Introduction
The majority of malignant brain tumors are
astrocytoma or glioma, which are often fatal with a median survival
time from diagnosis of ~15 months (1). Connexins (Cxs), particularly Cx43
(2) and certain microRNAs
(3,4) are important in the modulation of
astrocytoma and glioma. Since current clinical approaches,
including surgery, chemotherapy and radiotherapy do not have a
significant impact on patient survival (5), it is crucial to develop new
therapeutic methods and drugs for the treatment of these diseases.
Cx43 is the major Cx in astrocytes and is involved in cell cycle
regulation and growth modulation (2). Cx43 has inhibitory effects on
astrocytoma (6) and glioma
progression (4). In addition, Cx43
enhances apoptotic effects (7–9).
Cx43 expression is decreased in astrocytoma and glioma (10). In this regard, downregulation and
upregulation of Cx43 leads to enhanced and decreased development of
tumor malignancies, respectively (5,11,12).
miRNAs are small, non-coding RNAs of 19–24
nucleotides in length that regulate gene expression and are
significantly associated with either tumor development or
suppression (13). miRNAs have
been associated with several types of cancer, including glioma and
its aggressive glioblastoma subtype (14). A study concerning the effects of
miRNA and Cx43 expression demonstrated that miR-125b inhibits Cx43
expression and increases glioma cell development (15). However, overexpression of Cx43
inhibits the effects of miR-125b in glioma cell improvement and its
anti-apoptotic effects (15).
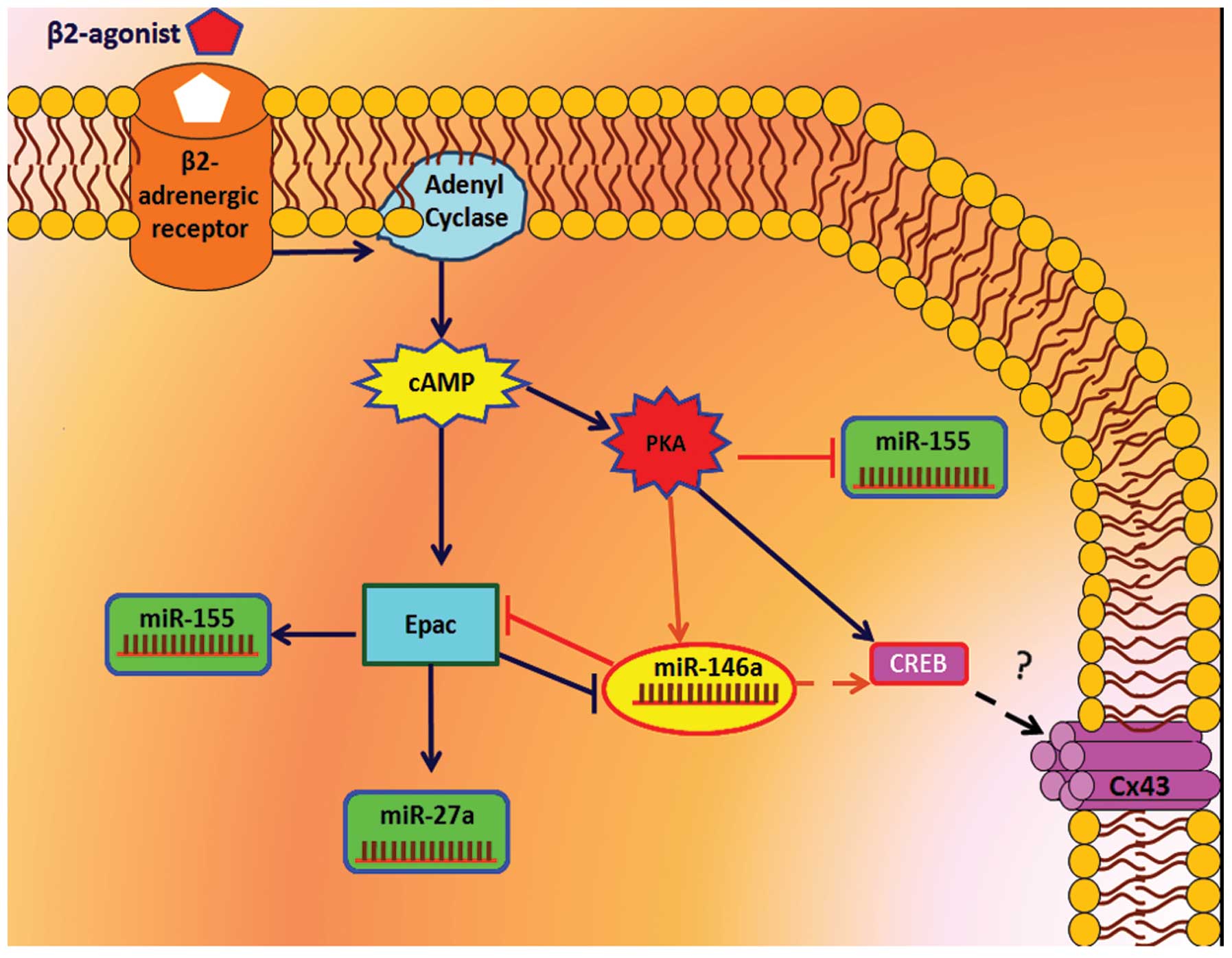
β2-adrenergic receptor (AR) signaling upregulates Cx43 expression
via cAMP/protein kinase A (PKA) and cAMP/exchange protein directly
activated by cAMP (Epac) pathways in myocytes (16). The cAMP/PKA pathway markedly
activates miR-335, which is required for differentiation therapy of
C6 glioma cells (17). In this
cell line, miR-146a is a glioma suppressor candidate (18), which has possible associations with
β2-AR signaling and Cx43 expression.
The current study aimed to identify the effect of
β2-AR signaling and the role of its downstream components, adenyl
cyclase inhibition, PKA activation and Epac on Cx43 expression in a
human-derived astrocytoma cell line (A-1321N1). In addition, the
effects of miR-146a on the overexpression of Cx43 were evaluated.
Finally, the effect of the pathways downstream of β2-AR signaling
(PKA and Epac) on the expression of miR-146a were investigated.
Materials and methods
Materials
Clenbuterol hydrochloride (Cln), selective β2
agonist (C5423), SQ22, 536 (SQ), adenyl cyclase inhibitor (ACi,
S153), 6-Bnz-cAMP sodium salt (6Bnz), specific PKA activator
(B4560), 8-(4-chlorophenylthio)-2′-O-meth-yladenosine-3′,5′-cyclic
monophosphate (8CPT), Epac-specific activator (C8988), Brefeldin
(BF) and Epac inhibitor (B7651) were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Antibodies, including mouse monoclonal
anti-connexin43/GJA1 (1:1,000; cat no. ab79010), mouse monoclonal
anti-β-actin (1:5,000; cat no. ab6276), rabbit polyclonal secondary
antibody to mouse IgG (1:10,000; cat no. ab6728) and goat
polyclonal secondary antibody to rabbit IgG (1:10,000; cat no.
ab6721) were all purchased from Abcam (Cambridge, MA, USA).
Cell culture and treatment
An A-1321N1 cell line was purchased from the
National Cell Bank of Iran (Tehran, Iran). microRNA-146a (miR-146a)
was overexpressed in this cell line. The cells were maintained in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA,
USA) containing 10% fetal bovine serum (FBS) supplemented with 50
mg/ml streptomycin and 100 U/ml penicillin (Gibco-BRL) in a
humidified atmosphere at 37°C and 5% CO2. Cells in a
confluent monolayer were routinely passaged by trypsinization and
the passaged cells were seeded into six-well plates. The following
day, the DMEM medium, containing 1% FBS, was changed prior to drug
exposure for 24 h. Subsequently, the cells were treated with Cln or
6Bnz for a further 24 h. The Cln-treated groups of cells were
pretreated in the presence or absence of SQ or BF (45 min prior to
treatment with Cln) for adenyl cyclase and Epac inhibition,
respectively. RNA and protein extraction were performed for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis, respectively.
Virus packaging and transduction
The HEK293T cell line was used for virus packaging
of PB-miR-146a containing green fluorescent protein (GFP) and
puromycin resistance genes. The cells were maintained in DMEM
containing 10% FBS supplemented with 50 mg/ml streptomycin and 100
U/ml penicillin in a humidified atmosphere at 5% CO2 and
37°C. A Lentivirus packaging system containing the psPAX2 plasmid
(encoding gag/pol), the pMDG plasmid (encoding VSV-G) and
PB-miR-146a (as well as empty vector) were used for lentivirus
production using the calcium-phosphate method (19). Subsequently, GFP expression was
assessed using an inverted fluorescence microscope (Nikon TEF2000;
Nikon, Tokyo, Japan) following 24 h of transfection. The medium
containing the produced viruses was collected 24, 48 and 72 h after
transfection and kept at 4°C. The medium was subsequently
concentrated by centrifuging at 40,000 x g at 4°C for 2.5 h.
