Introduction
Estrogen has protective effects in cardiovascular
function (1). The biological
effects of estrogen are mainly mediated by estrogen receptors
(ERs). Two classic nuclear ER isoforms, ERα and ERβ, are encoded by
separate genes and have differential distribution within tissues
and cells. These receptors have been demonstrated to be expressed
in both neonatal (2) and adult
(3) cardiac myocytes.
In addition to the classic ERs, a G protein-coupled
ER (GPER) has been found to be expressed in cardiomyocytes
(4,5). Notably, the majority of studies which
have examined ER localization with cardiac cells did not find a
difference in the distribution or abundance between males and
females (3,4). Recent studies have demonstrated the
existence of a novel G protein-coupled receptor 30, GPR30, here
referred to as G protein-coupled estrogen receptor (GPER), that
binds directly to estrogen and mediates its action (6–10).
Several studies have discovered coagulable cytolysis
around the ischemic myocardial infarction zone without the presence
of infiltration of inflammatory cells, a phenomenon similar to the
morphological changes of apoptosis (11). Furthermore, apoptotic cells began
to appear in the ischemic margin 1 h following ischemia, and the
number of the apoptotic cells increased with time and peaked at 5 h
following ischemia, suggesting apoptosis may be a main feature of
myocardial ischemia (12).
GPER activation improved functional recovery and
reduced the infarct size in isolated rat hearts following ischemia
and reperfusion (13). The
potential role and the mechanism of GPER activation in
cardioprotection is an important topic under investigation. The
aims of the present study were to examine the role of GPER
activation in cardiocyte apoptosis and SOD, TNF-α, ATPase
expression by using a specific GPER agonist (G1) (14) in H9C2 myocardial cells following
ischemia-reperfusion.
Materials and methods
Model preparation
H9C2 myocardial cells were subjected to 20 min of
ischemia (in a hypoxia chamber filled with 95% N2 and 5%
CO2 at 37°C) followed by 120 min of reperfusion (with
95% O2, 5% CO2 at 37°C). The cells were then
randomly assigned to three experimental groups (n=5/group): The
control G1 (a GPER-specific agonist; 10 nmol/l) and E2 (1 nmol/l)
groups (Sigma-Aldrich, St Louis, MO, USA). G1 or 17β-estradiol (E2)
was administered 5 min prior to ischemia.
GPER-knockout (KO) H9C2 cells were similarly
subjected to 20 min of ischemia and 120 min of reperfusion. They
were randomly assigned to five experimental groups (n=5/group);
control, empty vector, G1, GPER-KO, GPER-KO+G1.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted from H9C2 cell with the use
of TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA). Isolated RNA (10 µg) was reverse-transcribed using the
RevertAid First Strand cDNA Synthesis kit (therm k1622; Thermo
Scientific, Waltham, MA, USA). PCR amplification was conducted in a
total volume of 25 µl (QPS-201; Toyobo, Osaka, Japan): cDNA
2.5 µl, F (5 pmol/ml) 2 µl, R (5 pmol/ml) 2
µl, THUNDERBIRD™ SYBR Green pPCR Mix 12.5 µl and
H2O 6 µl, using an Applied Biosystems 7300
Real-Time PCR System (Applied Biosystems Life Technologies, Foster
City, CA, USA). The primer sequence for rat GADPH was as follows:
F, 5′-CGCTAACATCAAATGGGGTG-3′; and R, 5′-TTGCTGACAATCTTGAGGGAG-3′.
GAPDH was used as an internal control. Cycling conditions were:
95°C for 1 min (95°C for 15 sec, 58°C for 20 sec, 72°C for 20 sec)
for 40 cycles, and 82°C for 10 min. Data were calculated using the
2−ΔΔCt method.
