Introduction
Reperfusion of ischemic brain tissues is an
effective protocol for the treatment of stroke. However, the
aggravation of ischemia and hypoxia as well as the
necrosis/apoptosis of neurons in the peripheral zone destroys the
normal structure and function of brain tissues (1). The underlying mechanism of this
effect involves the release of oxygen free radicals and excitatory
amino acids as well as the activation of apoptosis-associated genes
(including an increase in the expression of pro-apoptotic and a
decrease in anti-apoptotic genes) (1). The destruction of the blood brain
barrier (BBB) further induces cerebral edema and normal
brain-tissue damage. These effects have markedly influenced the
efficacy of therapies for cerebral ischemia/reperfusion injury
(2). Numerous studies have shown
that during cerebral ischemia in the BBB, matrix
metalloproteinase-9 (MMP-9) and aquaporin-4 (AQP-4) have crucial
roles (3,4) in the development of cerebral edema.
MMP-9 is able to degrade the extracellular matrix and BBB,
influence the permeability of blood vessels and participate in the
induction of vasogenic brain edema (3). AQP-4 regulates water metabolism,
water distribution and osmotic pressure (4). Furthermore, the c-Jun N-terminal
kinase (JNK) pathway is involved in the activation of MMP-9 and
AQP-4 (5,6).
Propofol, which is clinically used as an anesthetic,
reduces cerebral blood flow, oxygen consumption and intracranial
pressure (7). A previous study has
indicated that propofol pre- and post-conditioning treatments may
protect tissues from cerebral ischemia-reperfusion injury. Propofol
inhibits the activation of proapoptotic genes, thereby reducing
neuronal apoptosis and improving prognosis (7–9).
However, previous studies have focused on the protective effects of
propofol on neurons and the mechanisms underlying these effects,
whereas there are few studies regarding the effects of propofol on
the BBB and its underlying mechanisms. To the best of our
knowledge, no study has been performed investigating the effects of
propofol on the expression of MMP-9, AQP-4 and phosphorylated JNK
following cerebral ischemia/reperfusion.
In the present study, an established rat model of
cerebral ischemia/reperfusion was treated with propofol
administered via intravenous injection. The condition of the BBB,
level of cephaloedema and neural functions of propofol-treated rats
were assessed and scored to evaluate the protective effects of
propofol post-conditioning. The expression levels of MMP-9 and
AQP-4 and the phosphorylation level of pJNK were also determined in
order to elucidate the mechanism underlying the protective effects
of propofol on the BBB.
Materials and methods
Drugs and reagents
The drugs and reagents used in the present study
included propofol (Diprivan; AstraZeneca, London, UK), Evans blue
(EB) and dimethylformamide (Sigma-Aldrich, St. Louis, MO, USA).
Antibodies included rabbit anti-mouse MMP-9 polyclonal antibody and
JNK/pJNK polyclonal antibody (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) rabbit anti-mouse AQP-4 antibody
(Chemicon®, EMD Millipore, Billerica, MA, USA) and
rabbit anti-mouse β-actin antibody (Sigma-Aldrich). A western
blotting kit was purchased from KPL (Gaithersburg, MD, USA).
Experimental animal selection and
grouping
Ninety-six healthy adult Wistar rats, weighing
260–300 g and aged 4–6 weeks (Experimental Animal Centre of
Zhongshan University, Guangzhou, China) were kept in a 12 h/12 h
light/dark cycle at 28–32°C with free access to food and water.
Rats were randomly divided into four groups: The sham operation
group (group S, n=24), the ischemia reperfusion group (group I,
n=24) and two propofol-treated groups, which included group P1 (20
mg/kg/h profopol, n=24) and group P2 (40 mg/kg/h profopol, n=24).
The study was approved by the Animal Care Committee of Sun Yat-sen
University (Guangzhou, China) and performed in strict accordance
with the National Institutes of Health Guide for the Use of
Laboratory Animals.
Preparation of cerebral
ischemia/reperfusion rat model and assessment of neural
function
The rats were subjected to fasting without drinking
for 12 h prior to surgery. Anesthesia was induced in a Plexiglass
chamber with 4% halothane (Sigma-Aldrich), followed by tracheal
intubation and mechanical ventilation with 1.3% halothane in 30%
O2/70% N2O. The left femoral artery was
cannulated to monitor blood pressure and spontaneous breath was
conserved. The rat was prepared for surgery by the removal of hair
in the middle of the neck in the supine position. Subsequently the
skin was prepared with 75% alcohol and a surgical incision was made
at the flank of the neck. The right common carotid artery, right
external carotid artery, right internal carotid artery and wing
palatal artery were isolated. The external carotid and wing palatal
arteries were ligatured and the right common carotid artery was
blocked by a bulldog clamp, while the right external carotid artery
was punctured 0.5 cm from the ligation at the proximal end to
heart. A nylon thread was inserted into the wound and pushed
through the right internal carotid artery in the direction of the
brain and the opposite side of the artery was pierced 18–20 mm
further down. The middle cerebral artery was then blocked.
Following surgery, cerebral blood flow was required to be reduced
to ~30%; otherwise, the rat model was considered unsuccessful and
removed (10). Two hours following
the arterial block, the thread was removed, the residual end of the
artery was fastened and the skin and subcutaneous tissue were
sutured. Once the rats regained consciousness, behavioral disorders
were assessed according to the Longa method (11) using the following scale: No visible
neural function loss, 0; left forepaw unable to straighten, 1;
walking impaired with an inclination towards ischemic hemisphere,
2; repeated left turning, 3; falling left, 4; unable to walk,
5.
