIzol
reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) according
to the manufacturer’s instructions. Gene expression was detected by
RT-qPCR using the standard SYBR Green RT-PCR kit (Takara Bio, Inc.,
Shiga, Japan). Briefly, the cDNA was synthesized using the
RevertAid First-Strand cDNA Synthesis kit (Fermentas, Vilnius,
Lithuania) according to the manufacturer’s instructions. The
specific primer pairs and the amplified products were as follows:
ILK (212 bp), sense 5′-TCCACCTGCTCCTCATCC-3′ and anti-sense
5′-CCTCATCAATCATTACACTACGG-3′ and GAPDH (121 bp), sense
5′-TGACTTCAACAGCGACACCCA-3′ and antisense
5′-CACCCTGTTGCTGTAGCCAAA-3′. The relative levels of gene mRNA
transcripts were normalized to the internal control GAPDH.
Relative gene expression was quantified using the GraphPad Prism
4.0 software (GraphPad Software, San Diego, CA, USA).
Western blotting
Cells were lysed in 0.1 ml lysis buffer (0.1% SDS,
1% NP-40, 50 mM HEPES, pH 7.4, 2 mM EDTA, 100 mM NaCl, 5 mM sodium
orthovanadate and 1% protease inhibitor mixture set I; Calbiochem,
San Diego, CA, USA) on ice for 30 min. Following centrifugation at
13,400 × g (Eppendorf centrifuge 5415 D; Eppendorf AG, Hamburg,
Germany) for 15 min, the supernatants were removed and total
protein concentration was determined using a bicinchoninic acid
protein assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA).
Proteins (20 μg) were separated in 10% sodium dodecyl
sulfate polyacrylamide gel electrophoresis and transferred onto a
polyvinylidene difluoride (PVDF) membrane at 400 mA for 2 h.
Following blocking at room temperature in 5% bovine serum albumin
for 1.5 h, PVDF membranes were incubated with the following
antibodies: Rabbit anti-ILK polyclonal antibody (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), rabbit
anti-phospho-glycogen synthase kinase (GSK)-3β-S9 monoclonal
antibody (1:1,000), rabbit anti-p-Akt-S473 poly-clonal antibody
(1:1,000), rabbit anti-MMP-9 (1:200), rabbit anti-activator protein
(AP-1; 1:800), rabbit anti-β-catenin polyclonal antibody (1:1,000),
rabbit anti-cyclin D1 monoclonal antibody (1:500), rabbit
anti-vascular endothelial growth factor polyclonal antibody (VEGF;
1:500), all purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA) and mouse anti-GAPDH monoclonal antibody (Santa
Cruz Biotechnology, Inc; 1:4,000), followed by incubation with the
correspondent peroxidase-conjugated secondary antibodies (goat
anti-rabbit lgG; 1:4,000; goat anti-mouse lgG; Santa Cruz
Biotechnology, Inc.; 1:4,000). Chemiluminescent detection was
performed with the enhanced chemiluminescence kit (Pierce
Biotechnology, Inc.).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay for assessment of proliferation and drug sensitivity
The experimental cells in the exponential phase of
growth were plated at a final concentration of 2×103
cells/well in 96-well culture plates for different culture time
periods. Varied concentrations of CDDP (5, 10, 15, 20 and 25%) were
added to each well and the cells were incubated for 72 h. MTT (10
μl, 5 mg/ml) was then added. Following an additional 4 h of
incubation, the reaction was terminated by removal of the
supernatant and addition of 100 μl dimethyl sulfoxide for 30
min. The optical density of each well was measured at 490 nm using
an ELISA reader (ELx808; Bio-Tek Instruments, Winooski, VT,
USA).
Detection of apoptosis by flow
cytometry
Cells were stained with fluorescein isothiocyanate
(FITC)-labeled annexin-V and simultaneously with propidium iodide
(PI), to discriminate intact cells (annexin-/PI-) from apoptotic
cells (annexin+/PI-) and necrotic cells (annexin+/PI+). A total of
1.0×106 cells were washed twice with ice-cold
phosphate-buffered saline (PBS) and incubated for 30 min in a
binding buffer (1 μg/ml PI and 1 μg/ml FITC-labeled
annexin-V), respectively. Fluorescence-activated cell sorting
analysis for annexin-V and PI staining was performed using a flow
cytometer (Beckman Coulter, Inc., Fullerton, CA, USA). All
experiments were performed in triplicate.
Cell cycle assay
Cells were seeded into a 6-well plate, harvested by
centrifugation at 13,40 × g for 5 min (Eppendorf centrifuge 5415 D;
Eppendorf AG). Following being washed twice in pre-cooled PBS (pH
7.4), cells were fixed in 70% alcohol. The percentage of cells in
each stage of the cell cycle was determined by staining with PI.