Western blot analysis
Protein (40 µg) from H9C2 cells was separated
oby SDS-PAGE and electrophoretically transferred onto
polyvinylidene fluoride membranes. The membranes were incubated
with Bcl-2 (rabbit monoclonal; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and Bax (rabbit monoclonal; Abcam, Cambridge, UK)
antibodies. The blots were then washed and incubated with
horseradish peroxidase-conjugated secondary antibody (KPL, Inc.,
Gaithersburg, MD, USA) and the blot was developed with a
supersignal enhanced chemiluminescence detection kit (Thermo
Scientific). The blots were scanned using an Epson V300 scanner
(Epson, Suwa, Japan) and the blot densities were analyzed with
AlphaEaseFC software (Alphalnnotech, San Leandro, CA, USA).
Identification of apoptotic cells with
Hoechst staining
Apoptotic cells were determined using Hoechst 33258
staining. The cells were washed in phosphate-buffered saline and
labeled with Hoechst 33258 (Invitrogen Life Technologies) at room
temperature in the dark for 10 min. The cell nuclei were observed
and visualized by an inverted fluorescence microscope (Nikon-Ti;
Nikon Corporation, Tokyo, Japan). The number of apoptotic nuclei
was determined in at least six randomly selected areas from three
coverslips of every experimental group. The data were expressed as
the percentage of apoptotic cells relative to the total number of
cells.
SOD
Measurement of SOD enzyme activation was assessed
with xanthine oxidase enzyme kit (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) following the manufacturer’s
instructions and measured with an autoanalyzer (752-P UV-visible
spectrophotometer; Xianguang Instrument Co., Ltd. Shanghai, China)
at 550 nm and 37°C.
TNF-α
TNF-α was measured using commercially available
quantitative sandwich ELISA kits according to manufacturer’s
instructions. The analyses were performed with 96-well microtiter
plate ELISA kits for TNF-α (eBioscience, San Diego, CA, USA).
Microtiter strips pre-coated with biotinylation antibodies
generated against the proteins were used for quantification.
ATPase
Measurement of ATPase activation was measured via
inorganic phosphorus activation. The samples were analyzed
according to the manufacturer’s instructions (Nanjing Jiancheng
Bioengineering Institute) and measured with the 752-P UV-visible
spectrophotometer at 636 nm and room temperature.
Statistical analysis
All of the data were subjected to analysis of
variance, followed by a Bonferroni correction for post-hoc t-test
using the SAS version 9.3 software (SAS Institute Inc., Cary, NC,
USA). P≤0.05 was considered statistically significant.
Results
Pretreatment with G1 and E2 increases
Bcl-2 mRNA and protein expression in H9C2 cells following I/R
In the G1 and E2 groups, in which the H9C2 cells had
been subjected to 20 min of ischemia followed by 120 min of
reperfusion, the Bcl-2 mRNA expression, as measured by qPCR, was
higher than that in the control group (Fig. 1A). To further confirm the relative
expression of Bcl-2 in both strains, western blot analysis was also
performed, and the Bcl-2 protein expression was significantly
enhanced in the G1 and E2 groups (Fig.
1B). To confirm that the G1-induced effects were due to the
specific activation of GPER, G1 was administered 5 min prior to
ischemia in the GPER-knockout H9C2 cells. No evident effects were
found in the GPER-knockout H9C2 cells (Fig. 1C and D). Therefore, it is evident
that Bcl-2 mRNA and protein expression were increased in the I/R
myocardial cells by G1 or E2, and the G1-induced activation was
dependent on activation of GPER.