Drug treatment
The P1 and P2 groups were treated with 20 mg/kg/h or
40 mg/kg/h propofol, respectively, for 30 min via the tail vein, 30
min following reperfusion. Group I was treated with an equal amount
of physiological saline (Sigma-Aldrich) and in group S, the
incision was made but no arterial occlusion was performed.
The water content of the ischemic hemisphere of the
brain was detected using the wet/dry weight ratio method with the
following formula: Water content (%)=100×[(wet weight)−(dry
weight)]/wet weight.
Detection of EB
The permeability of the BBB was determined according
to the protocol described by Uyama et al (12), by the detection of EB fluorescence.
Twenty-three hours following reperfusion, rats were anesthetized
with 10% chloral hydrate and 2% EB (3 ml/kg) was injected into the
femoral vein. One hour later, the ventriculus sinister was washed
with physiological saline for 0.5 h. The brain was removed by
decollation and surface moisture was blotted with filter paper. The
cerebral cortex of the ischemic side was excised and the wet weight
was measured with an electronic balance prior to incubation with 4
ml dimethylformamide (Sigma-Aldrich) at 50°C for 48 h followed by
centrifugation at 1509 × g for 15 min. The absorbency of the
supernatant at 620 nm was determined using a Hitachi U2001
(Hitachi, Ltd, Tokyo, Japan). The content of EB (µg/g) was
subsequently calculated according to the standard curve method
(12).
Immunohistochemical examination
Half of the animals in each group were randomly
selected to be sacrificed by administration of 5% halothane
(Sigma-Aldrich) and fixed in the dorsal position immediately
following behavioral testing. The rat hearts were exposed by
opening the chest from the xiphisternum and were perfused through
the ascending aorta with 150 ml saline followed by 400 ml 4%
paraformaldehyde. Following perfusion, the brain was removed, fixed
in 4% paraformaldehyde (Sigma-Aldrich) overnight and subsequently
transferred to 30% sucrose (Sigma-Aldrich) and incubated for 3–5
days. The damaged brain area was dissected into 1-mm coronal
sections. Sequential 10-µm coronal sections were cut from
the sections (from bregma −2.3 to −2.45 mm) by cryomicrotomy
(CM1900; Leica Microsystems GmbH, Wetzlar, Germany), the sections
were treated with 0.3% H2O2 (Sigma-Aldrich)
in methanol for 30 min, hydrated gradually in distilled water and
incubated for 2 h with 5% goat serum (Sigma-Aldrich) to block
nonspecific immune reactions. The sections were subsequently
incubated at 4°C overnight with rabbit polyclonal anti-MMP-9 or
anti-AQP-4 antibodies (1:2,000). Following washing with
Tris-buffered saline (Sigma-Aldrich), the sections were incubated
with biotinylated goat anti-rabbit immunoglobulin G (IgG) (1:200;
Zhongshan Golden Bridge Biotechnology Co., Ltd, Beijing, China) for
2 h followed by incubation with avidin-biotinperoxidase complex
(1:200; Zhongshan Golden Bridge Biotechnology Co.) for 2 h.
Finally, the sections were exposed to 0.01% 3,3′-diaminobenzidine
(Sigma-Aldrich) for 0.5–2 min, followed by examination under a
light microscope (Olympus BX51; Olympus Corp., Tokyo, Japan). The
optical density of MMP-9 and AQP-4 immunoreactivity was evaluated
in an area of 1×1 mm2 in the boundary zone.
Western blot analysis
Twenty-four hours following reperfusion, rats were
sacrificed. The brains were removed and immediately cut into two
2-mm sections on dry ice, excluding 4 mm of the rostral tissue.
Slices were cut into the right and left hemispheres, and the right
striatum and parietal cortex were isolated and homogenized in
tissue lysate buffer (10 ml/mg tissue) containing a mixture of
proteinase inhibitors (Thermo Fisher Scientific China Co., Ltd,
Beijing, China), prior to dispersion by sonication. The homogenate
was centrifuged at 2,250 × g for 30 min at 4°C, and the supernatant
was saved. A bicinchoninic acid assay (Pierce Biotechnology, Inc.;
Thermos Fisher Scientific, Rockford, IL, USA) was used to determine
the protein concentration. Proteins (30 µg/well) were
separated on using 10% SDS-PAGE and transferred to nitrocellulose
membranes (Sigma-Aldrich) at 100 mV. The membranes were blocked
with 5% non-fat milk (Sigma-Aldrich) in phosphate-buffered saline
with 0.1% Tween-20 (Sigma-Aldrich) for 1 h at room temperature and
incubated overnight at 4°C with rabbit anti-mouse MMP-9 polyclonal
antibody (1:2,000; Santa Cruz Biotechnology, Inc.), rabbit
anti-mouse AQP-4 antibody (1:2,000; Abcam, Cambridge, MA, USA) or
rabbit polyclonal anti-pJNK antibody (1:2,000; Santa Crux
Biotechnology, Inc.). Following three washes in Tris-buffered
saline for 5 min, the blots were incubated for 1 h at room
temperature with the appropriate horseradish peroxidase-conjugated
secondary antibody (1:3,000; Santa Cruz Biotechnology, Inc.).
MMP-9, AQP-4 and pJNK were subsequently visualized by incubation
with enhanced chemiluminescence reagent (Thermo Fisher Scientific,
Waltham, MA, USA) for 1 min, and exposure onto hyperfilm
(Sigma-Aldrich) for 1–10 min. Membranes which were positive for JNK
were re-blocked with 5% non-fat milk for 1 h at room temperature
and incubated with rabbit polyclonal anti-JNK antibody (Santa Cruz
Biotechnology, Inc.) overnight at 4°C. Visualization of JNK was
performed as described above. Image J 1.35 software (National
Institutes of Health, Bethesda, MD, USA) was used to analyze the
relative density values.