Finally, the two cell lines were infected with the concentrated
supernatants. At 24 h after transduction, the cells were treated
with puromycin (2 µg/ml) and this treatment continued for 3
days. Negative control groups were also treated with puromycin,
which, as expected, underwent cell death after 48 h. The
transfected group (PB-miR-146a) was resistant to puromycin and
survived following antibiotic selection (Fig. 1).
Primer design and probe for detecting
miRNAs
The sequences of miRNAs, including miR-146a, miR-155
and miR-27a were obtained from NCBI (http://www.ncbi.nlm.nih.gov/gene). Subsequently, an
miRNA specific stem-loop primer was designed by adding a few
complimentary nucleotides from the 3′ end of the miRNA to the 3′
end of the pre-evaluated stem-loop construction (20). qPCR was performed with an
miRNA-specific forward primer, universal reverse primer and
specific TaqMan probe.
RNA extraction and RT-qPCR for mRNA
expression
Relative quantification of gene expression of
various groups of A-1321N1 cells was performed. Total RNA was
isolated using QIAzol lysis reagent (Qiagen, Hilden, Germany)
according to the manufacturer’s instructions and used for cDNA
synthesis using reverse transcriptase (cat no. RTPL12; Vivantis,
Oceanside, CA, USA). Specific human primers, including GAPDH, Cx43
and cAMP response element-binding protein (CREB; Table. I) were used in these experiments
with Takara SYBR Premix Ex Taq (Takara Bio, Inc., Shiga, Japan).
Gene expression levels were measured using Rotor-Gene 6000 (Corbett
Life Science, Concorde, NSW, Australia) and the relative expression
ratio of each gene was compared with GAPDH. All data were
calculated using the relative expression software tool (REST
2009.2.0.13; Qiagen).
 | Table ISequences of human primers. |
Table I
Sequences of human primers.
| Gene | Accession
number | Forward primer | Reverse primer | Length (bp) | Conc
(µM) |
|---|
| Cx43 | NM_000165.3 |
GGATTGTCCTTAAGTCCCTG |
CACAAGTCCATTGACACCTG | 100 | 0.4 |
| CREB | XM_005246324.1 |
AACCAGCAGAGTGGAGATGCA |
GGCATAGATACCTGGGCTAATGTG | 101 | 0.4 |
| GAPDH | NM_002046 |
CTCTCTGCTCCTCCTGTTCG |
ACGACCAAATCCGTTGACTC | 114 | 0.4 |
Analyzing the expression of miRNAs using
stem-loop RT-qPCR
Total miRNAs and specially overexpressed miR146a
were extracted from A-1321N1 cells by QIAzol RNA extraction
(Qiagen). For this purpose, the time and speed of centrifugation
was increased to 15,000 x g for 15 min to increase the miRNA yield.
Finally, extracted miRNA was dissolved in RNase-free water and
stored at −80°C until use. The quality was confirmed using an
Eppendorf Spectrophotometer (Eppendorf AG, Hamburg, Germany). miRNA
was extracted from the immortalized cell line or the empty vector
(EV) control cells. Subsequently, 5 µl of extracted miRNA
was adjusted to contain no less than 5 µg total miRNA and
added to 1.5 µl stem-loop RT primer (50 nM) and 4 µl
distilled water. Reaction samples (10.5 µl) were incubated
in a Bio-Rad thermal cycler (MJ Mini; Bio-Rad, Hercules, CA, USA)
at 65°C for 10 min. The samples were kept on ice and 4 µl 5X
reverse transcriptase buffer (Roche Diagnostics GmbH, Mannheim,
Germany), 2 µl dNTPs (10 mM), 2 µl DTT (10 mM), 1
µl Expand reverse transcriptase (50 units; Roche Diagnostics
GmbH) and 0.5 µl RNase inhibitor (20 units) was added. cDNA
synthesis was conducted as follows: 25°C for 15 min, 37°C for 15
min, 42°C for 40 min and finally 95°C for 2 min to inactivate the
enzyme.
RT-qPCR was performed in a 12.5 µl PCR
mixture volume composed of 6.5 µl 2X Maxima®
Probe qPCR master mix (Fermentas, Vilnius, Lithuania), 0.2
µM of each oligonucleotide primer, 0.1 µM probe and 2
µl cDNA. The following conditions were used for
amplification: Enzyme activation step at 95°C for 10 min and 45
cycles of two thermal amplification steps: 94°C for 10 sec and 60°C
for 40 sec. Single fluorescence detection was used in each cycle at
the end of the 60°C step (extension phase) to elucidate the
positive samples. The specificity was confirmed using gel
electrophoresis. Reactions were set up manually. Amplification,
data acquisition and analysis were performed on the Rotor-Gene 6000
(Corbett Life Science) using Rotor-Gene 6.1 and REST software.
Samples were assessed in triplicate along with pertaining controls,
including positive, negative, no-template and RT-minus controls. A
previously approved and non-treated RNA, SNORD 47, was used as an
internal control (21). The Ct is
the fractional cycle number at which the fluorescence passes the
fixed threshold. REST software was used for statistical analysis of
relative miRNA expression.
Western blot analysis
Total cell lysates were prepared following
harvesting cells in ice-cold phosphate-buffered saline (Gibco-BRL)
and homogenization in cold lysis buffer (RIPA buffer; Gibco-BRL).
Protease inhibitor cocktails (cat no. S8820; Sigma-Aldrich) were
added to the lysis buffer. Lysates were centrifuged at 13,680 x g
for 10 min and the supernatant was transferred to a new tube.
Protein content was measured using the bicinchoninic acid method
(BCA protein kit; Pierce Biotechnology, Inc.). Subsequently, equal
quantities of protein (25 µg) were added to each well in
12.5% standard SDS-PAGE and a wet transfer was performed (Bio-Rad).
Proteins were transferred onto polyvinylidene difluoride membranes
(Gibco-BRL) and membranes were blocked with 2% skimmed milk in
Tris-buffered saline/Tween 20 (TBST 20; 0.5%; Gibco-BRL) for 2 h at
room temperature. Membranes were incubated overnight at 4̊C with
primary antibodies against Cx43 1:5,000 diluted in TBST (0.5%).
Membranes were washed three times with TBST 20 and subsequently
incubated for 1 h at room temperature with the corresponding
horseradish-peroxidase-conjugated secondary antibodies: Rabbit
polyclonal secondary antibody to mouse IgG. Following three washes
with TBST 20 (0.5%), detection was conducted by applying Pierce ECL
plus western blotting substrate (cat no. 32134; Pierce
Biotechnology, Inc.), emanating chemiluminescence from the membrane
for manual x-ray film development. Levels of the analyzed proteins
were normalized to β-actin levels.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
To evaluate the cytotoxic effect of Cln treatment on
the A-1321N1 cell line, cell viability was measured using the MTT
colorimetric assay (Sigma-Aldrich). Cells were seeded into 96 well
plates at a density of 0.5×104 cells/well and left
overnight to attach. The following day, the medium was changed to a
fresh medium containing different concentrations of Cln (1, 5, 10,
20 and 40 µg/ml). At 24 h after treatment, MTT reagent (5
mg/ml) was added and incubation was continued at 37°C for 3 h. The
medium was discarded and the formazan crystals were dissolved by
adding dimethyl sulf-oxide (100 µl/well; Gibco-BRL).
Finally, the optical density of each well was determined using a
spectrophotometer (ELx800; BioTek Instruments, Inc., Winooski, VT,
USA) at a wavelength of 570 nm against 630 nm.
Statistical analysis
The acquired data were analyzed by SPSS software 19
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference. Experiments were
performed in triplicate and data are presented as the mean ±
standard error of the means.