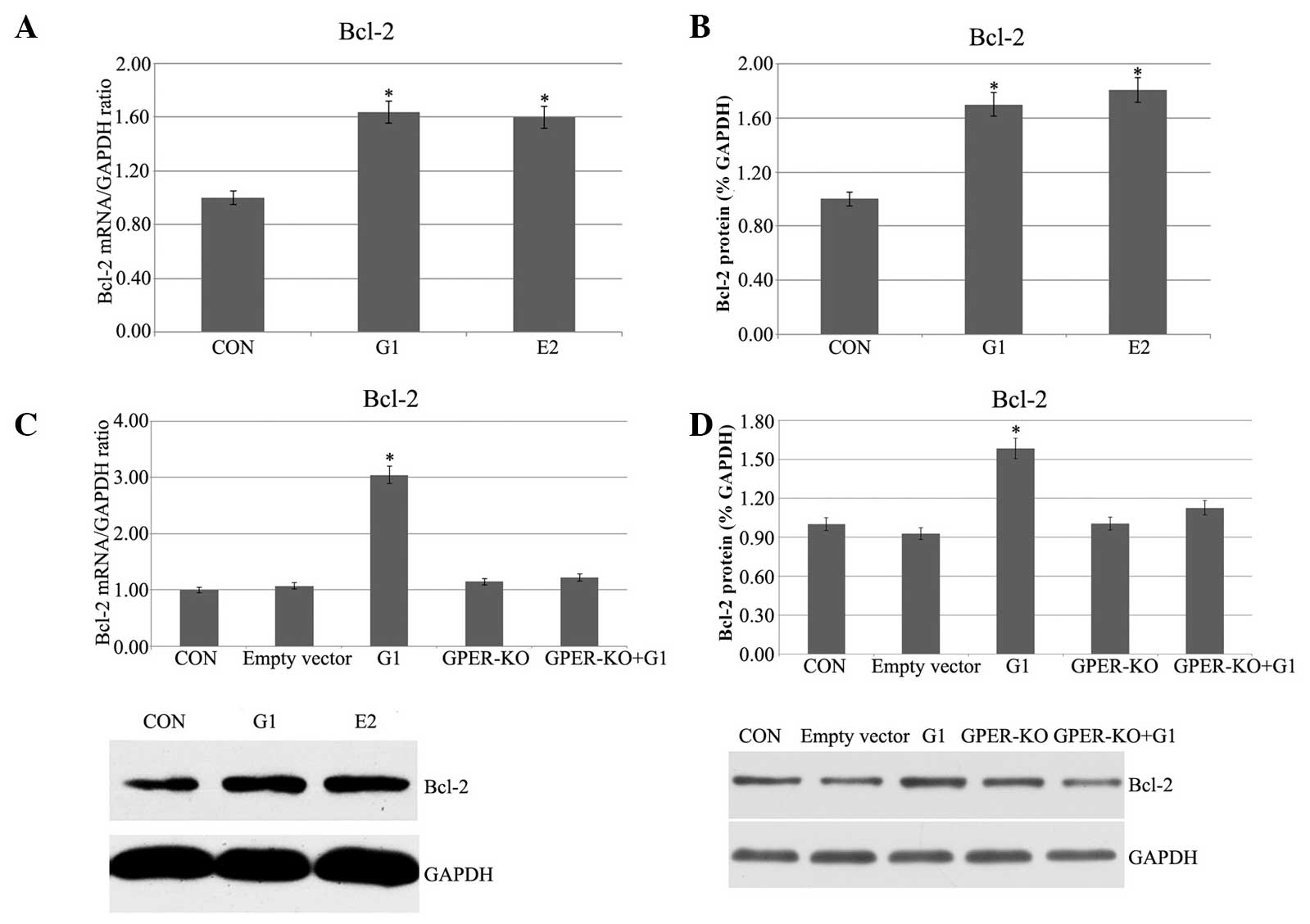 | Figure 1(A) Myocardial Bcl-2 mRNA expression.
CON, H9C2 cells were subjected to 20 min of ischemia followed by
120 min of reperfusion; G1, G1 (10 nmol/l) was administered 5 min
prior to ischemia; E2, E2 (1 nmol/l) was administered5 min prior to
ischemia. (B) Myocardial Bcl-2 protein expression. (C and D) Bcl-2
mRNA and protein expression, respectively, in GPER-KO H9C2 cells.
(P<0.05 vs. control). GPER, G protein-coupled estrogen receptor;
E2, 17β-estradiol; G1, GPER agonist; CON, control; KO, knockout;
Bcl-2, B-cell lymphoma 2. |
Pretreatment with G1 and E2 decreases Bax
mRNA and protein expression in H9C2 cells following I/R
Following administration of G1 or E2, a marked
decrement of Bax expression was observed as determined by qPCR and
western blotting experiments (Fig. 2A
and B). G1 had no evident effects on the GPER-knockout H9C2
cells (Fig. 2C and D). The
observed stable Bax expression suggested that G1 and E2 inhibited
Bax in I/R H9C2 cells, and the G1-induced activation was through
the activation of GPER.