The analysis of cell cycle distribution was performed by FACScan
flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA) in
accordance with the manufacturer’s instructions.
Colony-forming assay
Exponentially growing cells transfected with
ILK-RNAi-lentivirus and negative control lentivirus were suspended
in complete growth medium and seeded in 6-well plates at 200 cells
per well. The plates were maintained at 37°C in a humidified
incubator with 5% CO2 for 2 weeks. The visible colonies
were subsequently recorded under an inverted fluorescence
microscope (MicroPublisher 3.3RTV; Olympus). Following fixation in
paraformaldehyde, the colonies were subjected to Giemsa (Karyomax;
Gibco, Grand Island, NY, USA) staining for 10 min followed by
acquisition of images with an Olympus C5050 digital camera attached
to an Olympus CKX1 inverted microscope (Olympus).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analysis was performed using SPSS 11.0 software (SPSS,
Inc., Chicago, IL, USA). The difference between two groups was
analyzed using Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
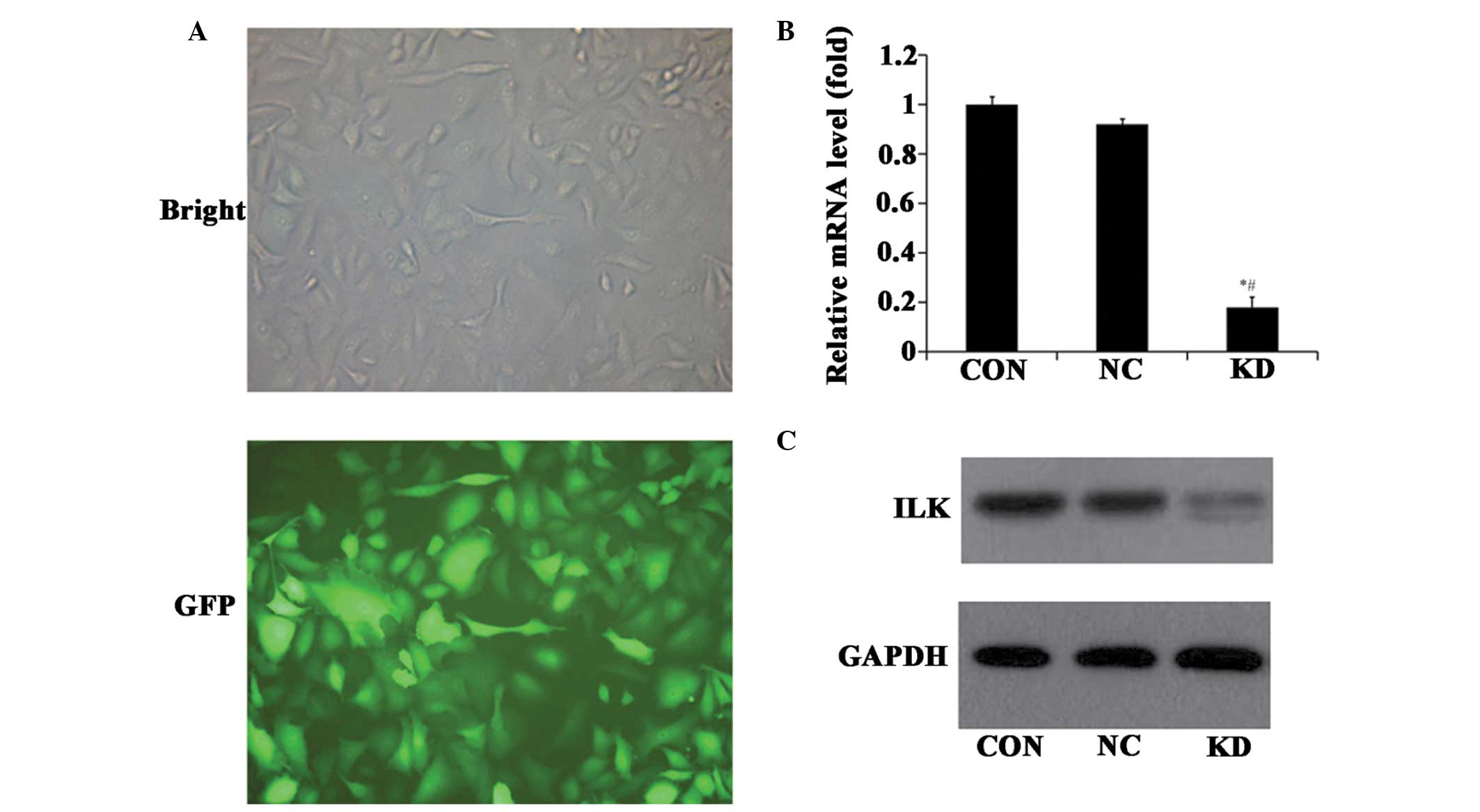
ILK expression in A549 cells following
treatment with lentivirus-mediated RNAi
At 3 days after transfection, A549 cells were
visualized using a fluorescence microscope and the mRNA and protein
levels of ILK were also analyzed. The results revealed that >80%
of cells had green fluorescent signals compared with the bright
field (Fig. 1A). The level of ILK
mRNA in ILK specific-RNAi infected cells was significantly
decreased by ~70% (P<0.05), compared with the control
RNAi-infected cells (Fig. 1B). In
accordance with the silencing of mRNA expression, ILK protein was
also down-regulated in ILK RNAi cells (Fig. 1C). These findings suggest that
ILK-specific RNAi may downregulate ILK expression efficiently.