Results
Expression of Cx43 by β2-AR signaling
agents
The specificity of cAMP signaling on Cx43 expression
in astrocytoma cells was assessed following 24 h treatment of cells
with drugs. Cln (10 µg/ml), a selective β2-AR agonist, led
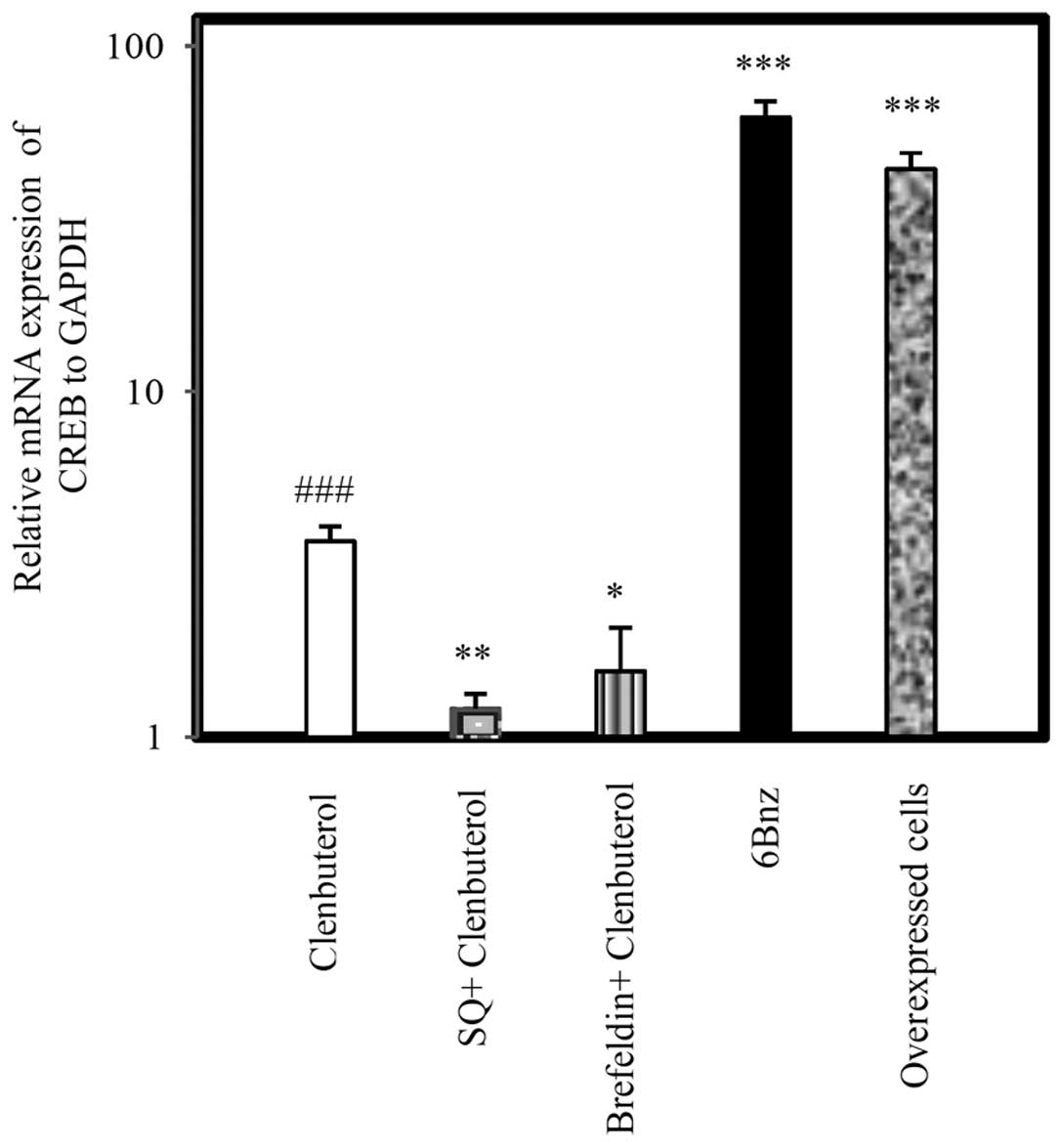
to a significant upregulation in Cx43 at the mRNA (Fig. 2; P<0.001) and protein levels
(Fig. 3; P<0.05). Pretreatment
with SQ (50 µg/ml) 45 min prior to Cln treatment
significantly inhibited Cx43 expression at the mRNA (Fig. 2; P<0.001) and protein levels
(Fig. 3; P<0.05). By contrast,
BF (100 µg/ml) administered 45 min prior to Cln treatment
increased Cx43 expression at the mRNA (Fig. 2; P<0.001) and protein level
(Fig. 3; P<0.05) compared with
the Cln alone group. Selective activation of PKA by 6Bnz (50
µg/ml) also resulted in significant upregulation of Cx43 at
the mRNA (Fig. 2) and protein
(Fig. 3) level (P<0.001). The
expression level of Cx43 mRNA was measured in miR-146a
overexpressing A-1321N1 cells (P<0.001). The results revealed
that Cx43 was significantly upregulated (Fig. 2; P<0.001) in a parallel manner
to 6Bnz treatment. Therefore, Cln may upregulate Cx43 via PKA
activation since 6Bnz, a PKA activator, upregulated Cx43 expression
significantly more than Cln and Epac inhibition did not prevent its
upregulation.
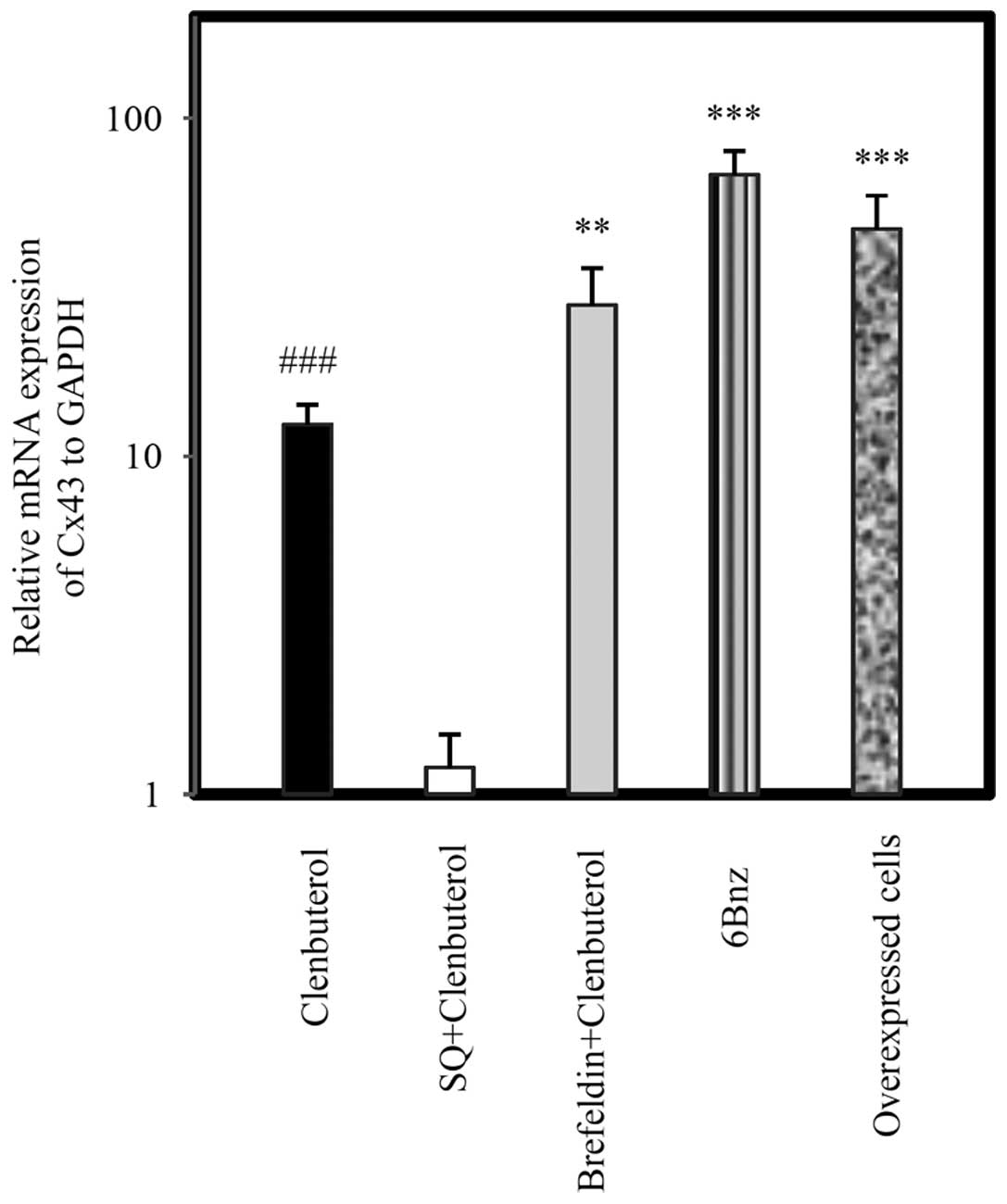 | Figure 2Relative mRNA expression of Cx43 in
A-1321N1 cells. A-1321N1 cells were treated with Cln (10
µg/ml), a selective β2 agonist; SQ (50 µg/ml), an
adenyl cyclase inhibitor + Cln (10 µg/ml); BF (50
µg/ml), an Epac inhibitor + Cln and 6Bnz (50 µg/ml),
a protein kinase A activator for 24 h and the relative mRNA
expression of Cx43 was measured. Cx43 was measured in miR-146a
overexpressing A-1321N1 cells. The results revealed that Cx43 was
significantly upregulated following treatment with Cln (10
µg/ml), BF (100 µg/ml) + Cln (10 µg/ml) and
6Bnz (100 µg/ml) in miR-146a overexpressing cells.
Pretreatment with SQ (50 µg/ml) completely inhibited Cx43
expression compared with the Cln group, however, Epac inhibition by
BF (100 µg/ml) increased its expression. Relative expression
of each group was normalized with GAPDH using a relative expression
software tool. Data obtained from three independent experiments
were analyzed similarly to the control group. Bar graphs show the
mean ± standard error of the mean from at least three individual
experiments. **P<0.01, ***P<0.001 and
###P<0.001, compared with the Cln treated and untreated group.