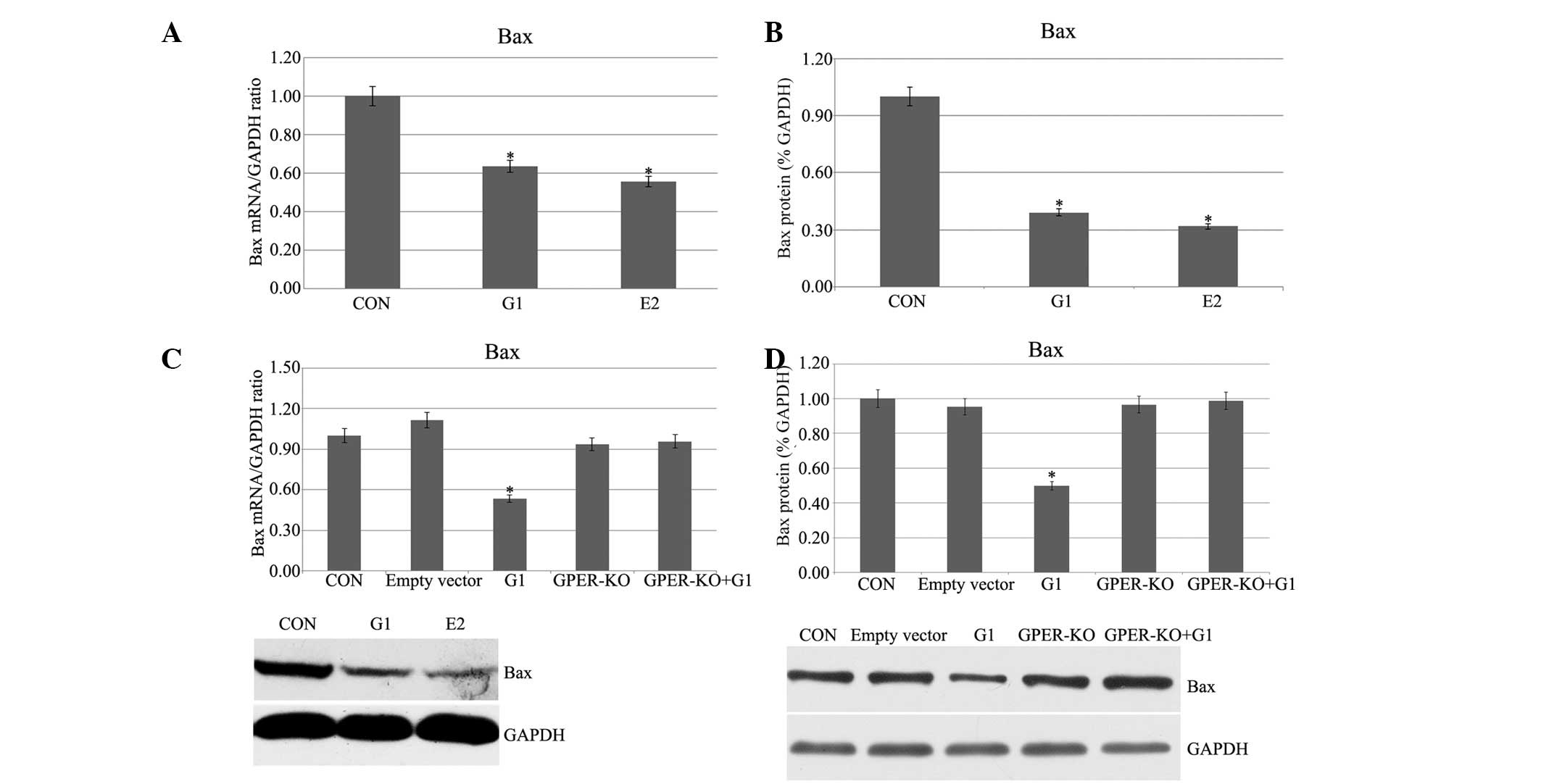 | Figure 2(A and B) Bax mRNA and protein
expression, respectively, in I/R H9C2 cells. (C and D) Bax mRNA and
protein expression, respectively, in GPER-knockout H9C2 cells.