Effect of ILK knockdown on cell
proliferation, clone forma- tion, the cell cycle and cell apoptosis
in vitro
To assess the effects of ILK knockdown on cell
proliferation of the cell line, A549, an MTT assay was performed.
As expected, cells with ILK RNAi exhibited an inhibited cell
proliferation ability compared with the control cells (Fig. 2A). In line with the MTT assay, the
quantity and size of the colony in the ILK RNAi-infected cells were
also significantly decreased compared with the control cells
(Fig. 2B). In order to elucidate
whether lentivirus-mediated ILK RNAi had any effects on the cell
cycle of A549 cells, all three groups of A549 cells were subjected
to a flow cytometry assay after 3 days of infection. The results
revealed that ILK-shRNA-lentivirus infected cells exhibited an
increased proportion in G0/G1 phase compared with the
control-shRNA-lentivirus infected cells (Fig. 2C). As shown in Fig. 3B and C, although levels of cell
apoptosis were higher in the ILK knockdown groups than that in the
normal and control groups, no significant differences were
observed, implying that downregulation of ILK alone is not an
optimal approach for the treatment of lung cancer.
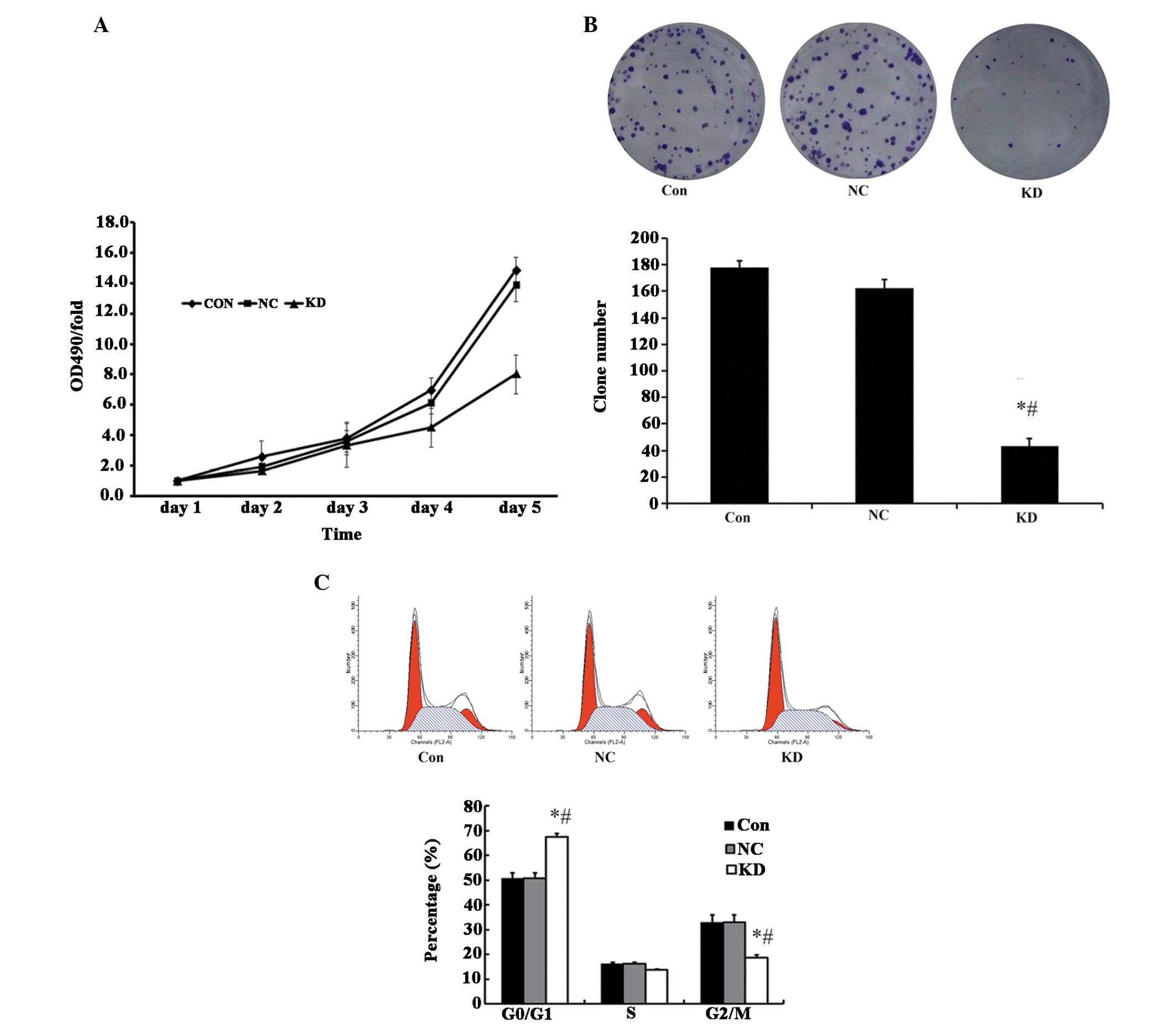 | Figure 2Functional effect of ILK knockdown.