GAPDH was used as a control. Cx43, connexin 43; Cln, clenbuterol
hydrochloride; 6Bnz, 6-Bnz-cAMP sodium salt; BF, Brefeldin; SQ,
SQ22,536. |
Expression of CREB
CREB level was measured following treatment with
different doses of Cln (10 µg/ml) and pre-inhibition with SQ
(50 µg/ml), ACi and BF (100 µg/ml). Cln treatment
upregulated CREB levels, while adenyl cyclase (P<0.01) and Epac
inhibition (P<0.05) downregulated CREB levels. CREB levels were
measured in the 6Bnz (50 µg/ml)-treated group, which
significantly upregulated the level of CREB (P<0.001). Epac
inhibition decreased CREB level compared with the Cln (P<0.05)
and 6Bnz (P<0.001) groups, however, its higher expression
compared with the adenyl cyclase inhibited group was not
significant. Expression of CREB in miR-146a overexpressing A-1321N1
cells was measured and the results shown in Fig. 4 demonstrated that its expression
significantly increased compared with GAPDH (P<0.001).
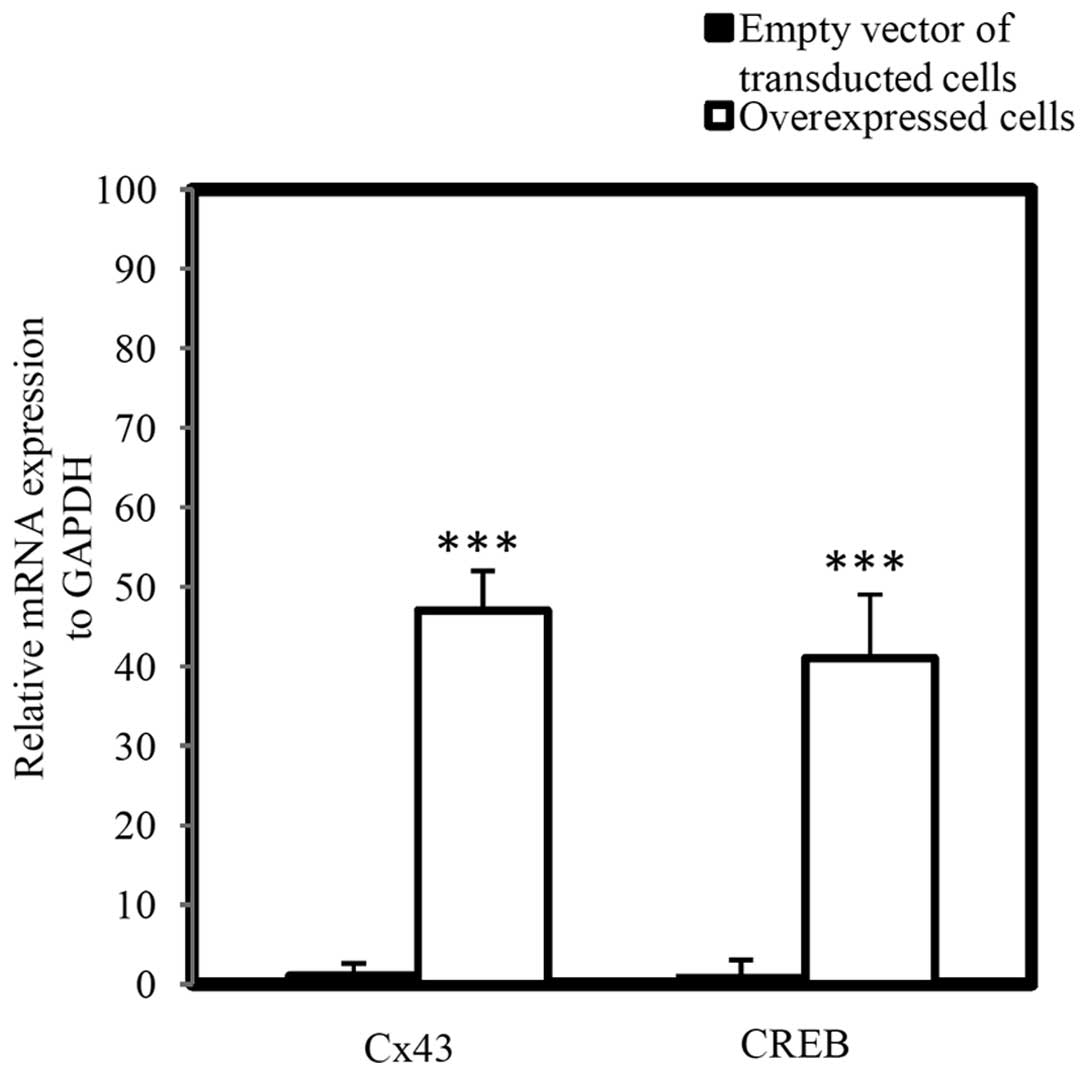
Additionally, the mRNA level of Cx43 and CREB in
miR-146a overexpressing A-1321N1 cells (P<0.001) was measured,
which demonstrated that Cx43 and CREB were significantly
upregulated in a parallel manner in miR-146a overexpressing cells
(Fig. 5; P<0.001).
Effects of β2-AR, PKA and Epac activation
on miRNA level
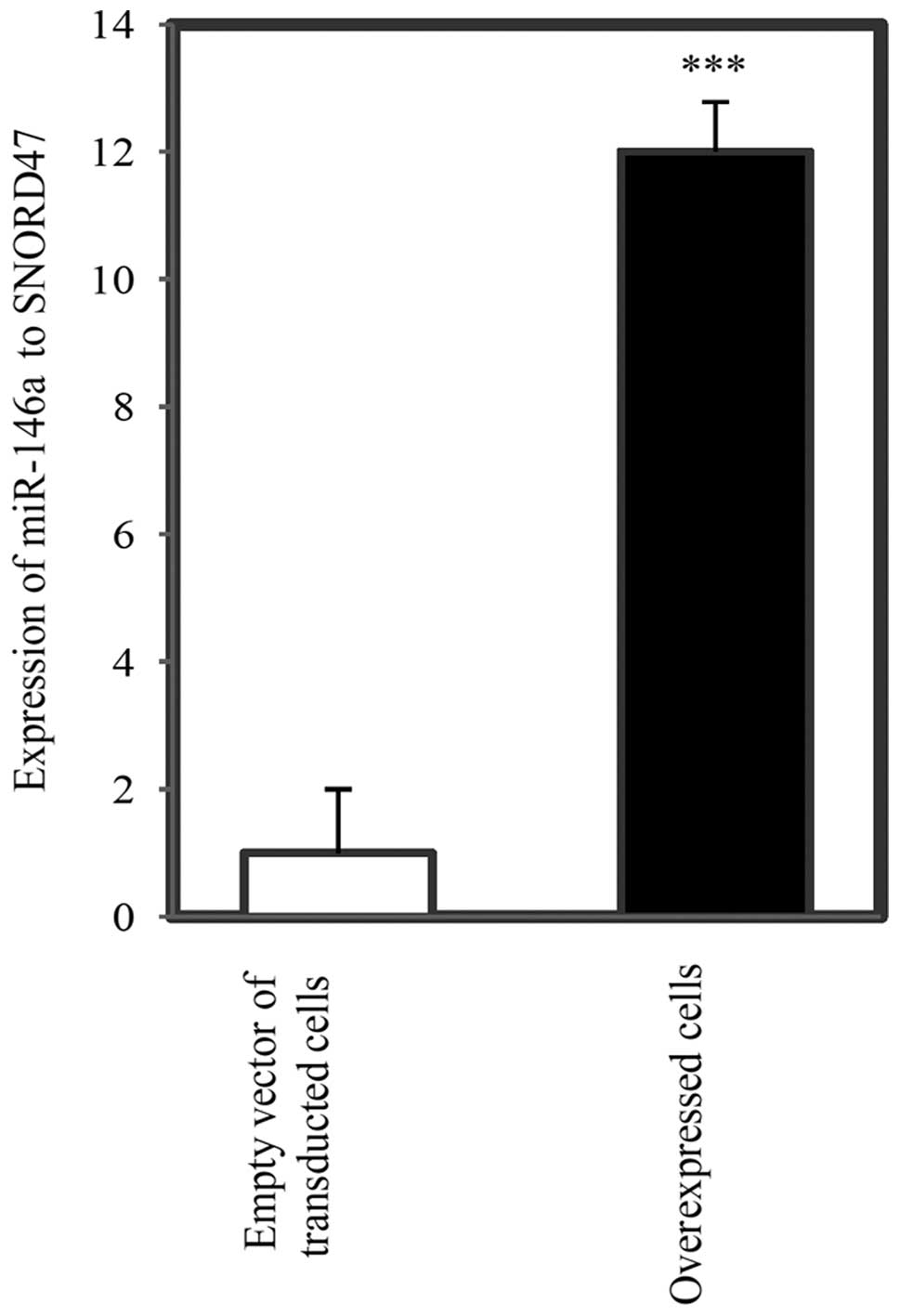
Fig. 6 shows the
miR-146a level in overexpressing astrocytoma cells and EV of
A-1321N1 cells. miR-146a was significantly upregulated in
overexpressing A-1321N1 cells compared with EV cells
(P<0.001).