Statistical analysis
Data were analyzed using SPSS 16.0 (SPSS, Inc.,
Chicago, IL, USA). Values are expressed as the mean ± standard
deviation. Differences between groups were analyzed using one-way
analysis of variance and multiple comparisons were made using the
least significant difference test. Data of skewed distribution are
presented as the median. P<0.05 was considered to indicate a
statistically significant difference between values.
Results
Propofol post-conditioning improves
neurobehavioral scores and decreases water- and EB content 24 h
following reperfusion
The assessment of neurological function indicated
that all experimental animals exhibited post-ischemic neurological
deficits. Rats in groups I, P1, and P2 scored significantly higher
than those in group S (P<0.05). Compared with the rats in group
I, those in groups P1 and P2 scored significantly lower
(P<0.05). The experimental groups demonstrated higher water
content and EB content in the ischemic hemisphere in comparison to
that of group S (P<0.05). The water and EB content were
significantly reduced in groups P1 and P2 compared to those in
group I (P<0.05; Table I).
 | Table IEffect of propofol post-conditioning
on neurobehavioral scores, water content and EB content 24 h
following reperfusion. |
Table I
Effect of propofol post-conditioning
on neurobehavioral scores, water content and EB content 24 h
following reperfusion.
| Group | Neurobehavioral
scores [M(Q), n=18] | Water content (%,
n=6) | EB content
(µg/g, n=6) |
|---|
| S | 0 | 77.41±0.19 | 0.54±0.31 |
| I | 3.0 (1.0)a | 86.51±0.53a | 12.65±3.14a |
| P1 | 2.0 (1.0)a,b | 82.72±0.86a,b | 5.72±2.78a,b |
| P2 | 2.0 (0.5)a,b | 81.25±0.67a,b | 4.76±2.17a,b |
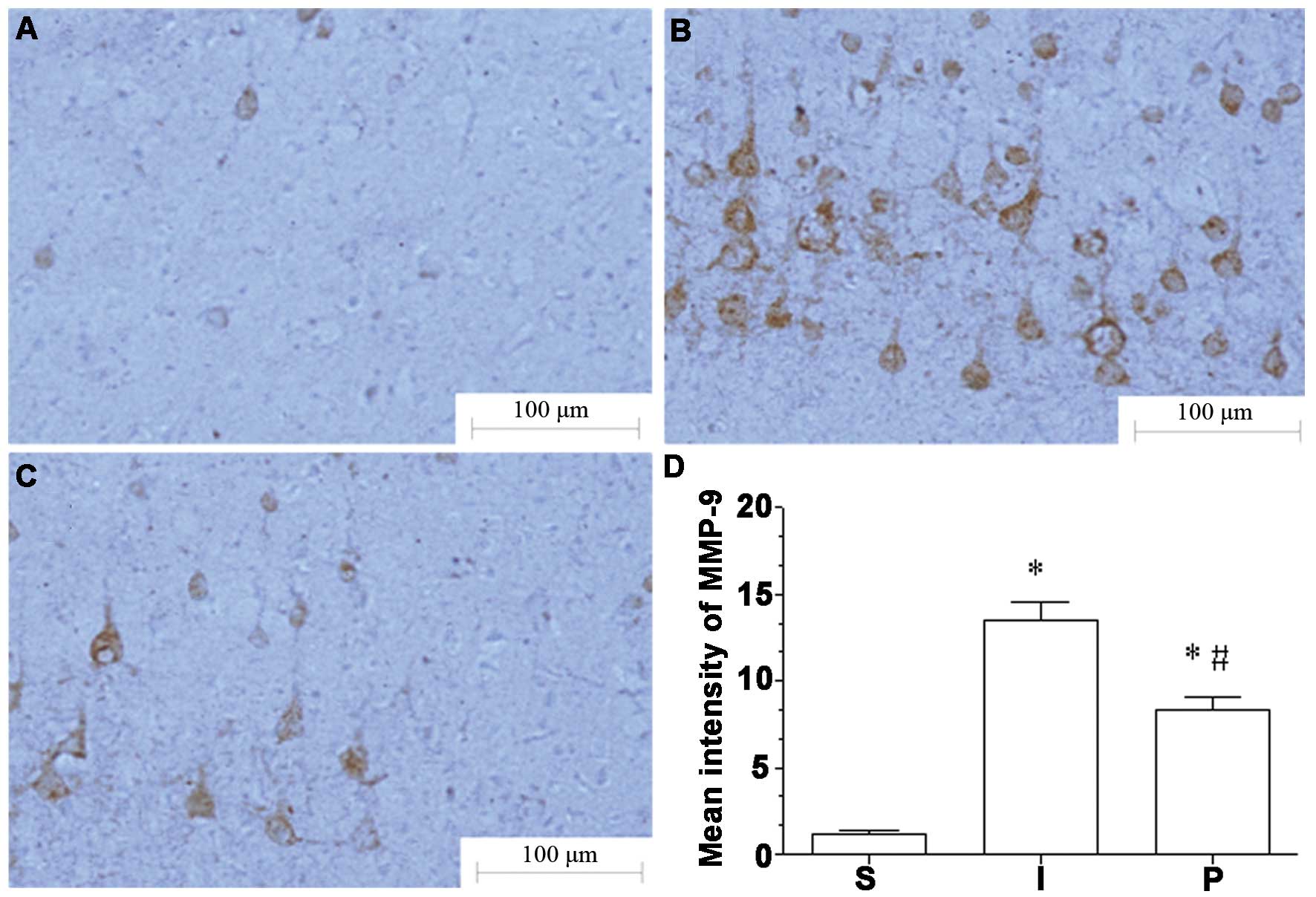
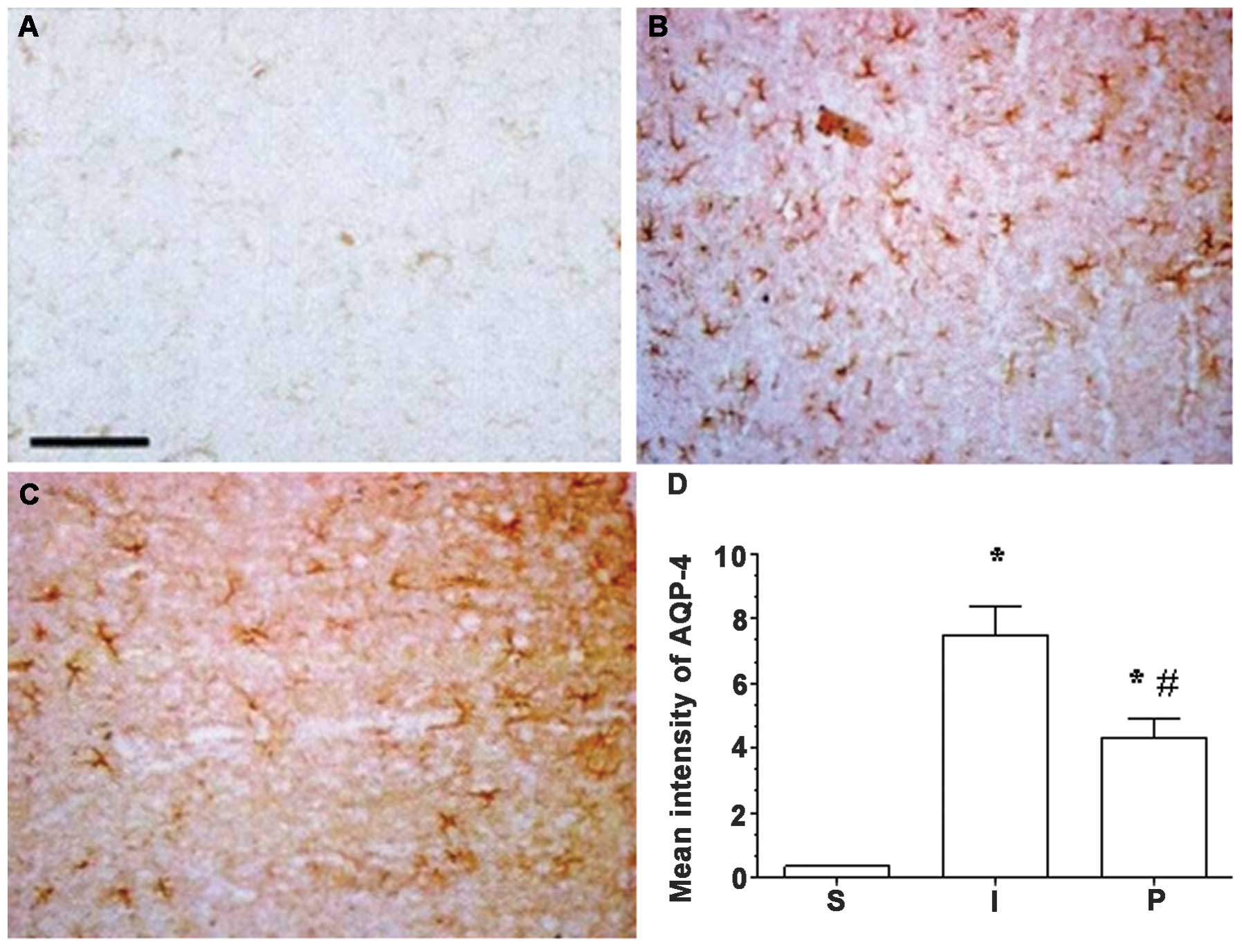
MMP-9 and AQP-4 expression in the
ischemic hemisphere are attenuated following propofol
treatment
Immunohistochemical analysis indicated that the
expression levels of MMP-9 and AQP-4 in ischemic brain tissue in
group I were significantly increased compared to those in group S
(P<0.05). Following propofol post-conditioning, the expression
levels of MMP-9 and AQP-4 were significantly decreased in groups P1
and P2 compared with those in group I (P<0.05), although
expression levels remained significantly higher than those in group
S (P<0.05; Figs. 1 and 2).
