Introduction
One of the major causes of acute renal failure (ARF)
is ischemia, which occurs in kidney transplantation, partial
nephrectomy, renal artery angioplasty, sepsis, accidental or
iatrogenic trauma, hydronephrosis, elective urological operations,
aortic bypass surgery, cardiopulmonary bypass, the use of
vasoconstricting drugs and certain hypotensive states (1,2). ARF
has a high incidence in intensive care units, representing an
isolated prognostic factor in patients with multiple organ
dysfunction syndrome (3). The
clinical significance of ARF is due to its high mortality, which
ranges between 30 and 70% (4).
Thus, novel therapies are required to prevent or alleviate ischemic
injury.
Previous studies have demonstrated that ischemic
preconditioning (IPR) and ischemic postconditioning (IPO) are two
important mechanical methods, which are able to improve the ability
of organs subjected to ischemia to tolerate injury (5,6).
Although IPR is effective at reducing ischemia-reperfusion injury
(IRI), its clinical application is limited as it must be initiated
prior to the ischemic period, which is unreasonable in a clinical
situation. IPO is a series of brief rapid intermittent cycles of
ischemia applied at the onset of reperfusion in the previously
ischemic tissue or organ (7).
Several studies have demonstrated that IPO was able to cause a
significant reduction in the systemic inflammatory response,
inhibit the expression of apoptosis-associated molecules and
activate endogenous protective molecules (8–10).
In renal IPO studies, major studies were based on animal models,
including our earlier studies using rat or canine models (11,12).
However, to the best of our knowledge, an in vitro
postconditioning model, which is able to effectively simulate the
process of IPO against IRI in the kidney, has not yet been
investigated. Based on a study using an in vitro model for
13), a novel IPO model, which simulates IPO in the kidney was
developed in the present study using a rat proximal tubular cell
line (NRK-52E cells). In addition, the molecular mechanism involved
in in vitro IPO of renal tubular epithelial cells was
analyzed.
Materials and methods
Cell culture
The renal tubular epithelial cell line, NRK-52E, was
purchased from the Cell Resource Center of the Shanghai Institutes
for Biological Sciences, Chinese Academy of Sciences (Shanghai,
China). The cells were cultured on culture dishes with 5%
CO2 and maintained at pH 7.4 and 37°C. The medium was
changed once every 3 days and the cells were used for experiments
at day 10 after seeding. Cells were cultured in serum-free medium
for 24 h prior to the experiments. Cells were seeded on 6-well
plates or culture dishes as appropriate.
In vitro IPO model
Prior to the experiment, the cells were placed in
serum-free medium for 24 h. Subsequently, all cell culture dishes
were randomly divided into nine groups (Fig. 1). For the normal group, the cells
were cultured in complete medium under normal conditions (5%
CO2, saturated humidity and 37°C) and 3 h later fresh
medium was added and cultured under the same conditions for 24 h.
For the control group, the cells were cultured in control
buffer(NaHCO3 24.0 mM, Na2HPO4 0.8
mM, NaH2PO4 0.2 mM, NaCl 86.5 mM, KCl 5.4 mM,
CaCl2 1.2 mM, MgCl2 0.8 mM, HEPES 20 mM and 5
mM glucose; pH adjustment to 7.4 with 1 N NaOH) (13) for 3 h and further cultured in
complete medium for 24 h. The cells in the ischemia/reperfusion
(I/R) group were washed with phosphate-buffered saline (PBS; Gibco
Life Technologies, Carlsbad, CA, USA) and placed in ischemic buffer
(NaHCO3 4.5 mM, Na2HPO4 0.8 mM,
NaH2PO4 0.2 mM, NaCl 106.0 mM, KCl 5.4 mM,
CaCl2 1.2 mM, MgCl2 0.8 mM and
morpholinoethanesulfonic acid 20 mM; pH 6.6) (13), and exposed to ischemic conditions
(5% CO2, 0.5% O2, saturated humidity and
37°C) using a tri-gas incubator for 3 h. Subsequently, the cells
were placed into the complete medium under normal conditions
representing the reperfusion period for 24 h. With respect to the
group simulating IPO, there were two approaches: i) Cells were
placed in the ischemic buffer and cultured under hypoxic conditions
for 3 h and cultured further in complete medium under normal
conditions for 10 min. This was followed by placing cells into
ischemic buffer and then growing them in ischemic conditions for 10
min. Thus, cells that had undergone one cycle of IPO were termed
the IPO1 group. Cells undergoing two or three cycles were termed
the IPO2 group and IPO3 group, respectively. Following this, cells
were cultured in complete medium under normal conditions for 24 h.
ii) Cells were placed in ischemic buffer and cultured under mimic
ischemic conditions for 3 h. Subsequently, the complete medium was
added and the cells were grown under normal conditions for 10 min.