Putative regulation of miR-146a was verified by
stem-loop RT-qPCR after 24 h treatment. Pharmacological
intervention of the cAMP/PKA pathway with different drugs
demonstrated that Cln at doses of 10 and 20 µg/ml
significantly upregulated miR-146a level in A-1321N1 cells
(P<0.001). 6Bnz (50 µg/ml), a PKA activator, had the same
effect as Cln (P<0.001). However, 8CPT, an Epac activator, (50
µg/ml) downregulated the level of miR-146a (P<0.001).
Adenyl cyclase (20 µg/ml) inhibition eliminated the effect
of Cln (10 and 20 µg/ml) on miR-146a expression at the two
doses (P<0.01). Taken together, the results suggested that
miR-146a expression was upregulated through the cAMP/PKA pathway
and was decreased via the cAMP-Epac pathways (Fig. 7).
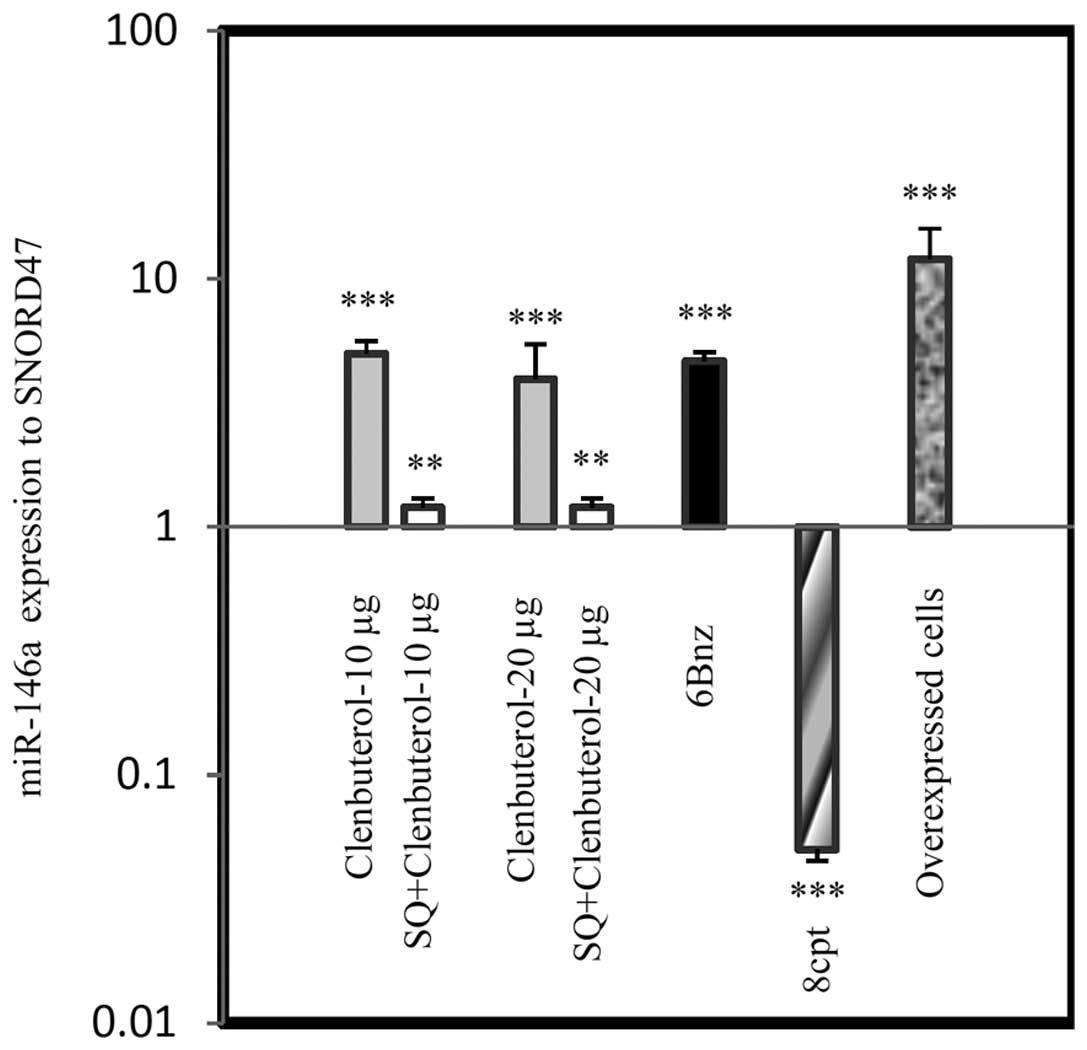 | Figure 7miR-146a expression via β2-AR
signaling. β2-AR signaling intervention modulated the relative
expression of miR-146a level in astrocytoma 1321N1. Cln at doses of
10 and 20 µg/ml increased miR-146a level and the adenyl
cyclase inhibitor, SQ (50 µg/ml), significantly suppressed
Cln treatment. 6Bnz (100 µg/ml; protein kinase A activator)
and 8CPT (50 µg/ml), an Epac activator, significantly
increased and decreased miR-146a expression, respectively.
Overexpressed miR-146a cells exhibited increased miR-146a levels
compared with all groups and SNORD47. **P<0.01 and
***P<0.001, compared with the untreated cells. Data
were normalized to the SNORD47, housekeeping gene. Cln, clenbuterol
hydrochloride; β2-AR, β2-adrenergic receptor; SQ, SQ 22,536; 6Bnz,
6-Bnz-cAMP sodium salt; 8CPT,
8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic
monophosphate. |
Fig. 8 illustrates
pharmacological intervention on miR-146a and two oncomiRs, miR-155
and miR-27a. Cln (10 µg/ml) and 6Bnz (50 µg/ml)
significantly upregulated miR-146a levels, while the level of
miR-155 was significantly downregulated. Treatment with Cln (10
µg/ml) and 6Bnz (50 µg/ml) also downregulated
miR-27a, however, this was not significant. Following treatment
with the Epac activator, 8CPT (20 µg/ml), the levels of
miR-155 and miR-27a were upregulated, while the levels of miR-146a
were significantly downregulated. By contrast, 6Bnz (50
µg/ml) significantly upregulated the level of miR-146a.
Therefore, β2-AR stimulation and PKA activation increased the
suppressant miR-146a levels and decreased the level of miR-155 and
miR-27a oncomiRs, while Epac activation downregulated miR-146a and
upregulated oncomiRs, miR-155 and miR-27a.