(A) Knockdown of ILK inhibited the cell proliferation of A549 cells
[measured using a
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide
assay], (B) reduced the colony formation ability (measured by
colony formation assay), (C) augmented the proportion of G0/G1
phase (the proportion of different cell cycle phases was
quantitated by propidium iodide staining followed by flow
cytometric analysis). *P<0.05, compared with the Con
group; #P<0.05, compared with the NC group. CON,
parent cells; NC, cells transfected with negative control RNAi; KD,
cells transfected with ILK specific RNAi; ILK, integrin-linked
kinase; OD, optical density. |
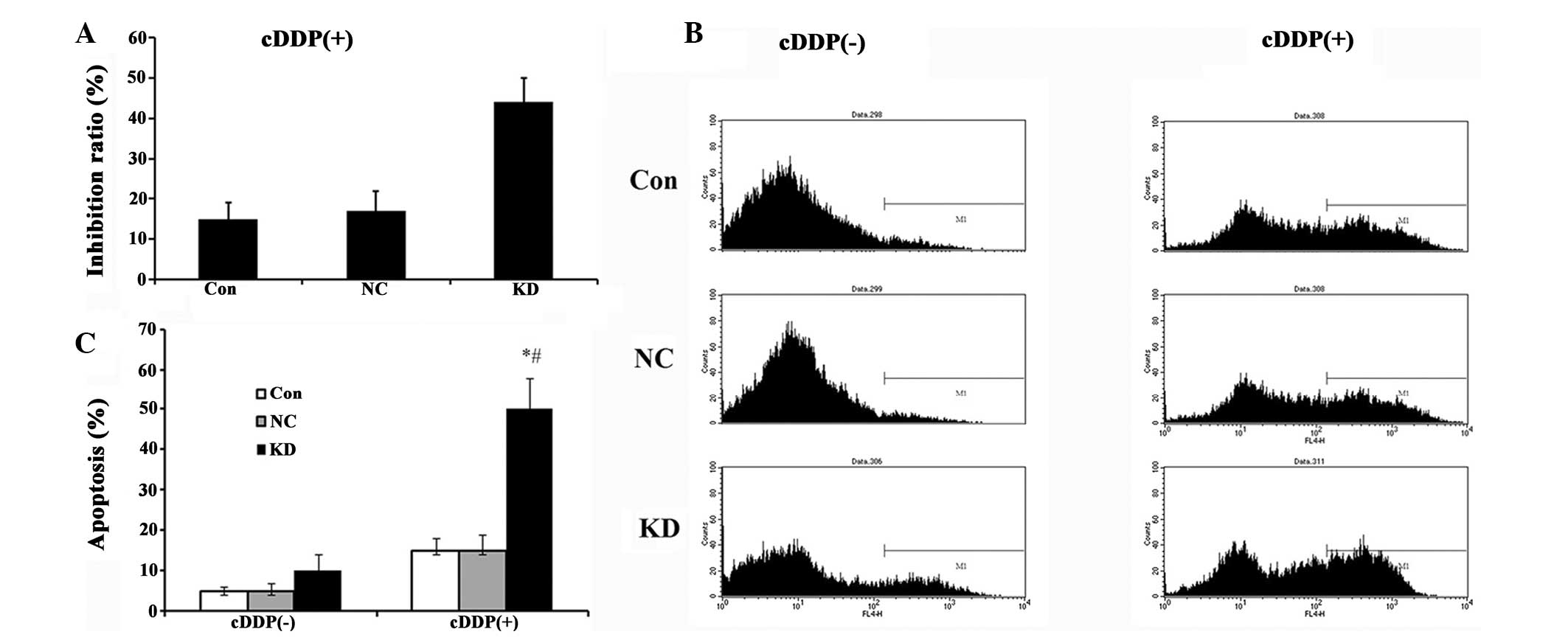 | Figure 3A549 cell proliferation and apoptosis
in cells by transfection with lentivirus expressing KD RNAi,
NC-RNAi or the parent A549 cells as CON combined with CDDP. (A)
Cytotoxicity measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Proliferation in A549 cells was significantly inhibited by ILK RNAi
and CDDP. (B) Apoptosis measured by flow cytometry. (C) Statistical
comparisons. The results demonstrated that transfection with KD and
CDDP treatment significantly enhanced cell apoptosis. Data are
presented as the mean ± standard deviation of a representative
experiment performed in triplicate (n=3). *P<0.05,
compared with the Con group; #P<0.05, compared with
the NC group. NC, cells transfected with negative control RNAi;
CON, control; KD, cells transfected with ILK specific RNAi; ILK,
integrin-linked kinase; CDDP, cisplatin. |
Synergistic effects between CDDP and ILK
RNAi on proliferation and apoptosis in A549 cells
Using drug sensitivity analysis, it was found that
10% CDDP treatment produced the maximal decrease in cell viability
(5% CDDP, 0.876±0.015 vs 10% CDDP, 0.921±0.009, P<0.05; no
difference between 10, 15, 20 and 25% treatment was observed,
0.921±0.009 vs 0.934±0.005, 0.934±0.004, 0.941±0.003; P>0.05).