(*P<0.05 vs. control). I/R, ischemia/reperfusion;
GPER, G protein-coupled estrogen receptor; E2, 17β-estradiol; G1,
GPER agonist; CON, control; KO, knockout; Bax, B-cell
lymphoma-associated X. |
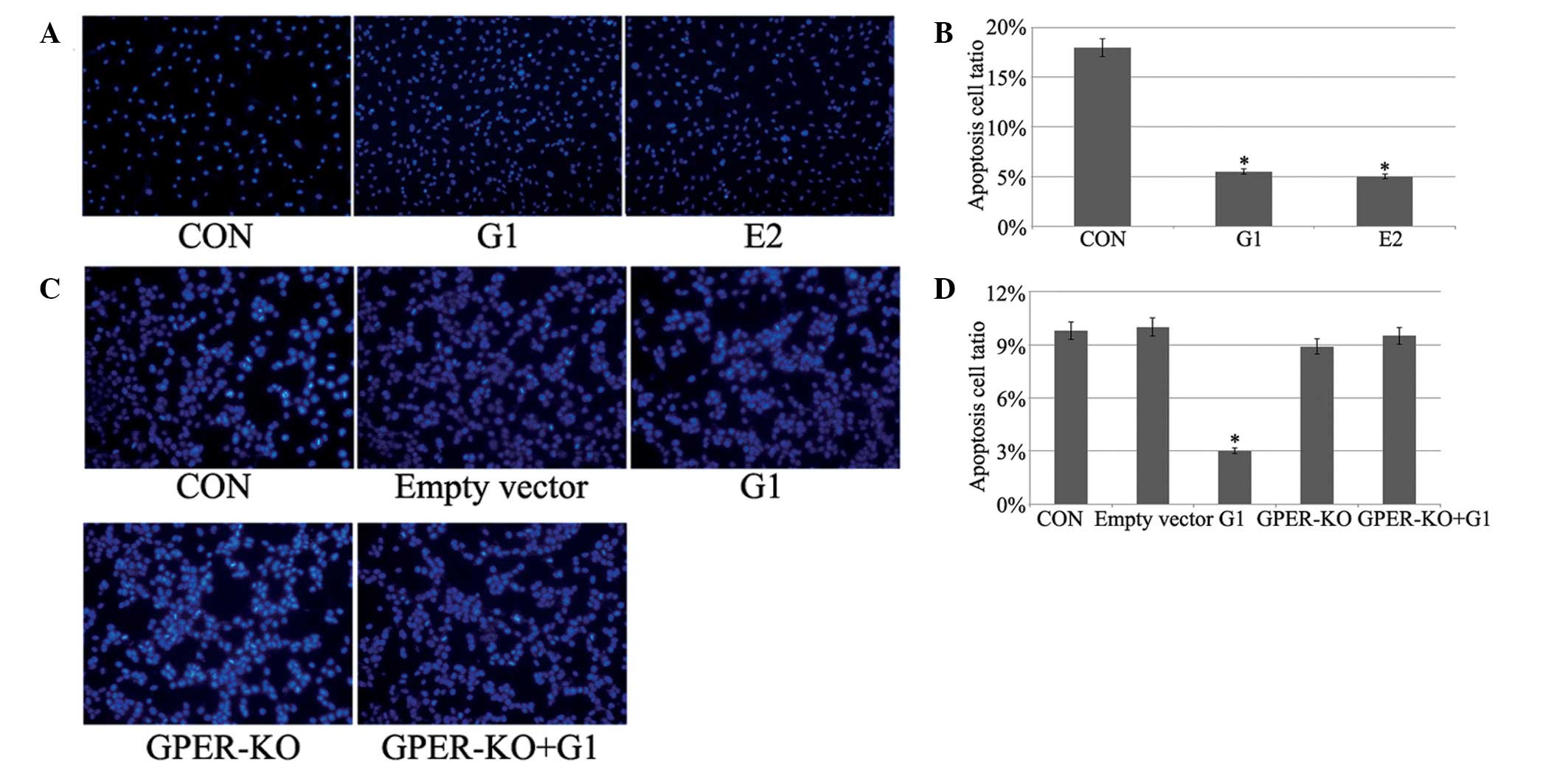
G1 and E2 protect H9C2 myocardial cells
from I/R-induced apoptosis
Apoptotic cells were determined by Hoechst 33258
staining, which allows determination and quantification of cells
with fragmented and condensed chromatin. Fig. 3 demonstrates that G1 or E2
treatment decreased apoptosis in H9C2 cells. No evident changes
were observed in the GPER-knockout H9C2 cells.