The medium was not replaced with buffer and was directly exposed to
ischemic conditions for 10 min. Cells undergoing one cycle were
termed the IPO1+ group. According to this, cells undergoing two or
three cycles were termed the IPO2+ group and IPO3+ group,
respectively. Cells were further placed in complete medium and
cultured under normal conditions for 24 h.
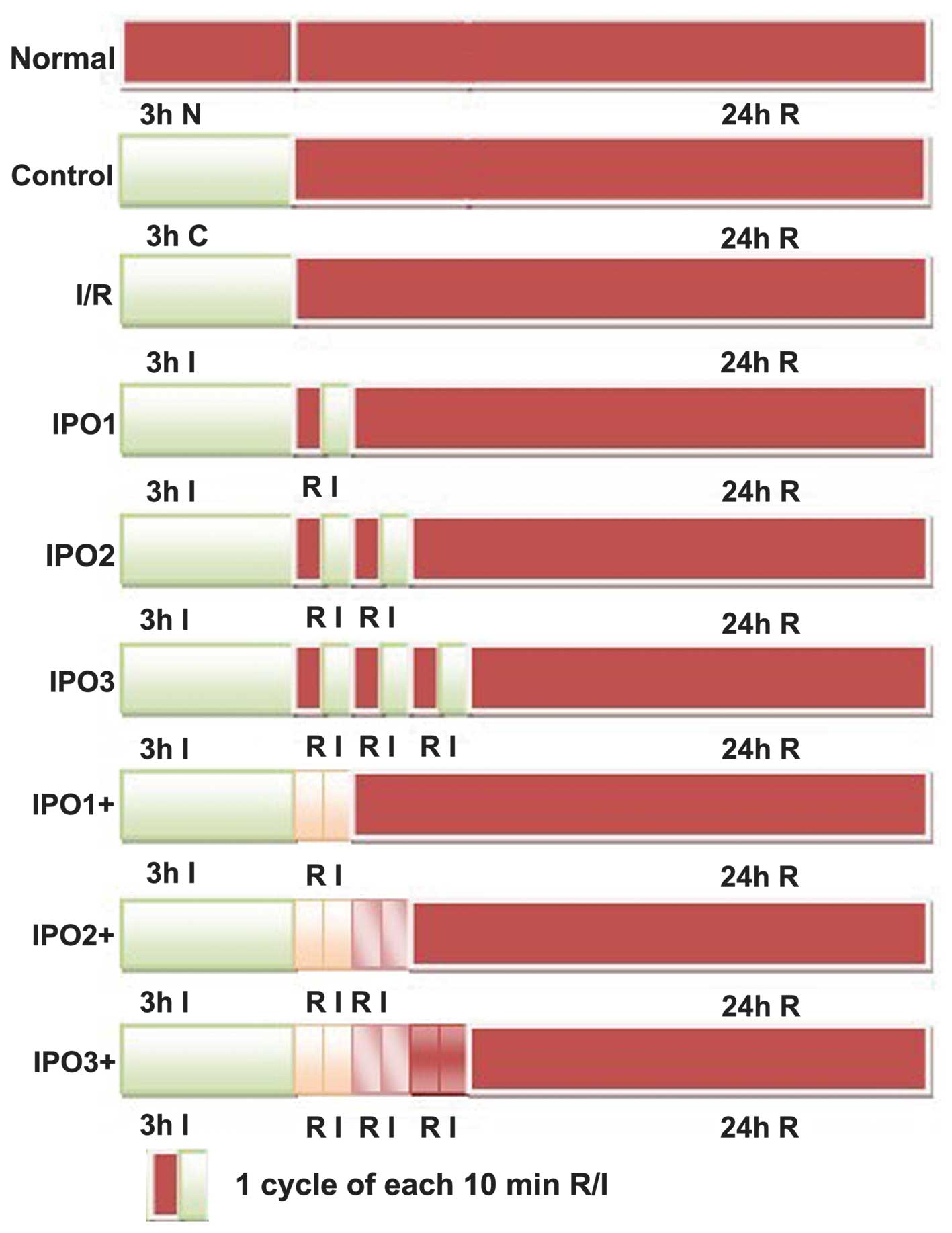 | Figure 1Experimental procedure used to
determine the effect of IPO following I/R in an in vitro
model. Normal, normal condition culture; control, cells cultured in
control medium followed by reperfusion; I/R, cells cultured in
ischemic conditions followed by reperfusion; IPO, cells cultured in
ischemic conditions for 3 h, then replaced with complete medium and
cultured under normal conditions for 10 min, followed by placing
cells in ischemic conditions for 10 min. IPO1 group, one cycle of
IPO; IPO2, two cycles of IPO; IPO3, three cycles of IPO. IPO+,
cells cultured under mimic ischemic conditions for 3 h. The
ischemic buffer was not changed and another 0.5 ml fresh complete
medium was added. The cells were grown under normal conditions for
10 min. Following this, the cells were exposed to ischemic
conditions for 10 min without changing the mixed medium. IPO1+, one
cycle of IPO+; IPO2+, two cycles of IPO+; IPO3+, three cycles of
IPO+. I, ischemia; R, reperfusion; IPO, ischemic
postconditioning. |
Analysis of apoptosis by flow
cytometry
For analysis of apoptosis, 5×105 NRK-52E
cells were cultured on a 6-well plate containing 2 ml serum-free
medium. After 24 h, all cells were processed in accordance with the
above grouping. NRK-52E cells were collected 24 h post-treatment
and stained with Analysis of apoptosis by flow cytometry
(FITC)-conjugated annexin V and propidium iodide (PI) according to
the manufacturer’s instructions of the apoptosis detection kit
(Annexin V/PI Apoptosis kit; Liankebio, Hangzhou, China). Flow
cytometry was performed for analysis of apoptosis (FACSAria; BD
Biosciences, Heidelberg, Germany).