Therefore, proliferation and apoptosis in A549 cells undergoing ILK
RNAi infection and 10% CDDP treatment was evaluated. Cells infected
with ILK specific-RNAi and addition of CDDP exhibited significantly
increased apoptotic levels and inhibited cell viability compared
with the control cells (Fig. 3A, B and
C), indicating a synergistic effect between CDDP and ILK
RNAi.
Effect of downstream gene expression of
ILK on the regulation of cell survival and apoptosis
In order to investigate how cell survival and
apoptosis were affected by ILK RNAi and CDDP, whether ILK RNAi
regulated downstream gene expression was investigated. It was found
that MMP-9, p-GSK3β, p-AKT, AP-1, β-catenin and cyclin D1 protein
levels were downregulated in the ILK specific-RNAi trans-fected
cells. VEGF exhibited no change following ILK knockdown (Fig. 4).
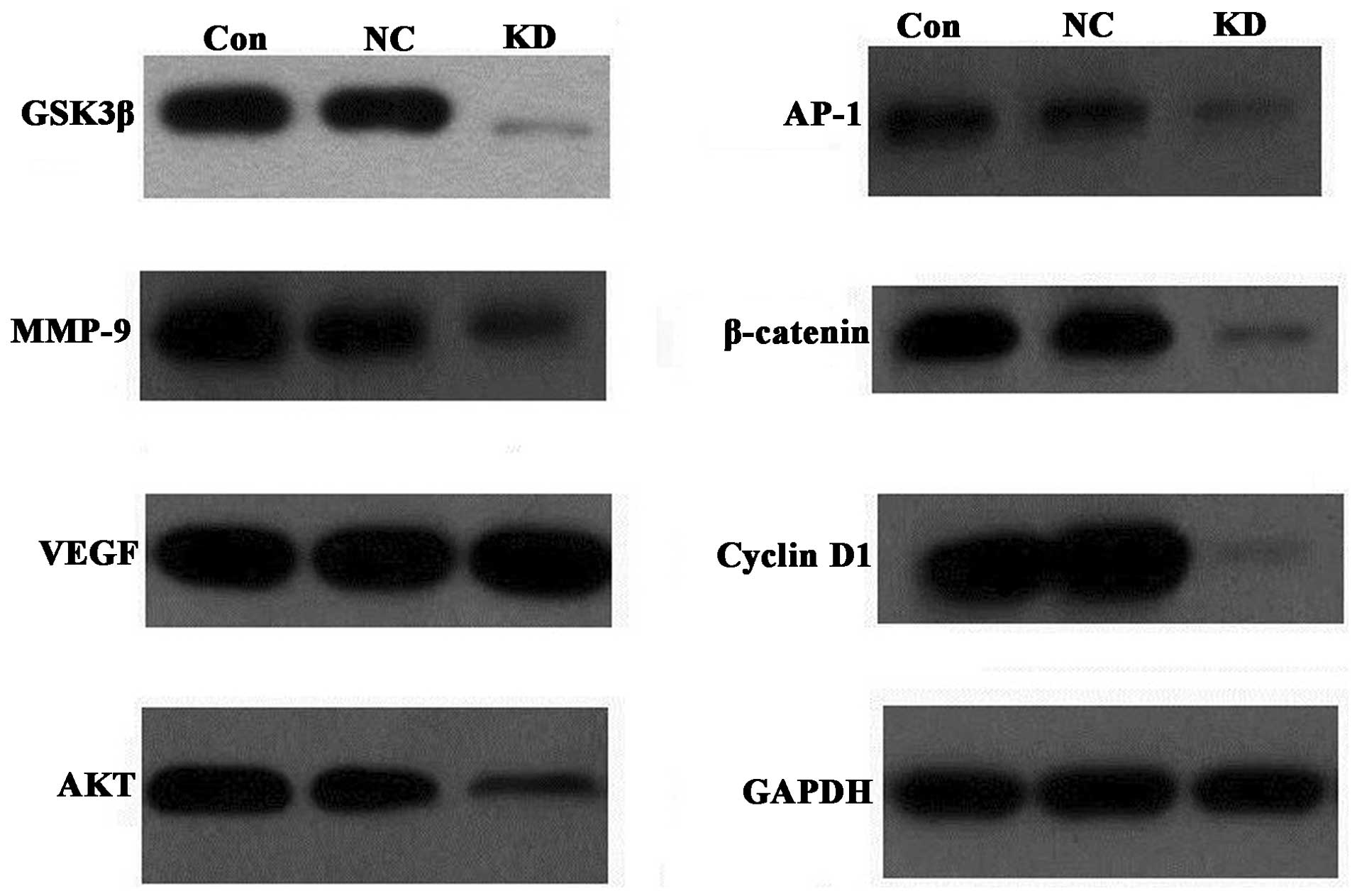 | Figure 4Effect of ILK knockdown on downstream
protein expression by western blotting. The protein expression of
GSK3β, AKT, AP-1, β-catenin, cyclin D1 and MMP-9 were found to be
downregulated in the ILK specific-RNAi transfected cells. VEGF
demonstrated no change following ILK knockdown. GAPDH was used as
an internal control. CON, parent cells; NC, cells transfected with
negative control RNAi; KD, cells transfected with ILK specific
RNAi; GSK, glycogen-synthase kinase; VEGF, vascular endothelial
growth factor; ILK, integrin-linked kinase; AP, activator protein;
MMP, matrix metalloproteinase. |
Discussion
A significant number of studies have identified that
ILK is a potential oncogene and inhibition of ILK by siRNA
(15,16) or small molecules (18) may be a potentially useful
therapeutic approach for treating cancer. However, whether the
knockdown of ILK affects growth and apoptosis of lung cancer cells
remains to be elucidated. In the present study, data revealed that
lentivirus-mediated ILK gene silencing may significantly inhibit
A549 cell proliferation and alter cell cycle progression. However,
treatment with ILK RNAi alone had significant effects on cell
apoptosis. CDDP-based combination chemotherapy is currently one of
the most active treatments for advanced lung cancer. The mechanism
of cisplatin-based chemotherapy is generally accepted as its