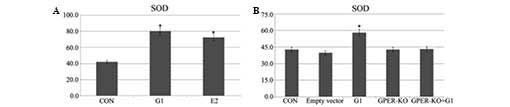
Pretreatment with G1 and E2 increases SOD
levels in H9C2 cells following I/R
Application of G1 or E2 5 min prior to ischemia
induced an increase in SOD levels in the H9C2 cells subjected to 20
min of ischemia followed by 120 min of reperfusion (Fig. 4A). In the GPER-knockout H9C2 cells,
the increase was not evident (Fig.
4B).
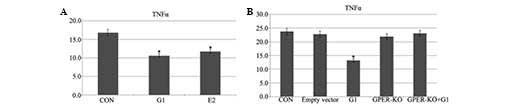
G1 and E2 decrease levels of TNF-α
following I/R
The ELISA demonstrated that administration of G1 or
E2 prior to ischemia-reperfusion decreased the levels of TNF-α as
compared with the control group (Fig.
5A). There were no further effects in the GPER-knockout H9C2
cells (Fig. 5B).
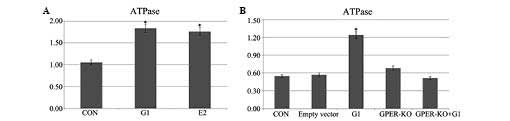
G1 and E2 increase levels of ATPase
following I/R
The administration of G1 or E2 in H9C2 cells induced
elevated ATPase levels following 20 min of ischemia and 120 min of
reperfusion (Fig. 6A). In the
GPER-knockout H9C2 cells, this elevation was no evident (Fig. 6B).
Discussion
The present study reported that the activation of
GPER by the specific agonist G1 protects the heart against I/R
injury, inhibiting cardiocyte apoptosis.
GPER has been demonstrated to be localized in the
endoplasmic reticulum (8,15) and plasma membrane (16) of reproductive organs, uterus and
mammary glands, as well as in hippocampal regions (17). The results of the present study
demonstrated the protection of H9C2 cells via activation of GPER.
Haas et al (18) concluded
that GPER contributes to the regulation of blood pressure and
vascular tone, suggesting the possibility that a number of the
known vasculoprotective effects of estrogen involve GPER
activation. Jean and Mansoureh (19) reported the acute G1 treatment
significantly reduced the infarct size following
ischemia-reperfusion in isolated hearts from male mice. In the
present study, it was identified that GPER activation inhibited
cardiocyte apoptosis, significantly increased SOD, ATP and
decreased the TNF-α expression level following
ischemia-reperfusion. Additionally, the G1-mediated effects were
eradicated in H9C2 cells. Taken together, these results provide
evidence that GPER is the primary receptor responsible for
long-term cellular changes leading to cardioprotection in H9C2
cells. Therefore the cardioprotective action of estrogen may
involve the activation of GPER.
Besides activation by E2, each of these ER isoforms
has specific pharmacological agonists. Bopassa et al
(19) have demonstrated that acute
G1 treatment considerably improves the recovery of cardiac function
following ischemia-reperfusion. Similarly, a recent study
demonstrated that G1 treatment of isolated rat hearts is
cardioprotective during ischemia-reperfusion (13).