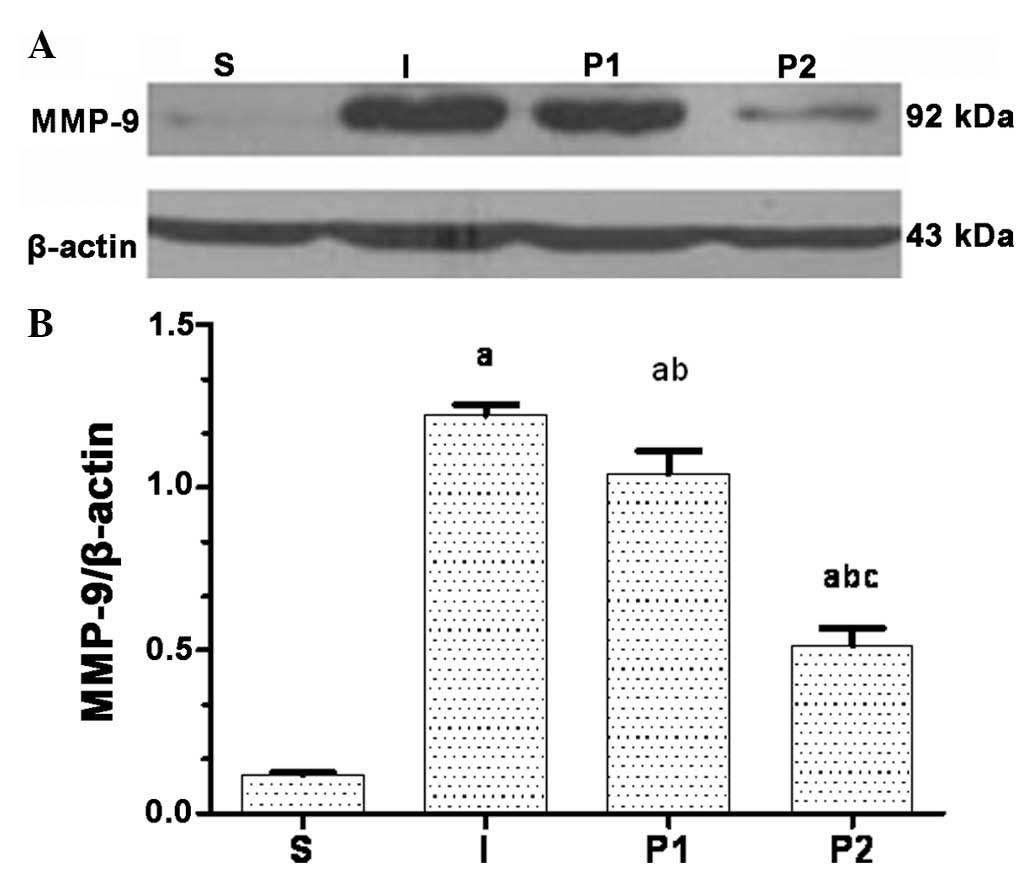
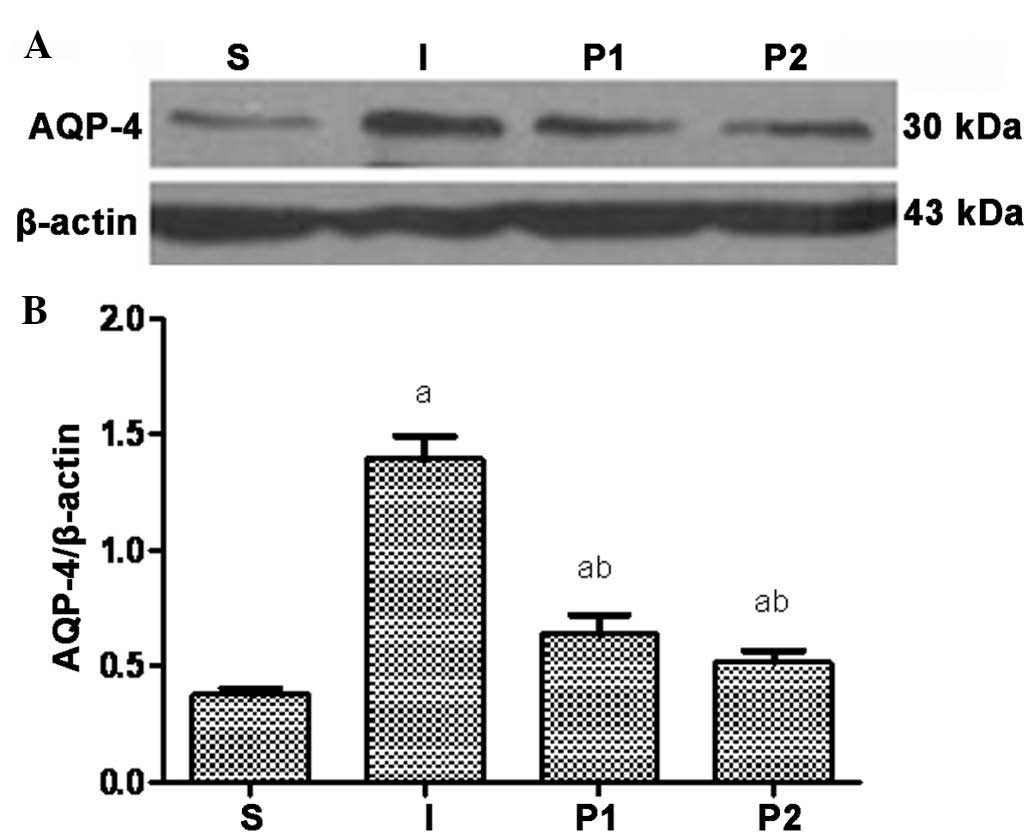
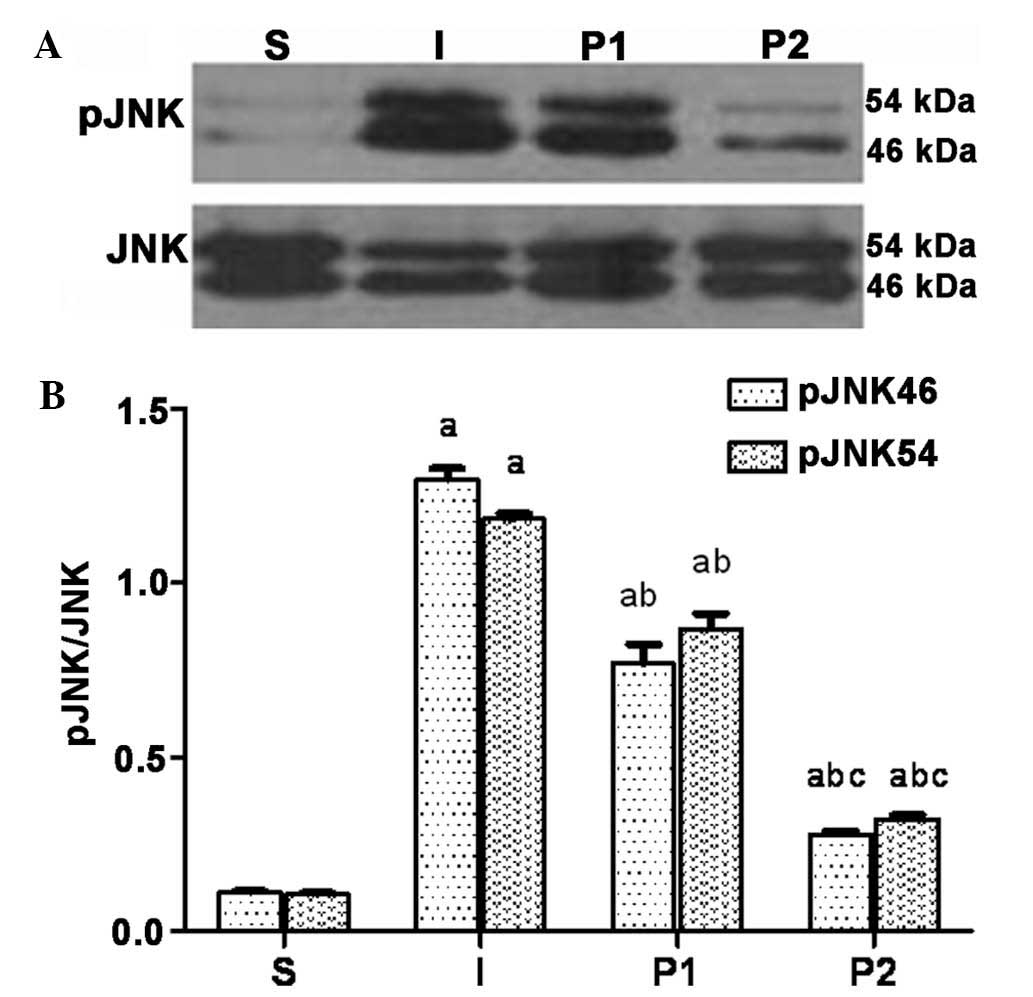
Propofol post-conditioning decreases the
expression of MMP-9, AQP-4 and pJNK in brain tissues
Twenty-four hours following ischemia/reperfusion,
the expression levels of MMP-9, AQP-4 and the levels of pJNK were
significantly increased. Following treatment with propofol, the
expression levels of MMP-9, AQP-4 and pJNK in groups P1 and P2 were
significantly decreased (P<0.05). Furthermore, the expression
levels of MMP-9, AQP-4 and pJNK were significantly decreased in
group P2 compared with those in group P1 (P<0.05; Figs. 3Figure 4–5).
Discussion
The middle cerebral artery is prone to ischemic
cerebrovascular disease. In a previous study, a model of cerebral
ischemia/reperfusion was established using the Longa method
(11), which was selected for the
high success rate, reproducibility and low risk of complications.
This established model effectively simulated the clinical and
pathological characteristics of cerebral infarction. In the present
study, rats with cerebral ischemia/reperfusion scored significantly
lower in the assessment of neural function. Cerebral infarction was
observed in the ischemic hemisphere, which indicated that the model
reflected the clinical and pathological characteristics of cerebral
infarction. Vascular occlusion resulted in cerebral circulatory
dysfunction, and therefore cerebral ischemia and hypoxia
irreversibly damaged neurons in the affected part of the brain.
Furthermore, damage to the BBB induced successive cerebral edema,
which further impaired brain tissues. A significant improvement in
neural function was observed following propofol post-conditioning
in cerebral ischemia/reperfusion rats and the water content of the
brain was also decreased. These results demonstrated that propofol
treatment may protect against cerebral ischemia/reperfusion damage,
which was in agreement with the results reported in a previous