ability to form adducts with DNA and cause DNA strand breaks in the
nucleus, which interferes with normal transcription and/or DNA
replication, leading to either repair of the DNA damage and cell
survival or activation of the irreversible cell death program as a
consequence (4,5). However, in the present study no
significant differences in apoptosis in parent A549 cells and
negative control-RNAi transfection cells with or without CDDP
treatment were identified. This may be attributed to cisplatin
insensitivity of A549 cell lines (19). In consideration of the mechanism of
the above two approaches, lentivirus-mediated ILK siRNA and
cisplatin-based chemotherapy were combined to investigate whether
there is a synergistic interaction between them to promote cell
apoptosis and inhibit cell growth. As expected, the present results
demonstrated that apoptosis was signifi-cantly increased in cells
infected with ILK specific-RNAi and addition of CDDP compared with
other groups (Fig. 3). These
findings suggest combined treatment modalities with
lentivirus-mediated ILK interference and cisplatin chemotherapy may
be more effective for cell apoptosis induction than
mono-chemotherapy or knockdown. This contrasted with the conclusion
drawn from the study by Kalra et al (20), who demonstrated that combination of
CDDP and QLT0267, an ILK inhibitor, produced antagonistic
interactions in a breast cancer model. This may result from the
different pharmacological effects of these two compounds.
Furthermore, the present results also revealed that
ILK siRNA may affect cell growth and apoptosis by regulating its
downstream genes, including p-GSK3β, p-AKT, AP-1, β-catenin, cyclin
D1 and MMP-9. Indirectly, it was also demonstrated that these
downstream genes may mediate cisplatin resistance in lung cancer
cells. These conclusions appeared to be in accordance with previous
studies: ILK kinase activity is rapidly stimulated by the
engagement of inte-grins to the extracellular matrix components.
These stimuli result in activation of protein kinase B/Akt,
suppression of apoptosis and promotion of cell survival. Thus,
targeting inhibition of ILK led to low expression of p-Akt and
promoted cell apoptosis (21,22).