Clinically, myocardial ischemia-reperfusion is an
acute and severe injury and the extensive apoptosis and necrosis of
cardiomyocytes at the earliest stage of reperfusion accounts for
the majority of clinical manifestations (20). For a long time, necrosis was
regarded as the sole cause of cell death in myocardial
ischemia-reperfusion injury. However, a recent study indicated that
apoptosis also has an important role in the process of
cardiomyocyte damage (21). Bcl-2
and Bax are two proteins that are directly involved in apoptosis
signaling. Therefore, in the present study, the contribution of
Bcl-2 and Bax to myocardial cell apoptosis induced by I/R was
examined. The mRNA and protein levels of Bcl-2 were evaluated 120
min following reperfusion subsequent to pretreatment with E2 or G1;
however, the mRNA and protein expression of Bax decreased 120 min
following reperfusion. The above cells were stained with Hoechst
33258 for detecting apoptotic cells. Compared with the cells
subjected to I/R, pretreatment with E2 and G1 inhibited cell
apoptosis. To confirm that the G1-induced effects were due to the
specific activation of GPER, G1 was administered 5 min prior to
ischemia in GPER-knockout H9C2 cells. No evident effects were found
in the GPER-knockout H9C2 cells.
It is well established that the activation of SOD
during I/R has an important role in the modulation of the
cardioprotection. Liesa et al (22) reported that increasing SOD shortly
prior to the onset of ischemia represents an improved strategy to
improve the functional recovery from I/R. The present study
demonstrated an increase in SOD levels induced by administering G1
and E2 in H9C2 cells prior to ischemia-reperfusion. In the
GPER-knockout H9C2 cells, no elevation was identified. These data
indicated that GPER activation by G1 induced cardioprotection.
Despite the appearance of numerous cytokines
following myocardial ischemia, the elevation of TNF-α expression
was shown to only be apparent during reperfusion (23,24).
TNF-α may have an important role during the activation of the
neutrophilic granulocytes (12).
Pro-inflammatory cytokines, including TNF-α, have emerged as
significant contributors to myocardial dysfunction (25). In the present study, it was
demonstrated that E2 and G1 treatment lead to the degradation of
TNF-α following I/R. By contrast, G1 had no evident effects on the
GPER-knockout H9C2 cells.
It was also identified that ATPase expression was
enhanced in E2- and G1-pretreated H9C2 cells following I/R, and
this effect of G1 was eliminated in GPER-knockout H9C2 cells.
In conclusion, the present study demonstrated that
GPER activation protected cadiocytes following
ischemia-reperfusion, which was evident from the inhibition of
apoptosis, elevation of SOD and ATPase as well as reduction of
TNF-α. These results enhance the understanding of the GPER
activation-induced cardioprotection following ischemia-reperfusion.
Additionally, the present study offers a potentially unique therapy
in terms of a selective estrogen receptor modulator that confers
cardio-protection without the increased risk for cancer. However,
the mechanism through which the activation of GPER inhibits
cardiocyte apoptosis remains to be investigated.
Acknowledgments
This study was supported financially by Dr Jiahong
Xia.
References
|
1
|
Murphy E and Steenbergen C:
Cardioprotection in females: a role for nitric oxide and altered
gene expression. Heart Fail Rev. 12:293–300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grohé C, Kahlert S, Löbbert K, et al:
Cardiac myocytes and fibroblasts contain functional estrogen
receptors. FEBS Lett. 416:107–112. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lizotte E, Grandy SA, Tremblay A, Allen BG
and Fiset C: Expression, distribution and regulation of sex steroid
hormone receptors in mouse heart. Cell Physiol Biochem. 23:75–86.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deschamps AM and Murphy E: Activation of a
novel estrogen receptor, GPER, is cardioprotective in male and
female rats. Am J Physiol Heart Circ Physiol. 297:H1806–H1813.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bopassa JC, Eghbali M, Toro L and Stefani
E: A novel estrogen receptor GPER inhibits mitochondria
permeability transition pore opening and protects the heart against
ischemia-reperfusion injury. Am J Physiol. 98:H16–H23. 2010.
|
|
6
|
Albanito L, Madeo A, Lappano R, et al: G
protein-coupled receptor 30 (GPR30) mediates gene expression
changes and growth response to 17beta-estradiol and selective GPR30
ligand G-1 in ovarian cancer cells. Cancer Res. 67:1859–1866. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carmeci C, Thompson DA, Ring HZ, Francke U
and Weigel RJ: Identification of a gene (GPR30) with homology to
the G-protein-coupled receptor superfamily associated with estrogen
receptor expression in breast cancer. Genomics. 45:607–617. 1997.