Hoechst 33258 staining
In order to distinguish apoptotic cells from
necrotic cells, the cells were stained with Hoechst 33258. The
cells from different groups were fixed using Carnoy’s fixative
(Beyotime Institute of Biotechnology, Haimen, China) for 10 min.
Cells were washed with PBS (pH 7.4), stained with Hoechst 33258 (10
μg/ml) for 5 min at room temperature and microscopically
examined (BX-53F; Olympus Corporation, Tokyo, Japan).
Western blot analysis
The protein expression levels of B-cell lymphoma 2
(Bcl-2), Bcl-2-associated X protein (Bax) caspase-3, caspase-8 and
cleaved caspase-3 were examined by western blotting. Briefly,
proteins were extracted from NRK-52E cells, separated on 10%
SDS-PAGE gels and transferred onto a nitrocellulose membrane
(Novex, San Diego, CA, USA). The membranes were blocked with 5%
non-fat milk in Tris-buffered saline (Boster Biological Technology,
Ltd., Wuhan, China) and Tween 20 buffer (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and incubated with the following rabbit
primary antibodies: Bax (1:1,000; cat. no. 2772, Cell Signaling
Technology, Inc., Danvers, MA, USA), Bcl-2 (1:1,000; cat. no. 3498,
Cell Signaling Technology, Inc.), caspase-3 (1:1,000; cat. no.
9662, Cell Signaling Technology, Inc.), cleaved caspase-8 (1:1,000;
cat. no. 9429, Cell Signaling Technology, Inc.) and cleaved-caspase
3 (1:1,000; cat. no. 9661, Cell Signaling Technology, Inc.). All of
the primary antibodies were polyclonal, except for the antibody
targeting Bcl-2, which was monoclonal. Subsequently, the membranes
were incubated with secondary horseradish peroxidase-conjugated
goat anti-rabbit IgG antibody (1:2,000; ZDR-5306; ZSGB-BIO,
Beijing, China). Specific bands were developed and visualized using
an enhanced chemiluminescence detection kit (Immobilon Western
Chemiluminescent HRP Substrate; Merck Millipore, Darmstadt,
Germany).
Statistical analysis
All experiments were repeated in triplicate. Data
are presented as the mean ± standard error of the mean. For
determining the number of apoptotic cells, the groups were compared
using one-way analysis of variance and Student-Newman-Keuls test.
P<0.05 was considered to indicate a statistically significant
difference. All the statistical tests were performed using GraphPad
Prism software version 5.0 (GraphPad Software, Inc., San Diego, CA,
USA).