Additionally, Akt activity is reported to be a determinant of CDDP
resistance (23–25). Therefore, reduced expression of
p-Akt may reduce this resistance, further inducing cell apoptosis.
In addition to regulating the activity of PKB/Akt, ILK also
inhibits the activity of GSK-3 by phosphorylation at Ser9 (26). Downregulation of ILK led to a
decrease in p-GSK3β and an increase in GSK-3 activity, which has
been demonstrated to facilitate the cell apoptosis pathway
(27–29). Further studies indicate that GSK-3
may be involved in cancer cell cycle arrest and apoptosis by
regulating cyclin D1 expression, nuclear translocation of β-catenin
and activation of the transcription factor AP-1 (26,30).
Cyclin D1 is frequently overexpressed in lung cancer patients
(31) and associated with poor
survival of patients with lung cancer (32). Non-small cell lung cancer cells
transfected with cyclin D1-targeted siRNA exhibited a marked
decrease in cell growth rate and invasive capacity (33). AP-1 is a major transcription factor
that regulates MMP-9 expression, which may contribute to the lower
invasiveness and growth potential of cancer cells (34,35).
In conclusion, the results from the present study
have identified for the first time, to the best of our knowledge,
that downregulating ILK expression inhibits proliferation and cell
cycle arrest in lung cancer cells. Downregulation of ILK expression
and CDDP treatment, in combination, is a more effective approach
for the treatment of lung cancer through affecting downstream gene
expression, including p-GSK3β, p-AKT, AP-1, β-catenin, cyclin D1
and MMP-9.
Acknowledgments
This study was supported by the National Science
Foundation (grant no. 81072739) and the Xinglin Scholar Program of
Shanghai University of Traditional Chinese Medicine (grant no.
2209–13–03).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Metro G, Chiari R, Mare M, et al:
Carboplatin plus pemetrexed for platinum-pretreated, advanced
non-small cell lung cancer: a retrospective study with
pharmacogenetic evaluation. Cancer Chemother Pharmacol.
68:1405–1412. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siddik ZH: Mechanisms of action of cancer
chemotherapeutic agents: DNA-interactive alkylating agents and
antitumour platinum-based drugs. (The Cancer Handbook). 1st
Edition. Nature Publishing Group; London: pp. 1295–1313. 2002
|
|
5
|
Rosell R, Lord RV, Taron M and Reguart N:
DNA repair and cisplatin resistance in non-small-cell lung cancer.
Lung Cancer. 38:217–227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bordin DL, Lima M, Lenz G, et al: DNA
alkylation damage and autophagy induction. Mutat Res. 753:91–99.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galluzzi L, Senovilla L, Vitale I, et al:
Molecular mechanisms of cisplatin resistance. Oncogene.
31:1869–1883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cetintas VB, Kucukaslan AS, Kosova B, et
al: Cisplatin resistance induced by decreased apoptotic activity in
non-small-cell lung cancer cell lines. Cell Biol Int. 36:261–265.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hannigan G, McDonald P, Walsh M and Dedhar
S: Integrin-linked kinase: not so ‘pseudo’after all. Oncogene.
30:4375–4385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takanami I: Increased expression of
integrin-linked kinase is associated with shorter survival in
non-small cell lung cancer. BMC Cancer. 5:12005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Watzka SB, Rauscher-Pötsch I, Stubenberger
E, et al: Immunoreactivity of integrin-linked kinase in primary
non-small-cell lung cancer and survival after curative resection.
Eur J Cardiothorac Surg. 38:254–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Posch F, Setinek U, Flores RM, et al:
Serum integrin-linked kinase (sILK) concentration and survival in
non-small cell lung cancer: a pilot study. Clin Transl Oncol.
16:455–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao M, Gao Y, Wang L, et al:
Overexpression of integrin-linked kinase promotes lung cancer cell
migration and invasion via NF-κB-mediated upregulation of matrix
metalloproteinase-9. Int J Med Sci. 10:995–1002. 2013. View Article : Google Scholar
|
|
14
|
Chen D, Zhang Y, Zhang X, et al:
Overexpression of integrin-linked kinase correlates with malignant
phenotype in non-small cell lung cancer and promotes lung cancer
cell invasion and migration via regulating epithelial-mesenchymal
transition (EMT)-related genes. Acta Histochem. 115:128–136. 2013.
View Article : Google Scholar
|
|
15
|
Gao J, Zhu J, Li HY, Pan XY, Jiang R and
Chen JX: Small interfering RNA targeting integrin-linked kinase
inhibited the growth and induced apoptosis in human bladder cancer
cells. Int J Biochem Cell Biol. 43:1294–1304. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu XY, Liu N, Liu W, Song SW and Guo KJ:
Silencing of the integrin-linked kinase gene suppresses the
proliferation, migration and invasion of pancreatic cancer cells
(Panc-1). Genet Mol Biol. 35:538–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song W, Jiang R and Zhao CM: Role of
integrin-linked kinase in multi-drug resistance of human gastric
carcinoma SGC7901/DDP cells. Asian Pac J Cancer Prev. 13:5619–5625.