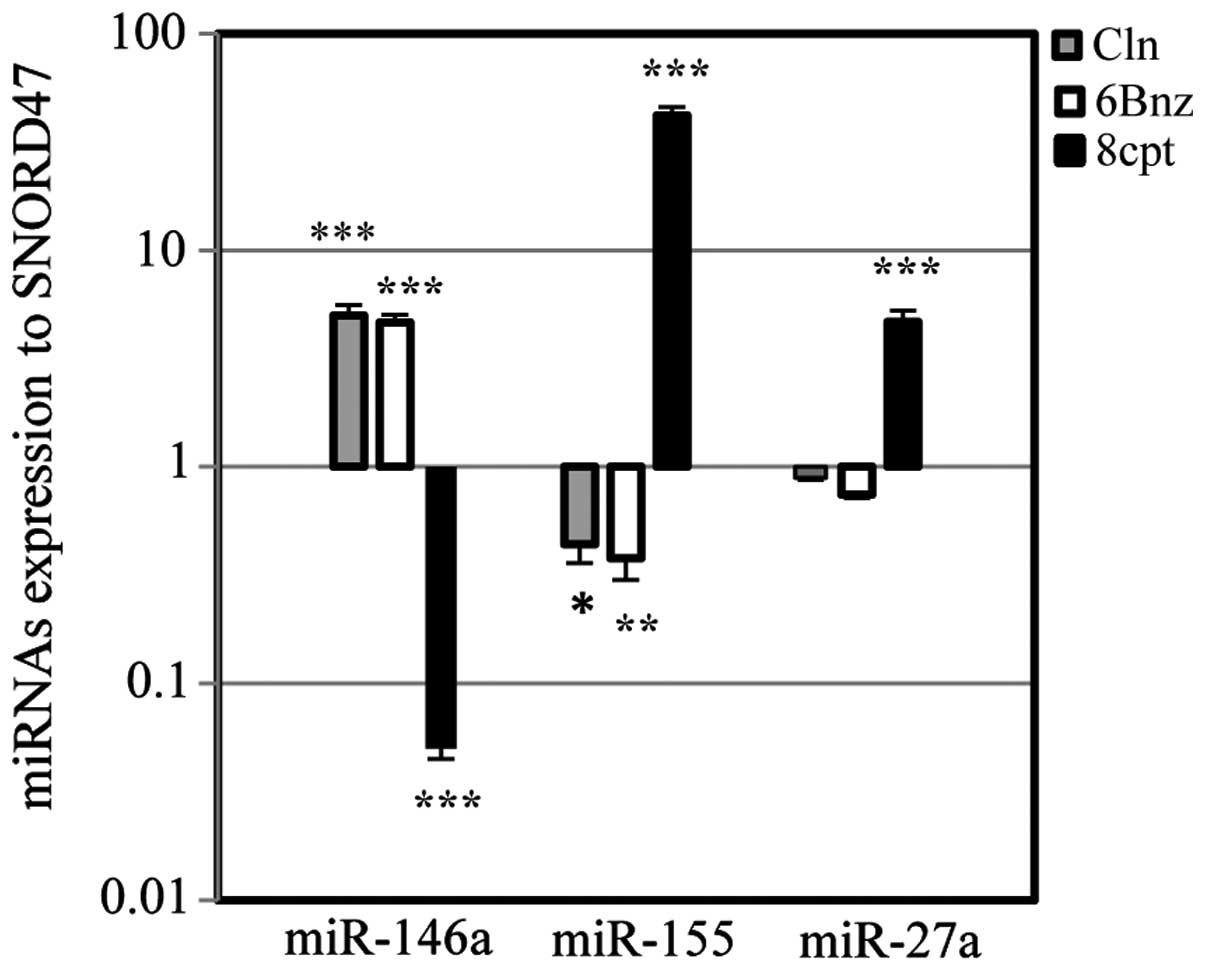 | Figure 8miRNA expression via protein kinase A
and Epac activation. The effect of Cln, 6Bnz and 8CPT treatment on
the levels of miR-146a, miR-155 and miR-27a in 1321N1 astrocytoma
cells. Cln (10 µg/ml) and 6Bnz (50 µg/ml) increased
miR-146a and decreased miR-155 significantly in addition to miR-27a
(not significant), however, 8CPT (20 µg/ml) decreased
miR-146a and increased miR-155 and miR-27a significantly. Data were
normalized to SNORD47, a housekeeping miRNA. *P<0.05,
**P<0.01 and ***P<0.001, compared with
the untreated cells. Cln, clenbuterol hydrochloride; 6Bnz,
6-Bnz-cAMP sodium salt; 8CPT,
8-(4-chlorophenylthio)-2′-O-meth-yladenosine-3′,5′-cyclic
monophosphate. |
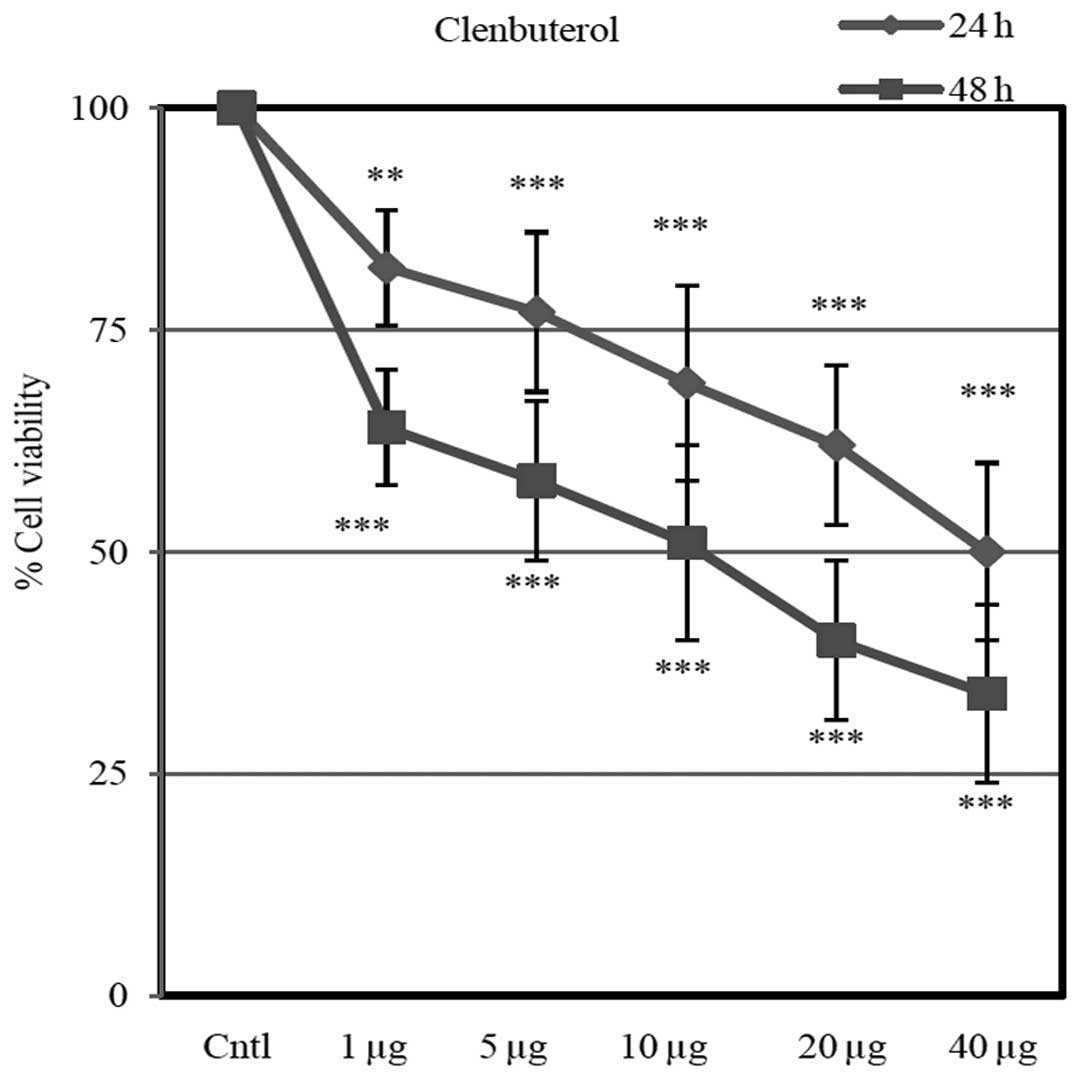
Cell viability
Different doses of Cln (1, 5, 10, 20 and 40
µg) inhibited cell proliferation in a dose-dependent manner
(Fig. 9). Cln significantly
inhibited cell proliferation after 24 and 48 h compared with the
control group.
Expression of Connexin 43 and miRNAs via
β2 adrenoceptor signaling in A-1321N1cell
β2 adrenoceptor activation via cAMP-PKA pathway
upregulated the expression levels of Cx43, CREB and miR-146a and
downregulated the expression of miR-155. By contrast, the β2
adrenoceptor upregulated the expression levels of miR-27a and
miR-155, and downregulated the expression of miR-146a via the
cAMP-Epac pathway (Fig. 10).
Discussion
Currently, molecular target therapy has become one
of the most useful strategies for disease treatment (22) and may be a promising approach for
astrocytoma and glioma therapy (23). The findings in the present study
exhibit novel Cx43 regulation in astrocyte models, that were
demonstrated by overexpression of miR-146a in the A-1321N1 cell
line and β2-AR stimulation in A-1321N1 cells. These results
revealed that β2-AR activation upregulates the expression of Cx43
in astrocytes via a PKA-regulated pathway, possibly involving CREB.
In this regard, the overexpression of miR-146a is also able to
cause upregulation of CREB and Cx43 in this cell line, in which
miR-146a was upregulated by PKA activation. Furthermore, β2-AR
adrenoceptor signaling activation decreases the severity of the
glioma as well as decreases the promotion of molecules, including
miR-155 (24–27) and miR-27a (28,29)
via its signaling pathway. PKA, activated by 6Bnz, acts as a
suppressor of the astrocytoma signaling pathway and glioma while
the Epac signaling pathway, activated by 8CPT acts as a modulator.
Therefore, β2-AR signaling and particularly PKA activation may be
useful strategies for astrocytoma and glioma therapy. Despite
considerable advances in miRNA research, there are various
obstacles for the use of miRNAs in clinics, including miRNA
delivery, which prevents the use of miRNAs as drugs. Therefore, the
drugs that may activate PKA or β2-AR signaling have modulating
potential on miRNA delivery in astrocytoma and Cx43 expression.
In the present study, the effects of β2-AR signaling
on Cx43 expression and its modulating effect on miR-146a level were
evaluated. In addition, the effects of high levels of miR-146a on
Cx43 expression were assessed by overexpressing miR-146a in
A-1321N1 cells. In further investigations, the activation of β2-AR
signaling was evaluated and the mechanism by which PKA and Epac
downstream pathways affected the two oncomiRs, miR-155 and miR-27a
was assessed. The expression of CREB, a pathway downstream of PKA,
in A-1321N1 cells overexpressing miR-146a and standard A-1321N1
cells was also evaluated, which were subject to pharmacological
intervention. These evaluations revealed that CREB level increased
in miR-146a overexpressing cells and in cells treated with Cln and
6Bnz.