Results
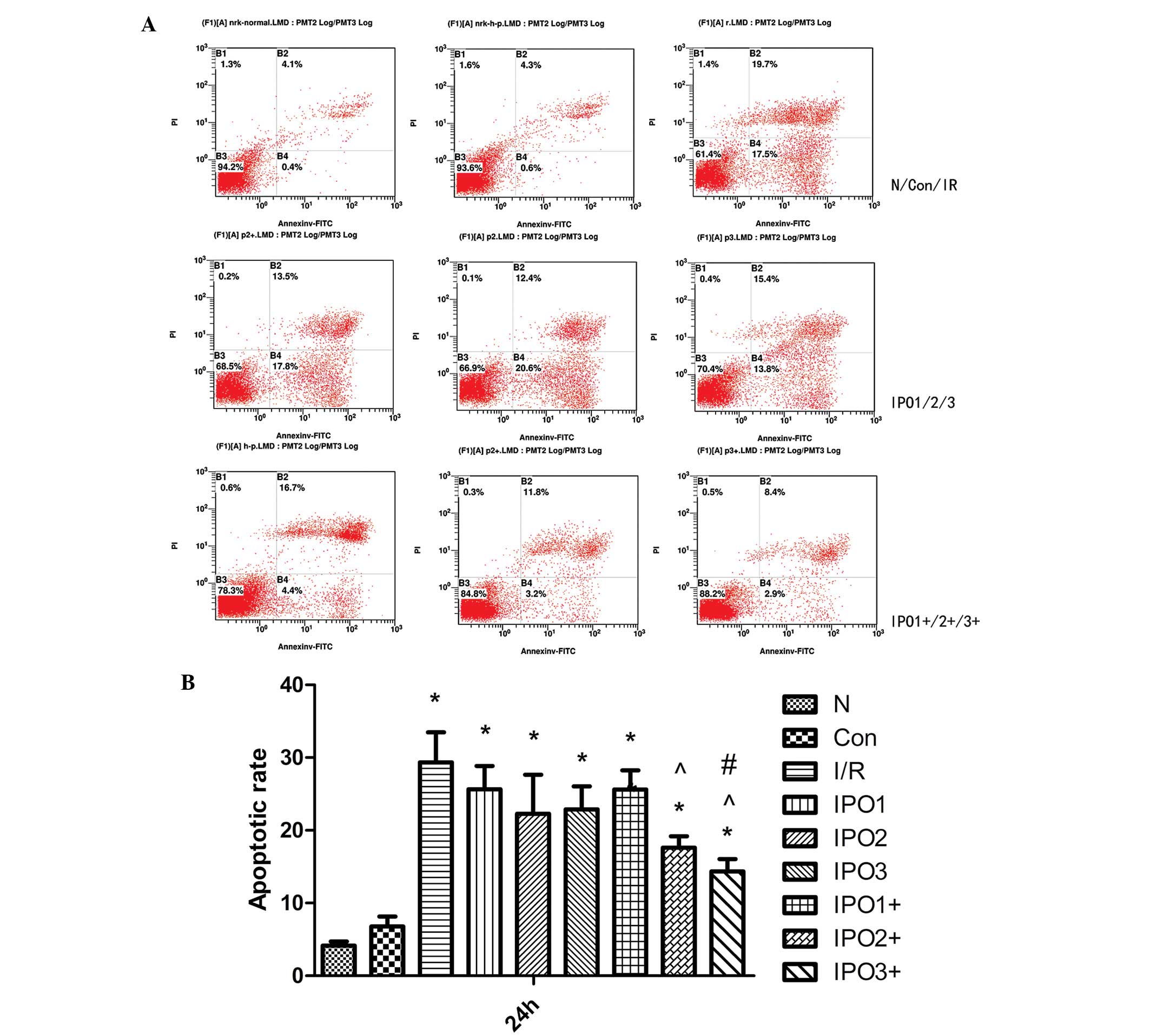
Apoptotic level in NRK-52E cells
following postconditioning
NRK-52E cells subjected to serum starvation were
treated in accordance with the grouping described earlier. After 24
h of culture, NRK-52E were collected and stained with
FITC-conjugated annexin V and propidium iodide for detecting
apoptosis. As shown in Fig. 2, the
rate of apoptosis in the control group was significantly lower than
in the I/R, IPO1, IPO2, IPO3 and IPO1+ groups (P<0.05). Compared
with the IPO2+ and IPO3+ group, the rate of apoptosis in the I/R,
IPO1 and IPO2 groups was significantly higher (P<0.05). In
addition, a significant difference between the IPO3 and IPO3+ group
(P<0.05) was observed. As the apoptotic rate of the IPO3+ group
was lower than in other postconditioning groups, this indicated
that this group was the most effective at reducing IRI. Therefore,
in the following experiments, the IPO3+ group was the only
post-processing method investigated.
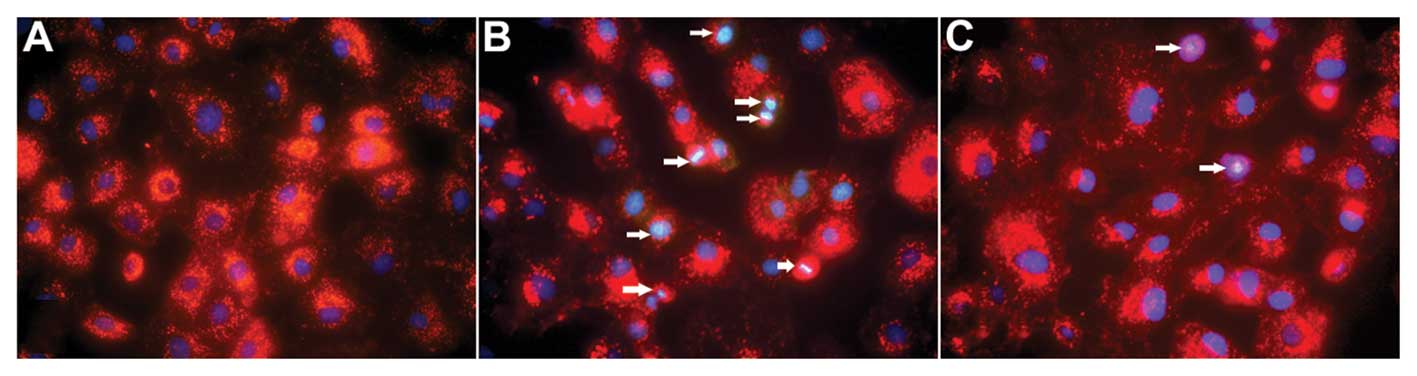
As shown in Fig. 3,
hoechst 33258 staining revealed that the nuclear chromatin was
affected. In the control group, faint blue fluorescence was
observed in the cell nuclei, which were homogenous. In the I/R
group, the blue emission was significantly brighter than in the
control group. In I/R cells, condensed chromatin was visible and
the formation of apoptotic bodies was observed. Compared with the
I/R group, the bright blue emission in the IPO3+ group was clearly
attenuated, suggesting that postconditioning was able to ameliorate
the injury caused by I/R.