2012. View Article : Google Scholar
|
|
18
|
Younes MN, Yigitbasi OG, Yazici YD, et al:
Effects of the integrin-linked kinase inhibitor QLT0267 on squamous
cell carcinoma of the head and neck. Arch Otolaryngol Head Neck
Surg. 133:152007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dolfen D, Schottler K, Valiahdi SM, et al:
Synthesis, structures and in vitro cytotoxicity of some platinum
(II) complexes containing thiocarbamate esters. J Inorg Biochem.
102:2067–2071. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kalra J, Warburton C, Fang K, et al:
QLT0267, a small molecule inhibitor targeting integrin-linked
kinase (ILK) and docetaxel can combine to produce synergistic
interactions linked to enhanced cytotoxicity, reductions in P-AKT
levels, altered F-actin architecture and improved treatment
outcomes in an orthotopic breast cancer model. Breast Cancer Res.
11:R252009. View
Article : Google Scholar
|
|
21
|
Persad S, Attwell S, Gray V, et al:
Inhibition of integrin-linked kinase (ILK) suppresses activation of
protein kinase B/Akt and induces cell cycle arrest and apoptosis of
PTEN-mutant prostate cancer cells. Proc Natl Acad Sci USA.
97:3207–3212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Troussard AA, McDonald PC, Wederell ED,
Mawji NM, et al: Preferential dependence of breast cancer cells
versus normal cells on integrin-linked kinase for protein kinase
B/Akt activation and cell survival. Cancer Res. 66:393–403. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar
|
|
24
|
Yang X, Fraser M, Moll UM, Basak A and
Tsang BK: Akt-mediated cisplatin resistance in ovarian cancer:
modulation of p53 action on caspase-dependent mitochondrial death
pathway. Cancer Res. 66:3126–3136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peng DJ, Wang J, Zhou JY and Wu GS: Role
of the Akt/mTOR survival pathway in cisplatin resistance in ovarian
cancer cells. Biochem Biophys Res Commun. 394:600–605. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maydan M, McDonald PC, Sanghera J, et al:
Integrin-linked kinase is a functional Mn2+-dependent
protein kinase that regulates glycogen synthase kinase-3β (GSK-3β)
phosphorylation. PloS One. 5:e123562010. View Article : Google Scholar
|
|
27
|
Beurel E and Jope RS: The paradoxical
pro-and anti-apoptotic actions of GSK3 in the intrinsic and
extrinsic apoptosis signaling pathways. Prog Neurobiol. 79:173–189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Watcharasit P, Thiantanawat A and
Satayavivad J: GSK3 promotes arsenite-induced apoptosis via
facilitation of mitochondria disruption. J Appl Toxicol.
28:466–474. 2008. View
Article : Google Scholar
|
|
29
|
Ngok-Ngam P, Watcharasit P, Thiantanawat A
and Satayavivad J: Pharmacological inhibition of GSK3 attenuates
DNA damage-induced apoptosis via reduction of p53 mitochondrial
translocation and Bax oligomerization in neuroblastoma SH-SY5Y
cells. Cell Mol Biol Lett. 18:58–74. 2013. View Article : Google Scholar
|
|
30
|
Ban JO, Kwak DH, Oh JH, et al: Suppression
of NF-κB and GSK-3β is involved in colon cancer cell growth
inhibition by the PPAR agonist troglitazone. Chem Biol Interact.
188:75–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kosacka M, Piesiak P, Kowal A, Gołecki M
and Jankowska R: Galectin-3 and cyclin D1 expression in non-small
cell lung cancer. J Exp Clin Cancer Res. 30:101–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li R, An SJ, Chen ZH, et al: Expression of
cyclin D1 splice variants is differentially associated with outcome
in non-small cell lung cancer patients. Hum Pathol. 39:1792–1801.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang H, Hu YD, Li N and Zhu Y: Inhibition
of tumor growth and metastasis by non-small cell lung cancer cells
transfected with cyclin D1-targeted siRNA. Oligonucleotides.
19:151–162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Troussard AA, Costello P, Yoganathan TN,
Kumagai S, Roskelley CD and Dedhar S: The integrin linked kinase
(ILK) induces an invasive phenotype via AP-1 transcription
factor-dependent upregulation of matrix metalloproteinase 9
(MMP-9). Oncogene. 19:5444–5452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lakka SS, Gondi CS, Yanamandra N, et al:
Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma
cell line via RNA interference reduces tumor cell invasion, tumor
growth and angiogenesis. Oncogene. 23:4681–4689. 2004. View Article : Google Scholar : PubMed/NCBI
|