paper (9).
The aquaporins are a family of water-selective cell
membrane transporters. AQP-4 widely exists in the central nervous
system, and localizes to the membrane of astrocytes, subarachnoid
space, glial cells surrounding vessels, ependymal cells and the
choroid plexus (13). AQP-4 is a
critical component of integrated water and potassium homeostasis
(14), and has important roles in
water metabolism, localization and osmotic pressure (4). AQP-4 knock-out rats were found to
have improved cephaloedema following cerebral ischemic stroke
(15). MMP-9 is a type of matrix
metalloproteinase that is able to degrade the basal lamina and
disrupt the BBB. MMP-9 is involved in the pathophysiological
process of cerebral ischemia via the degradation of tight junctions
between the basal lamina and endothelial cells, which results in
the destruction of the BBB (16).
MMP-9 has a pivotal role in BBB proteolysis (17), and increased MMP-9 expression and
activation increases the permeability of the BBB and may therefore
exacerbate cephaloedema (3,18,19).
Furthermore, during cerebral ischemia hypoxia-inducible factor-1α
may damage the BBB by activating the expression of AQP-4 and MMP-9
and aggravating cerebral edema. Inhibitors of AQP-4 and MMP-9 may
protect the BBB to a certain extent (20). The extent of brain edema observed
in cerebral ischemia/reperfusion animals was significantly reduced
following inhibition, and the animals demonstrated an improved
prognosis (21–23). These results indicated that the
mechanism underlying BBB protection may be associated with the
inhibition of AQP-4 and MMP-9 function. Studies have additionally
demonstrated that propofol inhibited the expression of AQP-4 in
astroglia in vitro (23)
and decreased the expression of MMP-9 in vivo (24). In the present study, the expression
levels of AQP-4 and MMP-9 were markedly lower in groups P1 and P2
than those in group S, indicating that treatment with propofol
inhibited the expression of these proteins.