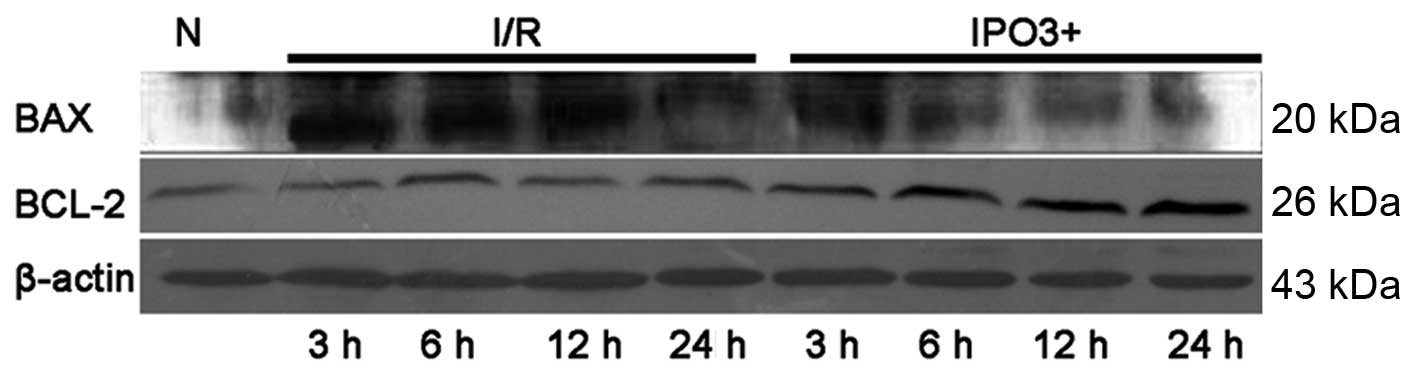
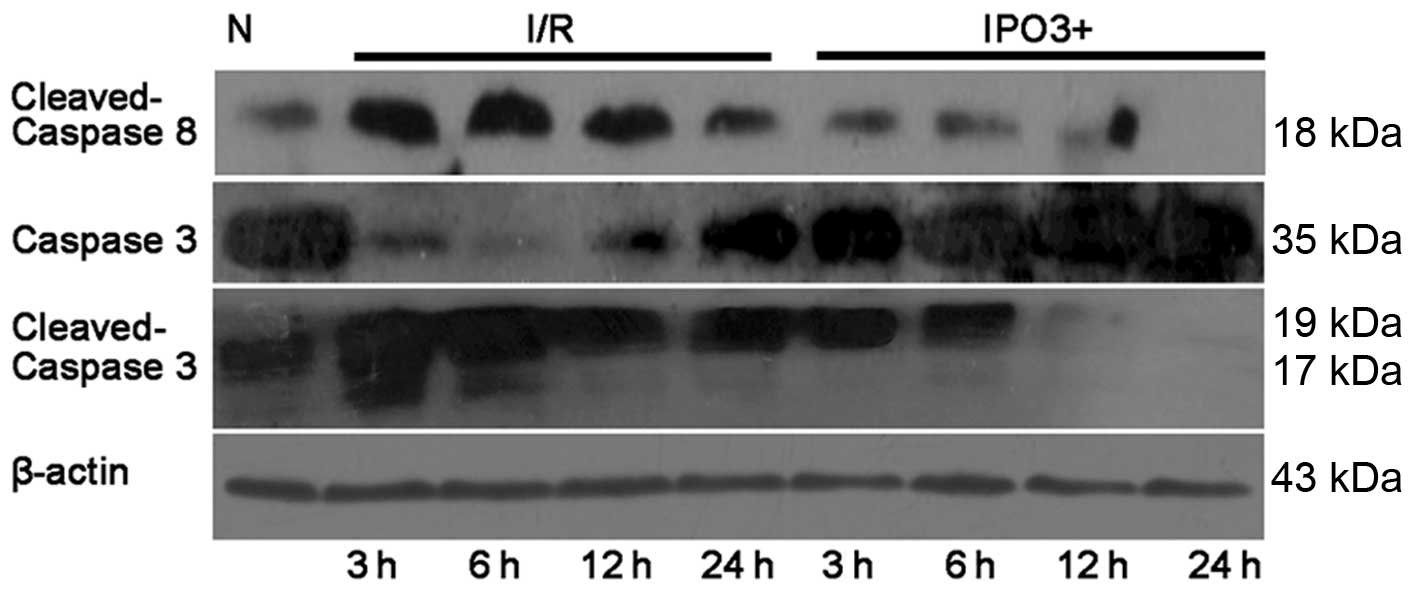
Expression level of apoptotic proteins in
NRK-52E cells due to postconditioning
The protein expression levels of Bax, Bcl-2,
pro-caspase-3, caspase-8 and cleaved caspase were examined by
western blotting (Figs. 4 and
5). The expression of Bcl-2, a
crucial inhibitor of the apoptotic process was upregulated in the
IPO3+ group, when compared with the control group and I/R group. In
addition, with the increase in postconditioning time, the
expression of Bcl-2 was increased in the IPO3+ group. Cleaved
caspase-3, which is the activated product of pro-caspase-3, has
catalytic activity in cell apoptosis. Western blot analysis
demonstrated a significant increase in cleaved caspase-3 in the I/R
group compared with the control group and with increases in time
this trend continued. In the IPO3+ group, the levels of cleaved
caspase-3 were clearly restored compared with those in the I/R
group. The expression of pro-caspase-3 was decreased in the I/R
group and restored in the IPO3+ group. Caspase-8, known to be
involved in apoptosis, was significantly increased in the I/R and
IPO3+ groups. However, the protein level of caspase-8 was
significantly lower in the IPO3+ group compared with the I/R
group.
Discussion
IPO, which was first reported by Zhao, has been
demonstrated to be an effective strategy against IRI (14). Previous studies have focused on
observing the protective effect of IPO on IRI in vivo.
However, few studies have focused on this effect in in vitro
conditions. Although it is not possible to use an in vitro
model to create an accurate replica of the in vivo
environment, in vitro models have several advantages over
in vivo as they provide an environment where the specific
stimuli can be controlled, isolated and assessed to determine their
contribution and effect on physiological or pathophysiological
events (15). Thus, in the present
study, an in vitro postconditioning model was created, which
was as close to the in vivo IPO pathophysiological
environment as possible, using a rat proximal tubular cell line
(NRK-52E cells) based on the model used by Sauvant et al
(13). In this model, all cellular
injury caused by reperfusion, including ‘hypoxia, hypercapnia