Since the aim of the present study was to
demonstrate the effects of β2-AR signaling on Cx43 and miR-146a,
the A-1321N1 cell line, which is a high grade astrocytoma model,
appropriate for signaling studies was utilized (30). miRNA detection was based on the
stem-loop method, which is recognized for producing high quality
data. The current results present a method for enhancing the level
of miR-146a and Cx43 in the astrocytoma cell line via PKA
activation and β2-AR stimulation.
Higher grade human brain tumors are associated with
lower adenyl cyclase activity and/or cellular cAMP levels (31), in which signaling is modulated by
the β2-AR signaling pathway (1).
cAMP and its associated downstream pathways, including 8-Chloro
cAMP (17) and PKA (18) have been observed to inhibit the
proliferation of tumor cells (20,23).
Additionally, β2-AR knockout astrocytes exhibited higher
proliferation rates compared with wild-type cells (32). Increasing the level of adenyl
cyclase activity has been associated with decreased glioma cell
proliferation (31). By contrast,
Somekawa et al (16)
demonstrated that PKA activation predominantly contributes to the
functional neoformation of gap junctions and that Epac is also
responsible for gap junction neoformation. In cultured astrocytes,
Salameh et al (33)
demonstrated that PKA and mitogen-activated protein kinase are
involved in Cx43 expression via activator protein 1 and CREB
through β2-AR stimulation. Lu et al (34) demonstrated that β2-AR blockers
decreased miR-1 level via inhibition of the β2-AR-cAMP-PKA pathway
in myocytes, which led to downregulation of Cx43 expression. In the
present study, it was observed that β2-AR stimulation, via
activation of PKA downstream pathways, upregulated the expression
of Cx43 and CREB as well as miR-146a level in A-1321N1 cells via
β2-AR activation. The current results implied that β2-AR activation
and PKA stimulation upregulate Cx43 and CREB expression. Activation
of another β2-AR downstream pathway, Epac, decreased miR-146a level
and overexpression of miR-146a had an enhancing effect on Cx43,
which was also demonstrated in the present study. This demonstrates
that the activation of Epac may negatively effect Cx43 expression.
In the present study, Epac was deactivated in order to analyze its
effect on Cx43 expression, which as expected, was enhanced. The
present study is in accordance with other studies regarding Cx43
modulation in astrocytes via PKA-CREB activation (33,35).
Certain studies have revealed that PKA activation in myocytes had
weak (16) or inhibitory effects
(36) on Cx43 expression. Although
in these studies, miR-1 was investigated in myocytes, which
directly targets Cx43, whereas in the present study, the microRNA
investigated (miR-146a) had an indirect role in the upregulation of
Cx43 in the A-1321N1 cell line.
Epidermal growth factor (EGF) signals and their
downstream phosphatidylinositide 3-kinase (PI3K)-Akt pathways are
established as astrocytoma and glioma promoting pathways (18,23,37).
Receptor tyrosine kinases (RTKs) also possess roles in activation
of these pathways (23). Our
bioinformatics evaluation demonstrated that miR-146a targets
Epidermal growth factor receptor (EGFR) and RTK signaling
(unpublished data). In addition, miR-146b inhibits EGFR expression
and decreases in vitro invasion and migration of astrocytoma
and glioma cell lines (38). By
contrast, there is a significant link between the overexpression of
EGFR and Cx43 downregulation. In rat cortical astrocytes, Cx43 was
downregulated by EGF signaling stimulation (39). In addition, in the present study,
it was demonstrated that β2-AR stimulation or PKA activation
upregulates miR-146a; therefore, it is possible that miR-146a
induced the downregulation of Cx43 via targeting EGFR pathways.
Expression of Cx43 enhanced tumor suppressant
function (37) and
anti-proliferative effects on astrocytoma cells. Cx43 upregulation
and PI3K-Akt pathway inhibition was able to modulate the
proliferation of astrocytoma cells (38,39).
Upregulation of miR-155 directly targets Foxo3a (24), which inhibits cell-cycle
progression at the G1/S transition via regulating transcription of
the cyclin-dependent kinase inhibitor p27 (Kip1), which is
recurrently downregulated in human glioma (40). In astrocytoma and glioma cells, the
level of miR-155 is higher than normal, which is known to be an
oncomiR in glioma development (24,41).
Thus, downregulation of miR-155 was able to decrease its inhibitory
effect on Foxo3a and consequently, Foxo3a inhibits the PI3K-Akt
pathway and its proliferative effects. It is evident that Epac
activation may have an opposite role since it was revealed in the
present study that it downregulates miR-146a and upregulates
miR-155 levels. Since miR-27a downregulation via β2-AR signaling
and the PKA pathway was not significant, this suggests that it is
not important in A-1321N1 cells. miR-155 is elevated in primary and
secondary glioblastoma and promotes glioma development (41). It has been established as one of
the prognostic and predictive markers in glioblastoma patients
(42). The present results
indicated that Cln and PKA activation decreased miR-155 level and
Epac activation increased its level. Thus, PKA downregulates
miR-155 and acts as a tumor suppressant pathway, while Epac
increases miR-155 as a tumor inducer pathway. miR-27a is an
oncogenic and multidrug resistant miRNA overexpressed in glioma,
which promotes cell proliferation (28). This microRNA is predicted to be
involved in glioma progression and initiation via different
signaling pathways, including adherens junction, focal adhesion,
the mitogen-activated protein kinase signaling pathway, the p53
signaling pathway and the apoptotic signaling pathway (29). miR-27a is upregulated by Epac
activation, however, PKA activation downregulates its level,
although Cln was not able to significantly alter its levels.
miRwalk and miRrecords predicted (unpublished data) that miR-155
and miR-27a targeted CREB expression. In adipocytes, it is
validated that CREB was targeted by miR-155 induced by tumor
necrosis factor-α (43). The
present study revealed that 6Bnz and Cln increased CREB expression,
thus, it is possible that the downregulation of miR-155 via 6Bnz
and Cln increases CREB levels and Cx43 expression indirectly.
The studies conducted by Toll et al (1) and Johnstone et al (44) support the present results. In these
studies, it was suggested that cAMP elevation induced by β2-AR
agonists has a role in antiproliferation and Cx43 in glioma growth
inhibition. Toll et al (1)
demonstrated that cAMP-associated inhibition of cell growth in
astrocytoma is able to reduce cell growth by preventing the growth
factor-mediated cell proliferation signaling pathways; for
instance, extracellular-signal-regulated kinase increases the
levels of cell-cycle inhibitor proteins p21cip1 and p27kip1
(1). Johnstone et al
(44) suggested that enhancement
of Cx43 expression prolonged mitotic periods in order to correspond
with a G1 delay in cell cycle, which was associated with an
enhancement in the expression of the cell cycle inhibitor p21cip1
in HeLa-43 and HFF cells (44).
Huang et al (45)
demonstrated that Cx43 acts as a tumor suppressor gene, which
reduced cell proliferation in vitro and in vivo in
Cx43-transfected glioma cell lines (45). The present study revealed that Cln
upregulates Cx43 expression and decreases cell viability in a
dose-dependent manner and its anti-proliferative effect may be
associated with Cx43 enhancement.
The current results indicated that β2-AR signaling
via the downstream cAMP-PKA pathway led to enhanced expression of
Cx43 and miR-146a in A-1321N1 cells. PKA is the most important
downstream pathway with a potential effect on the expression of
Cx43 and miR-146a for the treatment of astrocytoma and
glioblastoma. The current findings suggest that the β2-AR pathway
results in the upregulation of Cx43, CREB and miR-146a via the PKA
pathway. By contrast, it was demonstrated that miR-146a
overexpression may also result in upregulation of CREB and Cx43.
Furthermore, the activation of the PKA pathway caused a
downregulation in the expression of the miR-155 oncomiR. This
demonstrates the tumor suppressor role of the PKA pathway. However,
the Epac pathway decreases the expression of miR-146a, the glioma
suppressing microRNA, which also possesses a significant function
in the overexpression of Cx43. The Epac pathway also induces the
overexpression of miR-155 and miR-27a oncomiRs. Overall, the PKA
pathway decreased tumor-associated features in A-1321N1 cells.
These findings not only enhance understanding of certain aspects of
the molecular biology of astrocytoma and glioma, but may also
clarify views on new prospective therapeutic and drug targets.
Acknowledgments
This study was supported by The Deputy of Research,
Tehran University of Medical Sciences. The authors would like to
thank the Stem Cell Technology Research Center (Tehran, Iran) for
their support. In addition, they would like to thank Mrs. Fatemeh
Kohram and Dr Abolreza Ardeshirylajimi for technical
assistance.
References
|
1
|
Toll L, Jimenez L, Waleh N, et al:
β2-adrenergic receptor agonists inhibit the proliferation of 1321N1
astrocytoma cells. J Pharmacol Exp Ther. 336:524–532. 2011.
View Article : Google Scholar :
|
|
2
|
Vinken M, Decrock E, De Vuyst E, et al:
Connexins: sensors and regulators of cell cycling. Biochim Biophys
Acta. 1815:13–25. 2011.
|
|
3
|
Gabriely G, Yi M, Narayan RS, et al: Human
glioma growth is controlled by microRNA-10b. Cancer Res.