The activity of AQP-4 and MMP-9 is influenced by
numerous factors. Studies have indicated that damage to the central
nervous system may activate the expression of MMP-9 and AQP-4 via
the JNK pathway (5,6). JNK is a member of the
mitogen-activated protein kinase (MAPK) family, which are involved
in mediating the inflammatory response following cerebral ischemia,
cell factors, cell proliferation and differentiation as well as the
process of oxidant stress (5,6). The
activation of JNK induces the phosphorylation of downstream
factors, including c-Jun, protein 53 and activator protein 1, as
well as modulating gene expression, protein synthesis and neuronal
apoptosis (25–28). Animal experiments have revealed
that the phosphorylation of JNK is involved in neuronal apoptosis,
and that the inhibition of JNK phosphorylation is beneficial for
reducing the infarct area and improving prognosis following
cerebral ischemia (29). In
vitro experiments demonstrated that propofol inhibited the
activation of JNK and reduced the levels of cell death induced by
oxidative stress (30). Previous
studies demonstrated that intraperitoneal administration of l50
mg/kg propofol or intravenous administration of 20 mg/kg propofol
significantly alleviated cerebral edema, decreased the infarction
size during ischemia/reperfusion injury and improved the National
Institutes of Health Stroke Scale (NIHSS) score in a rat model
(9). Therefore, the dosage of
propofol selected for use in the present study was reasonable and
effective in accordance with previous experiments. The results of
the present study indicated that the levels of pJNK in groups P1
and P2 were markedly lower than those in the ischemia/reperfusion
group. Due to their similar chemical structures, propofol has
analogous biochemical functions to those of Vitamin E, including
anti-oxidative, mitochondrial protective and free-radical
scavenging activity (31–33). Furthermore, propofol inhibits the
phosphorylation of JNK (30,34).
These results therefore indicated that the mechanism underlying the
inhibitory effect of propofol on AQP-4 and MMP-9 may be via
inactivation of the JNK pathway.
The function of the BBB is to maintain the central
nervous system and following ischemia/reperfusion, the structure
and function of the BBB is altered. When the BBB is damaged, its
permeability is increased, which allows factors that cannot cross
the barrier under physiological conditions to cross the BBB
(2). This process may induce or
aggravate cephaloedema. The increased permeability of the BBB will
additionally further increase leukocyte infiltration in the
ischemic brain tissue, enhancing inflammation (35). EB is a classical indicator of BBB
permeability (12). The complex of
EB and plasma albumin (ESA) is unable to cross the BBB under normal
conditions; however, when the BBB is damaged, ESA is able to invade
and stain the brain. The infiltration of EB across the BBB is
positively correlated with the extent of BBB destruction (12). Propofol decreased the expression of
AQP-4 and MMP-9 in ischemia/reperfusion rats and downregulated the
phosphorylation of JNK, therefore protecting the integrity of the
BBB. The lower water content of groups P1 and P2 compared with that
of group I indicated that propofol treatment may protect the brain
by reducing the damage to the BBB and the extent of
cephaloedema.
Propofol reduced the levels of cephaloedema and the
destruction of the BBB following ischemia/reperfusion and improved
the score on the NIHSS. The underlying mechanism involved the
inhibition of AQP-4, MMP-9 and phosphorylation of JNK. In
conclusion, the results of the present study indicated that
propofol exerts a neuroprotective effect via protection of the
BBB.
Acknowledgments
The present study was supported by the Guangdong
Province Science and Technology Plan Project, Guangzhou, China (no.
2010B080701004) and the Guangdong Province Medical Research Fund
(no. B2010092).
References
|
1
|
Ginsberg MD: Role of free radical
reactions in ischaemic brain injury. Drug News Perspect. 14:81–88.
2001.
|
|
2
|
Pluta R: Pathological opening of the
blood-brain barrier to horseradish peroxidase and amyloid precursor
protein following ischemia-reperfusion brain injury. Chemotherapy.
51:223–226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tu XK, Yang WZ, Shi SS, et al:
5-lipoxygenase inhibitor zileuton attenuates ischaemic brain
damage: involvement of matrix metalloproteinase 9. Neurol Res.
31:848–852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Papadopoulos MC and Verkman AS:
Aquaporin-4 and brain edema. Pediatr Nephrol. 22:778–784. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vikman P, Ansar S, Henriksson M, et al:
Cerebral ischemia induces transcription of inflammatory and
extracellular-matrix-related genes in rat cerebral arteries. Exp
Brain Res. 183:499–510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yatsushige H, Ostrowski RP, Tsubokawa T,
et al: Role of c-jun N-terminal kinase in early brain injury after
subarachnoid hemorrhage. J Neurosci Res. 85:1436–1448. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Engelhard K, Werner C, Eberspächer E, et
al: Influence of propofol on neuronal damage and apoptotic factors
after incomplete cerebral ischemia and reperfusion in rats: a
long-term observation. Anesthesiology. 101:912–917. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang H, Luo M, Li C and Wang G: Propofol
post-conditioning induced long-term neuroprotection and reduced
internalization of AMPAR GluR2 subunit in a rat model of focal
cerebral ischemia/reperfusion. J Neurochem. 119:210–219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang HY, Wang GL, Yu YH and Wang Y: The
role of phosphoinositide-3-kinase/Akt pathway in propofol-induced
postconditioning against focal cerebral ischemia-reperfusion injury
in rats. Brain Res. 1297:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xing B, Chen H, Zhang M, et al: Ischaemic
post-conditioning protects brain and reduces inflammation in a rat
model of focal cerebral ischemia/reperfusion. J Neurochem.