induced acidosis, limited nutrient availability and waste removal
impairment’ were taken into account.
Previous studies have established an in vitro
model to simulate the ischemic environment, one of them using
NRK-52E cells with mineral oil overlay (16,17).
Although this method was able to approximately establish an in
vitro model of ischemia, the accuracy of simulation of the
ischemic environment was not effective and the protective effect of
postconditioning for ischemia was not able to be investigated. A
simple model of hypoxia and reoxygenation can be used to simulate
cells in IRI (18,19). Nutrients and oxygen
deprived/regeneration of the normal culture atmosphere system could
simulate ischemia-reperfusion (20,21).
However, in the present study, two different methods were used to
create an in vitro model of ischemia and postconditioning
and were compared.
There were two methods to achieve the
postconditioning process in the present study. For one approach,
following the completion of the pretreatment (24 h serum starvation
and 3 h mimic ischemia), 1 ml of complete medium was replaced at
each time point during the ischemia-reperfusion cycle. This
postconditioning method had certain effects on reducing cell damage
and inhibiting cell apoptosis, and compared with the normal group,
a significant difference in the apoptotic rate (P<0.05) was
observed. For the second approach, following the completion of the
pretreatment (24 h serum starvation and 3 h mimic ischemia), the
consumed medium was not removed and 0.5 ml fresh medium was added
to the dish at each reperfusion cycle. Flow cytometric analysis
results indicated that the rate of apoptosis in the IPO+ group was
lower than in the IPO group and a significant difference between
the IPO3 and IPO3+ group (P<0.05) was observed. This suggested
that the IPO+ treatment methods were superior to the IPO approach
and the IPO3+ group was selected as the postconditioning group for
Hoechst and western blot analysis. The result of Hoechst staining
indicated that the IPO3+ group was able to effectively reduce the
damage caused by ischemia and reperfusion.
The proteins of the Bcl-2 family, which are crucial
regulatory factors, can either promote cell survival, for example
Bcl-2, or cell death, for example Bax, by apoptosis (22–24).