View Article : Google Scholar
|
|
8
|
Otto C, Rohde-Schulz B, Schwarz G, et al:
G protein-coupled receptor 30 localizes to the endoplasmic
reticulum and is not activated by estradiol. Endocrinology.
149:4846–4856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shanmuganathan S, Hausenloy DJ, Duchen MR
and Yellon DM: Mitochondrial permeability transition pore as a
target for cardioprotection in the human heart. Am J Physiol Heart
Circ Physiol. 289:H237–H242. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bopassa JC, Eghbali M, Toro L and Stefani
E: A novel estrogen receptor GPER inhibits mitochondria
permeability transition pore opening and protects the heart against
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
298:H16–H23. 2010. View Article : Google Scholar :
|
|
11
|
Yuan SM and Jing H: Insights into the
monomers and single drugs of Chinese herbal medicine on myocardial
preservation. Afr J Tradit Complement Altern Med. 8:104–127.
2011.
|
|
12
|
Hu BJ, Chen JG, Xu XH, Chen Yc and Zhu JZ:
Experimental study on the cardiomyocyte apoptosis in the early
myocardial ischemia. Chin J Forens Med. 17:349–351. 2001.
|
|
13
|
Deschamps AM and Murphy E: Activation of a
novel estrogen receptor, GPER, is cardioprotective in male and
female rats. Am J Physiol Heart Circ Physiol. 297:H1806–H1813.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bologa CG, Revankar CM, Young SM, et al:
Virtual and biomolecular screening converge on a selective agonist
for GPR30. Nat Chem Biol. 2:207–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Revankar CM, Cimino DF, Sklar LA,
Arterburn JB and Prossnitz ER: A transmembrane intracellular
estrogen receptor mediates rapid cell signaling. Science.
307:1625–1630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Filardo E, Quinn J, Pang Y, et al:
Activation of the novel estrogen receptor G protein-coupled
receptor 30 (GPR30) at the plasma membrane. Endocrinology.
148:3236–3245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsuda K, Sakamoto H, Mori H, et al:
Expression and intracellular distribution of the G protein-coupled
receptor 30 in rat hippocampal formation. Neurosci Lett. 441:94–99.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haas E, Bhattacharya I, Brailoiu E, et al:
Regulatory role of G protein-coupled estrogen receptor for vascular
function and obesity. Circ Res. 104:288–291. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bopassa JC, Eghbali M, Toro L and Stefani
E: A novel estrogen receptor GPER inhibits mitochondria
permeability transition pore opening and protects the heart against
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
298:H16–H23. 2010. View Article : Google Scholar :
|
|
20
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ishihara Y and Shimamoto N: Sulfaphenazole
attenuates myocardial cell apoptosis accompanied with cardiac
ischemia-reperfusion by suppressing the expression of BimEL and
Noxa. J Pharmacol Sci. 119:251–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liesa M, Luptak I, Qin F, et al:
Mitochondrial transporter ATP binding cassette mitochondrial
erythroid is a novel gene required for cardiac recovery after
ischemia/reperfusion. Circulation. 124:806–813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hansson GK: Immune and inflammatory
mechanisms in the development of atherosclerosis. Br Heart J.
69(Suppl): S38–S41. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herskowitz A, Choi S, Ansari AA and
Wesselingh S: Cytokine mRNA expression in postischemic/reperfusion
myocardium. Am J Pathol. 146:419–428. 1995.PubMed/NCBI
|
|
25
|
Feldman AM, Combes A, Wagner D, Kadakomi
T, Kubota T, et al: The role of tumor necrosis factor in the
pathophysiology of heart failure. J Am Coll Cardiol. 35:537–544.
2000. View Article : Google Scholar : PubMed/NCBI
|