105:1737–1745. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniotomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uyama O, Okamura N, Yanase M, et al:
Quantitative evaluation of vascular permeability in the gerbil
brain after transient ischemia using Evans blue fluorescence. J
Cereb Blood Flow Metab. 8:282–284. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zador Z, Stiver S, Wang V and Manley G:
Role of aquaporin-4 in cerebral edema and stroke. Handb Exp
Pharmacol. 190:159–170. 2009.
|
|
14
|
Ho JD, Yeh R, Sandstrom A, et al: Crystal
structure of human aquaporin 4 at 1.8 A and its mechanism of
conductance. Proc Natl Acad Sci USA. 106:7437–7442. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Manley GT, Fujimura M, Ma T, et al:
Aquaporin-4 deletion in mice reduces brain edema after acute water
intoxication and ischaemic stroke. Nat Med. 6:159–163. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heo JH, Lucero J, Abumiya T, Koziol JA,
Copeland BR and del Zoppo GJ: Matrix metalloproteinases increase
very early during experimental focal cerebral ischemia. J Cereb
Blood Flow Metab. 19:624–633. 2009.
|
|
17
|
Barr TL, Latour LL, Lee KY, et al:
Blood-brain barrier disruption in humans is independently
associated with increased matrix metalloproteinase-9. Stroke.
41:e123–e128. 2010. View Article : Google Scholar
|
|
18
|
Lee K, Lee JS, Jang HJ, et al: Chlorogenic
acid ameliorates brain damage and edema by inhibiting matrix
metalloproteinase-2 and 9 in a rat model of focal cerebral
ischemia. Eur J Pharmacol. 689:89–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hartz AM, Bauer B, Soldner EL, et al:
Amyloid-β contributes to blood-brain barrier leakage in transgenic
human amyloid precursor protein mice and in humans with cerebral
amyloid angiopathy. Stroke. 43:514–523. 2012. View Article : Google Scholar
|
|
20
|
Wang Z, Meng CJ, Shen XM, et al: Potential
contribution of hypoxia-inducible factor-1α, aquaporin-4, and
matrix metalloproteinase-9 to blood-brain barrier disruption and
brain edema after experimental subarachnoid hemorrhage. J Mol
Neurosci. 48:273–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Igarashi H, Huber VJ, Tsujita M and Nakada
T: Pretreatment with a novel aquaporin 4 inhibitor, TGN-020,
significantly reduces ischaemic cerebral edema. Neurol Sci.
32:113–116. 2011. View Article : Google Scholar :
|
|
22
|
Hu Q, Chen C, Khatibi NH, et al:
Lentivirus-mediated transfer of MMP-9 shRNA provides
neuroprotection following focal ischaemic brain injury in rats.
Brain Res. 1367:347–359. 2011. View Article : Google Scholar
|
|
23
|
Wang Z, Leng Y, Tsai LK, et al: Valproic
acid attenuates blood-brain barrier disruption in a rat model of
transient focal cerebral ischemia: the roles of HDAC and MMP-9
inhibition. J Cereb Blood Flow Metab. 31:52–57. 2011. View Article : Google Scholar :
|
|
24
|
Zhu SM, Xiong XX, Zheng YY and Pan CF:
Propofol inhibits aquaporin 4 expression through a protein kinase
C-dependent pathway in an astrocyte model of cerebral
ischemia/reoxygenation. Anesth Analg. 109:1493–1499. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deegan CA, Murray D, Doran P, et al:
Anesthetic technique and the cytokine and matrix metalloproteinase
response to primary breast cancer surgery. Reg Anesth Pain Med.
35:490–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tripathi A and Sodhi A: Growth
hormone-induced production of cytokines in murine peritoneal
macrophages in vitro: role of JAK/STAT, PI3K, PKC and MAP kinases.
Immuno Biology. 214:430–440. 2009. View Article : Google Scholar
|
|
28
|
Park HS, Huh SH, Kim MS, et al: Nitric
oxide negatively regulates c-jun N-terminal kinase/stress-activated
protein kinase by means of S-nitrosylation. Proc Natl Acad Sci USA.
97:14382–14387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeng XW, Li MW, Pan J, et al: Activation
of c-jun N-terminal kinase 1/2 regulated by nitric oxide is
associated with neuronal survival in hippocampal neurons in a rat
model of ischemia. Chin Med J (Engl). 124:3367–3372. 2011.
|
|
30
|
Murata Y, Fujiwara N, Seo JH, et al:
Delayed inhibition of c-jun N-terminal kinase worsens outcomes
after focal cerebral ischemia. J Neurosci. 32:8112–8115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shu L, Li T, Han S, et al: Inhibition of
neuron-specific CREB dephosphorylation is involved in propofol and
ketamine-induced neuroprotection against cerebral ischaemic
injuries of mice. Neurochem Res. 37:49–58. 2012. View Article : Google Scholar
|
|
32
|
De La Cruz JP, Villalobos MA, Sedeno G, et
al: Effect of propofol on oxidative stress in an in vitro model of
anoxia-reoxygenation in the rat brain. Brain Res. 800:136–144.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shibata H, Katsuki H, Okawara M, et al:
c-jun N-terminal kinase inhibition and alpha-tocopherol protect
midbrain dopaminergic neurons from
interferon-gamma/lipopolysaccharide-induced injury without
affecting nitric oxide production. J Neurosci Res. 83:102–109.
2006. View Article : Google Scholar
|
|
34
|
Corcoran TB, Engel A, Sakamoto H, et al:
The effects of propofol on neutrophil function, lipid peroxidation
and inflammatory response during elective coronary artery bypass
grafting in patients with impaired ventricular function. Br J
Anaesth. 97:825–831. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ouyang YB, Voloboueva LA, Xu LJ and
Giffard RG: Selective dysfunction of hippocampal CA1 astrocytes
contributes to delayed neuronal damage after transient forebrain
ischemia. J Neurosci. 27:4253–4260. 2007. View Article : Google Scholar : PubMed/NCBI
|