Increased Bcl-2 can enhance cell survival and evidence indicates
that the increased level of Bcl-2 exerts protective effects against
apoptosis (25). However, Bax, a
key regulator of programmed cell death, is an apoptotic protein and
acts by activating caspases (26).
The ratio of Bcl-2 to Bax determines the cellular susceptibility to
apoptotic stimuli (26–30). The present study found that, as the
expression of Bcl-2 gradually increased and Bax gradually
decreased, the ratio of the expression of Bcl-2 with Bax was
increased in the IPO3+ group compared with the control group and
I/R group demonstrating an increased resistance to apoptosis. In
addition, with increasing time, this trend became more apparent. It
indicated that the postconditioning method in the IPO3+ group was
able to inhibit the apoptotic process in NRK-52E cells subjected to
simulative ischemia.
Several studies have demonstrated that the caspase
family is able to promote and implement cell apoptosis in mammalian
cells and caspase-3 is the most crucial downstream apoptotic
protease in the caspase cascade (31,32).
A number of extracellular signals activate caspase-8 through the
Fas receptor pathway and the activation of caspase-8 then promotes
caspase-3 activation, which hydrolyzes cell-specific proteins, and
poly-ADP ribose polymerase, thus inducing apoptosis (33,34).
Pro-caspase-3 itself does not have catalytic activity and it
divides into two fragments in the activation process to produce the
active form of caspase-3. In the present study, the expression of
caspase-3 and caspase-8 was examined by western blot analysis. The
result of cleaved caspase-3 and caspase-8 demonstrated a
significant decrease in the IPO3+ group compared with the I/R
group. In addition, with increasing time this trend became more
apparent. In the IPO3+ group subjected to 24 h of culture following
postconditioning, the expression of cleaved caspase-3 and caspase-8
decreased. This indicated that this postconditioning method was
able to inhibit the activation of the caspase pathway in NRK-52E
cells subjected to simulated ischemia.
In in vivo conditions, the process of IPO
involves repeated renal artery occlusion and opening. The
metabolism of the ischemic organ and the nutrient supply are slowly
restored to pre-ischemic levels, which are the essence of IPO. In
ex vivo conditions in the present study, 0.5 ml fresh
complete medium was added into the ischemic buffer three times,
which leads to gradual restoration of pre-ischemic levels in the
extracellular environments (oxygen, PH, nutrient, waste removal
impairment) and thus mimic the process of IPO in vivo.
However, the duration of protective effects of IPO and the exact
number of optimal intervals and cycles remain to be elucidated. In
addition, in vitro models do not fully represent in
vivo conditions. Therefore, more studies are required to
investigate this further.
In the present study, an in vitro
postconditioning model was established, which was able to
effectively simulate the process of IPO against IRI in the kidney.
In this model, cells were effectively protected from IRI by IPO.
Furthermore, this protection was achieved by inhibiting caspase
activation, thereby reducing cell apoptosis. In conclusion, IPO of
NRK-52E cells in vitro offers a potentially valuable
strategy to investigate the protective mechanism of IPO against
IRI.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 30901494, 30901552
and 2013RMFH012) and the Hubei Provincial Natural Science
Foundation (grant no. 2009CDB382).
References
|
1
|
Yun Y, Duan WG, Chen P, et al: Ischemic
postconditioning modified renal oxidative stress and lipid
peroxidation caused by ischemic reperfusion injury in rats.
Transplant Proc. 41:3597–3602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barri YM, Sanchez EQ, Jennings LW, et al:
Acute kidney injury following liver transplantation: definition and
outcome. Liver Transpl. 15:475–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schrier RW, Wang W, Poole B and Mitra A:
Acute renal failure: definitions, diagnosis, pathogenesis and
therapy. J Clin Invest. 114:5–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kellum JA: Acute kidney injury. Crit Care
Med. 36(Suppl 4): S141–S145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen X, Liu X, Wan X, Wu Y, Chen Y and Cao
C: Ischemic preconditioning attenuates renal ischemia-reperfusion
injury by inhibiting activation of IKKbeta and inflammatory
response. Am J Nephrol. 30:287–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Serviddio G, Romano AD, Gesualdo L, et al:
Postconditioning is an effective strategy to reduce renal
ischaemia/reperfusion injury. Nephrol Dial Transplant.
23:1504–1512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao ZQ, Corvera JS, Halkos ME, et al:
Inhibition of myocardial injury by ischemic postconditioning during
reperfusion: comparison with ischemic preconditioning. Am J Physiol
Heart Circ Physiol. 285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xing B, Chen H, Zhang M, et al: Ischemic
postconditioning inhibits apoptosis after focal cerebral
ischemia/reperfusion injury in the rat. Stroke. 39:2362–2369. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gyurkovics E, Aranyi P, Stangl R, et al:
Postconditioning of the lower limb - protection against the
reperfusion syndrome. J Surg Res. 169:139–147. 2011. View Article : Google Scholar
|
|
10
|
Liu XH, Zhang ZY, Sun S and Wu XD:
Ischemic postconditioning protects myocardium from
ischemia/reperfusion injury through attenuating endoplasmic
reticulum stress. Shock. 30:422–427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen H, Xing B, Liu X, et al: Ischemic
postconditioning inhibits apoptosis after renal
ischemia/reperfusion injury in rat. Transpl Int. 21:364–371. 2008.