71:3563–3572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castro MG, Cowen R, Williamson IK, et al:
Current and future strategies for the treatment of malignant brain
tumors. Pharmacol Ther. 98:71–108. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin JH, Takano T, Cotrina ML, et al:
Connexin 43 enhances the adhesivity and mediates the invasion of
malignant glioma cells. J Neurosci. 22:4302–4311. 2002.PubMed/NCBI
|
|
6
|
Sin WC, Crespin S and Mesnil M: Opposing
roles of connexin43 in glioma progression. Biochim Biophys Acta.
1818:2058–2067. 2012. View Article : Google Scholar
|
|
7
|
Huang RP, Hossain MZ, Huang R, Gano J, Fan
Y and Boynton AL: Connexin 43 (cx43) enhances chemotherapy-induced
apoptosis in human glioblastoma cells. Int J Cancer. 92:130–138.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang R, Liu YG, Lin Y, Fan Y, Boynton A,
Yang D and Huang RP: Enhanced apoptosis under low serum conditions
in human glioblastoma cells by connexin 43 (Cx43). Mol Carcinog.
32:128–138. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pu P, Xia Z, Yu S and Huang Q: Altered
expression of Cx43 in astrocytic tumors. Clin Neurol Neurosurg.
107:49–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soroceanu L, Manning TG Jr and Sontheimer
H: Reduced expression of connexin-43 and functional gap junction
coupling in human gliomas. Glia. 33:107–117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mesnil M: Connexins and cancer. Biol Cell.
94:493–500. 2002. View Article : Google Scholar
|
|
12
|
Sánchez-Alvarez R, Paíno T,
Herrero-González S, Medina JM and Tabernero A: Tolbutamide reduces
glioma cell proliferation by increasing connexin43, which promotes
the upregulation of p21 and p27 and subsequent changes in
retinoblastoma phosphorylation. Glia. 54:125–134. 2006. View Article : Google Scholar
|
|
13
|
Maatouk D and Harfe B: MicroRNAs in
development. ScientificWorldJournal. 6:1828–1840. 2006. View Article : Google Scholar
|
|
14
|
Novakova J, Slaby O, Vyzula R and Michalek
J: MicroRNA involvement in glioblastoma pathogenesis. Biochem
Biophys Res Commun. 386:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin Z, Xu S, Yu H, Yang B, Zhao H and Zhao
G: miR-125b inhibits Connexin43 and promotes glioma growth. Cell
Mol Neurobiol. 33:1143–1148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Somekawa S, Fukuhara S, Nakaoka Y, Fujita
H, Saito Y and Mochizuki N: Enhanced functional gap junction
neoformation by protein kinase A-dependent and Epac-dependent
signals downstream of cAMP in cardiac myocytes. Circ Res.
97:655–662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shu M, Zhou Y, Zhu W, et al: MicroRNA 335
is required for differentiation of malignant glioma cells induced
by activation of cAMP/protein kinase A pathway. Mol Pharmacol.
81:292–298. 2012. View Article : Google Scholar
|
|
18
|
Mei J, Bachoo R and Zhang CL:
MicroRNA-146a inhibits glioma development by targeting Notch1. Mol
Cell Biol. 31:3584–3592. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barde I, Salmon P and Trono D: Production
and titration of lentiviral vectors. Curr Protoc Neurosci.
2010.Chapter 4. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen C, Ridzon DA, Broomer AJ, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mohammadi-Yeganeh S, Paryan M and Mirab
Samiee S: Development of a robust, low cost stem-loop real-time
quantification PCR technique for miRNA expression analysis. Mol
Biol Rep. 40:3665–3674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takano S, Yamashita T and Ohneda O:
Molecular therapeutic targets for glioma angiogenesis. J Oncol.
2010:3519082010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bai RY, Staedtke V and Riggins GJ:
Molecular targeting of glioblastoma: Drug discovery and therapies.
Trends Mol Med. 17:301–312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ling N, Gu J, Lei Z, et al: microRNA-155
regulates cell proliferation and invasion by targeting FOXO3a in
glioma. Oncol Rep. 30:2111–2118. 2013.PubMed/NCBI
|
|
25
|
Lages E, Guttin A, El Atifi M, et al:
MicroRNA and target protein patterns reveal physiopathological
features of glioma subtypes. PLoS One. 6:e206002011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chaudhry MA, Sachdeva H and Omaruddin RA:
Radiation-induced micro-RNA modulation in glioblastoma cells
differing in DNA-repair pathways. DNA Cell Biol. 29:553–561. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Poltronieri P, D’Urso PI, Mezzolla V and
D’Urso OF: Potential of anti-cancer therapy based on anti-miR-155
oligonucleotides in glioma and brain tumours. Chem Biol Drug Des.
81:79–84. 2013. View Article : Google Scholar
|
|
28
|
Feng SY, Dong CG, Wu WK, Wang XJ, Qiao J
and Shao JF: Lentiviral expression of anti-microRNAs targeting
miR-27a inhibits proliferation and invasiveness of U87 glioma
cells. Mol Med Rep. 6:275–281. 2012.PubMed/NCBI
|
|
29
|
Yang S, Wang K, Qian C, et al: A predicted
miR-27a-mediated network identifies a signature of glioma. Oncol
Rep. 28:1249–1256. 2012.PubMed/NCBI
|
|
30
|
Blum AE, Walsh BC and Dubyak GR:
Extracellular osmolarity modulates G protein-coupled
receptor-dependent ATP release from 1321N1 astrocytoma cells. Am J
Physiol Cell Physiol. 298:C386–C396. 2010. View Article : Google Scholar :
|
|
31
|
Racagni G, Pezzotta S, Giordana MT, et al:
Cyclic nucleotides in experimental and human brain tumors. J
Neurooncol. 1:61–67. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mostafavi H, Khaksarian M, Joghataei MT,
et al: Selective β2 adrenergic agonist increases Cx43 and miR-451
expression via cAMP-Epac. Mol Med Rep. 9:2405–2410. 2014.PubMed/NCBI
|
|
33
|
Salameh A, Krautblatter S, Karl S, et al:
The signal transduction cascade regulating the expression of the
gap junction protein connexin43 by beta-adrenoceptors. Br J
Pharmacol. 158:198–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu Y, Zhang Y, Shan H, et al: MicroRNA-1
downregulation by propranolol in a rat model of myocardial
infarction: a new mechanism for ischaemic cardioprotection.
Cardiovasc Res. 84:434–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hanstein R, Trotter J, Behl C and Clement
AB: Increased connexin 43 expression as a potential mediator of the
neuro-protective activity of the corticotropin-releasing hormone.
Mol Endocrinol. 23:1479–1493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Curcio A, Torella D, Iaconetti C, et al:
MicroRNA-1 down-regulation increases connexin 43 displacement and
induces ventricular tachyarrhythmias in rodent hypertrophic hearts.
PLoS One. 8:e701582013. View Article : Google Scholar
|
|
37
|
Hegi ME, Rajakannu P and Weller M:
Epidermal growth factor receptor: a re-emerging target in
glioblastoma. Curr Opin Neurol. 25:774–779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Katakowski M, Zheng X, Jiang F, Rogers T,
Szalad A and Chopp M: MiR-146b-5p suppresses EGFR expression and
reduces in vitro migration and invasion of glioma. Cancer Invest.
28:1024–1030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ueki T, Fujita M, Sato K, Asai K, Yamada K
and Kato T: Epidermal growth factor down-regulates connexin-43
expression in cultured rat cortical astrocytes. Neurosci Lett.
313:53–56. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi J, Zhang L, Shen A, et al: Clinical
and biological significance of forkhead class box O 3a expression
in glioma: mediation of glioma malignancy by transcriptional
regulation of p27kip1. J Neurooncol. 98:57–69. 2010. View Article : Google Scholar
|
|
41
|
D’Urso PI, D’Urso OF, Storelli C, et al:
miR-155 is up-regulated in primary and secondary glioblastoma and
promotes tumour growth by inhibiting GABA receptors. Int J Oncol.
41:228–234. 2012.
|
|
42
|
Qiu S, Lin S, Hu D, Feng Y, Tan Y and Peng
Y: Interactions of miR-323/miR-326/miR-329 and
miR-130a/miR-155/miR-210 as prognostic indicators for clinical
outcome of glioblastoma patients. J Transl Med. 11:102013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu S, Yang Y and Wu J: TNFα-induced
up-regulation of miR-155 inhibits adipogenesis by down-regulating
early adipogenic transcription factors. Biochem Biophys Res Commun.
414:618–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Johnstone SR, Best AK, Wright CS, Isakson
BE, Errington RJ and Martin PE: Enhanced connexin 43 expression
delays intra-mitotic duration and cell cycle traverse independently
of gap junction channel function. J Cell Biochem. 110:772–782.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang RP, Fan Y, Hossain MZ, Peng A, Zeng
ZL and Boynton AL: Reversion of the neoplastic phenotype of human
glioblastoma cells by connexin 43 (cx43). Cancer Res. 58:5089–5096.
1998.PubMed/NCBI
|