View Article : Google Scholar
|
|
12
|
Jiang B, Liu X, Chen H, et al: Ischemic
postconditioning attenuates renal ischemic/reperfusion injury in
mongrel dogs. Urology. 76:e1–e7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sauvant C, Schneider R, Holzinger H,
Renker S, Wanner C and Gekle M: Implementation of an in vitro model
system for investigation of reperfusion damage after renal
ischemia. Cell Physiol Biochem. 24:567–576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao H: The protective effect of ischemic
postconditioning against ischemic injury: from the heart to the
brain. J Neuroimmune Pharmacol. 2:313–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Russ AL, Haberstroh KM and Rundell AE:
Experimental strategies to improve in vitro models of renal
ischemia. Exp Mol Pathol. 83:143–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Witzgall R: The proximal tubule phenotype
and its disruption in acute renal failure and polycystic kidney
disease. Exp Nephrol. 7:15–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meldrum K, Meldrum DR, Hile KL, Burnett AL
and Harken AH: A novel model of ischemia in renal tubular cells
which closely parallels in vivo inury. J Surg Res. 99:288–293.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cavdar Z, Oktay G, Egrilmez MY, et al: In
vitro reoxygenation following hypoxia increases MMP-2 and TIMP-2
secretion by human umbilical vein endothelial cells. Acta Biochim
Pol. 57:69–73. 2010.PubMed/NCBI
|
|
19
|
Sáenz-Morales D, Escribese MM, Stamatakis
K, et al: Requirements for proximal tubule epithelial cell
detachment in response to ischemia: role of oxidative stress. Exp
Cell Res. 312:3711–3727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sáenz-Morales D, Conde E, Escribese MM, et
al: ERK1/2 mediates cytoskeleton and focal adhesion impairment in
proximal epithelial cells after renal ischemia. Cell Physiol
Biochem. 23:285–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basnakian AG, Ueda N, Hong X, Galitovsky
VE, Yin X and Shah SV: Ceramide synthase is essential for
endonuclease-mediated death of renal tubular epithelial cells
induced by hypoxia-reoxygenation. Am J Physiol Renal Physiol.
288:F308–F314. 2005. View Article : Google Scholar
|
|
22
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ness JM, Harvey CA, Strasser A, Bouillet
P, Klocke BJ and Roth KA: Selective involvement of BH3-only Bcl-2
family members Bim and Bad in neonatal hypoxia-ischemia. Brain Res.
1099:150–159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gibson ME, Han BH, Choi J, et al: BAX
contributes to apoptotic-like death following neonatal
hypoxia-ischemia: evidence for distinct apoptosis pathways. Mol
Med. 7:644–655. 2001.
|
|
25
|
Martinou JC, Frankowski H, Missotten M,
Martinou I, Potier L and Dubois-Dauphin M: Bcl-2 and neuronal
selection during development of the nervous system. J Physiol
Paris. 88:209–211. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Golstein P: Controlling cell death.
Science. 275:1081–1082. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gillardon F, Lenz C, Waschke KF, et al:
Altered expression of Bcl-2, Bcl-X, Bax and c-Fos colocalizes with
DNA fragmentation and ischemic cell damage following middle
cerebral artery occlusion in rats. Brain Res. Mol Brain Res.
40:254–260. 1996. View Article : Google Scholar
|
|
28
|
Krajewski S, Mai JK, Krajewska M, Sikorska
M, Mossakowski MJ and Reed JC: Upregulation of bax protein levels
in neurons following cerebral ischemia. J Neurosci. 15:6364–6376.
1995.PubMed/NCBI
|
|
29
|
Gillardon F, Wickert H and Zimmermann M:
Up-regulation of bax and down-regulation of bcl-2 is associated
with kainate-induced apoptosis in mouse brain. Neurosci Lett.
192:85–88. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prabhakar G, Vona-Davis L, Murray D,
Lakhani P and Murray G: Phosphocreatine restores high-energy
phosphates in ischemic myocardium: implication for off-pump cardiac
revascularization. J Am Coll Surg. 197:786–791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qi L, Pan H, Li D, Fang F, Chen D and Sun
H: Luteolin improves contractile function and attenuates apoptosis
following ischemia-reperfusion in adult rat cardiomyocytes. Eur J
Pharmacol. 668:201–207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho BB and Toledo-Pereyra LH:
Caspase-independent programmed cell death following ischemic
stroke. J Invest Surg. 21:141–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|